
DNA Methylation of Igf2r Promoter CpG Island 2 Governs Cis-Acting Inheritance and Gene Dosage in Equine Hybrids



Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preparation
2.2. Genomic DNA and RNA Extraction
2.3. Single-Nucleotide Variant (SNV) Identification
2.3.1. SNV Analysis
2.3.2. Experimental SNV Validation
2.4. Allele-Specific Expression (ASE) Quantification
2.5. DNA Methylation Analysis
2.5.1. CpG Island Prediction and Bisulfite Conversion
2.5.2. Bisulfite PCR and Cloning
2.6. Absolute Quantification of Igf2r in Tissues
2.7. Statistical Analysis
3. Results
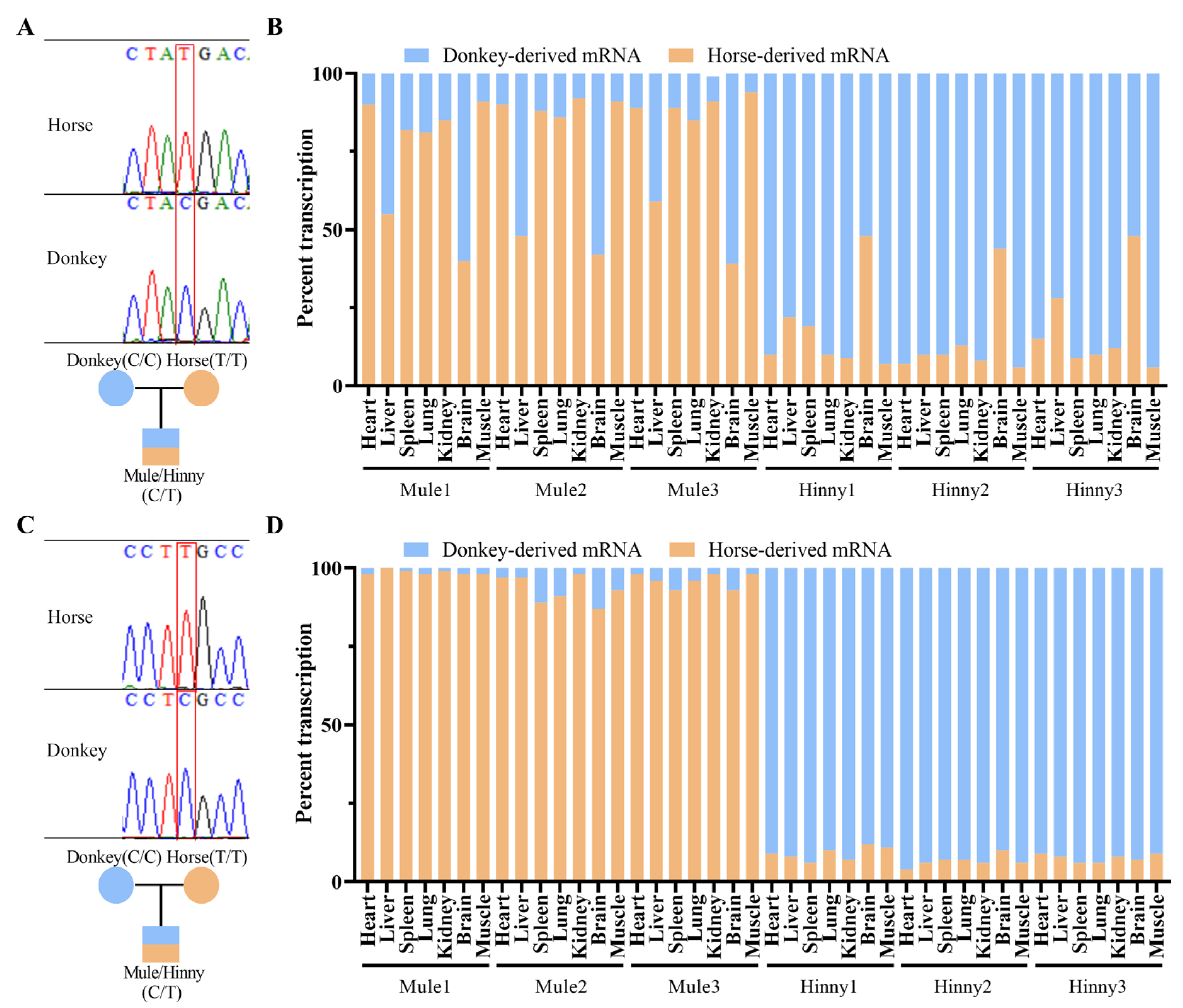
3.1. Igf2r Is Maternally Allele-Specifically Expressed in Adult Tissues of Mule and Hinny, Except for in the Brain of Mule and Hinny and the Liver of Mule
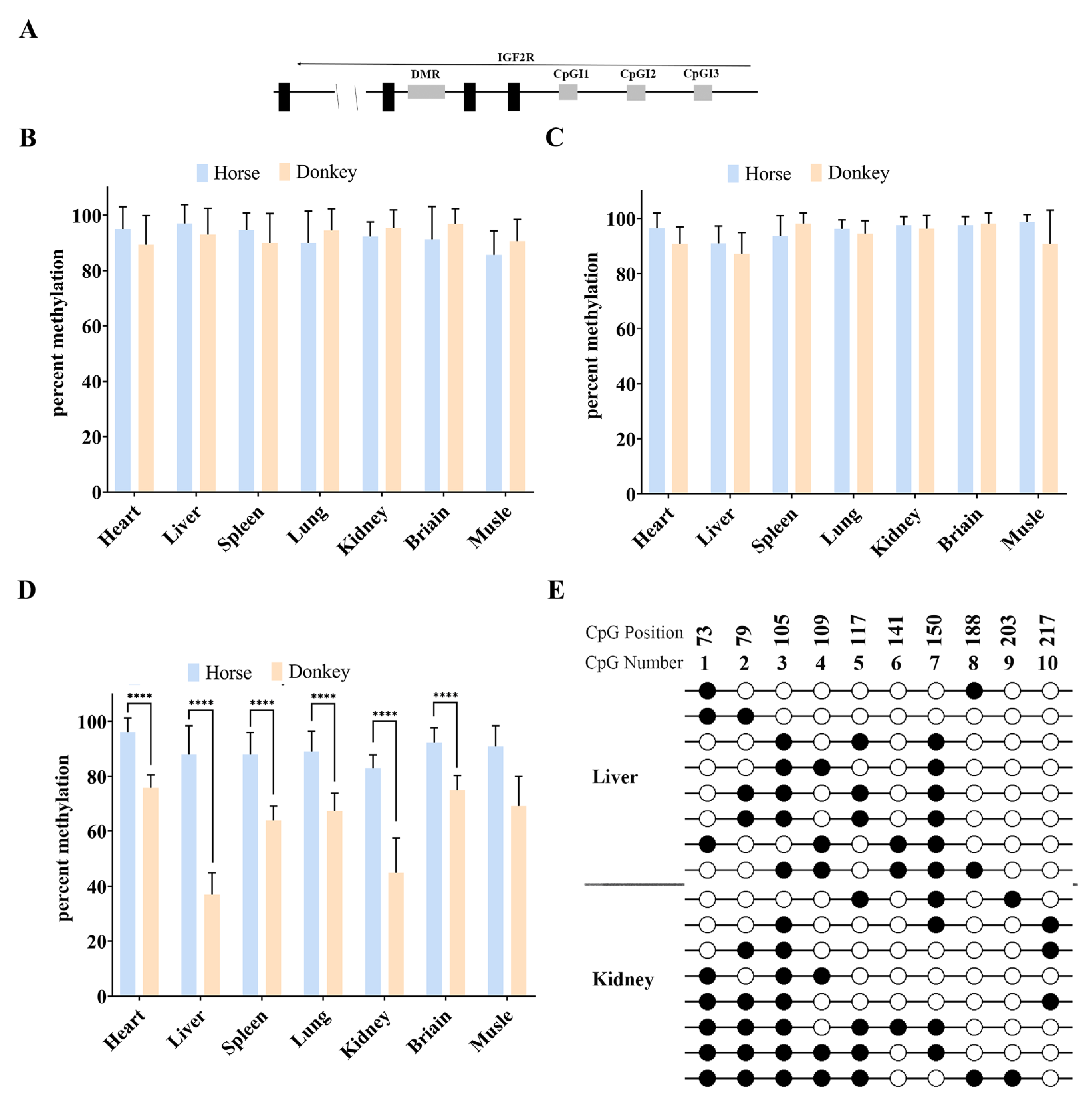
3.2. There Is No DMR in the Equine Promoter Region of Igf2r
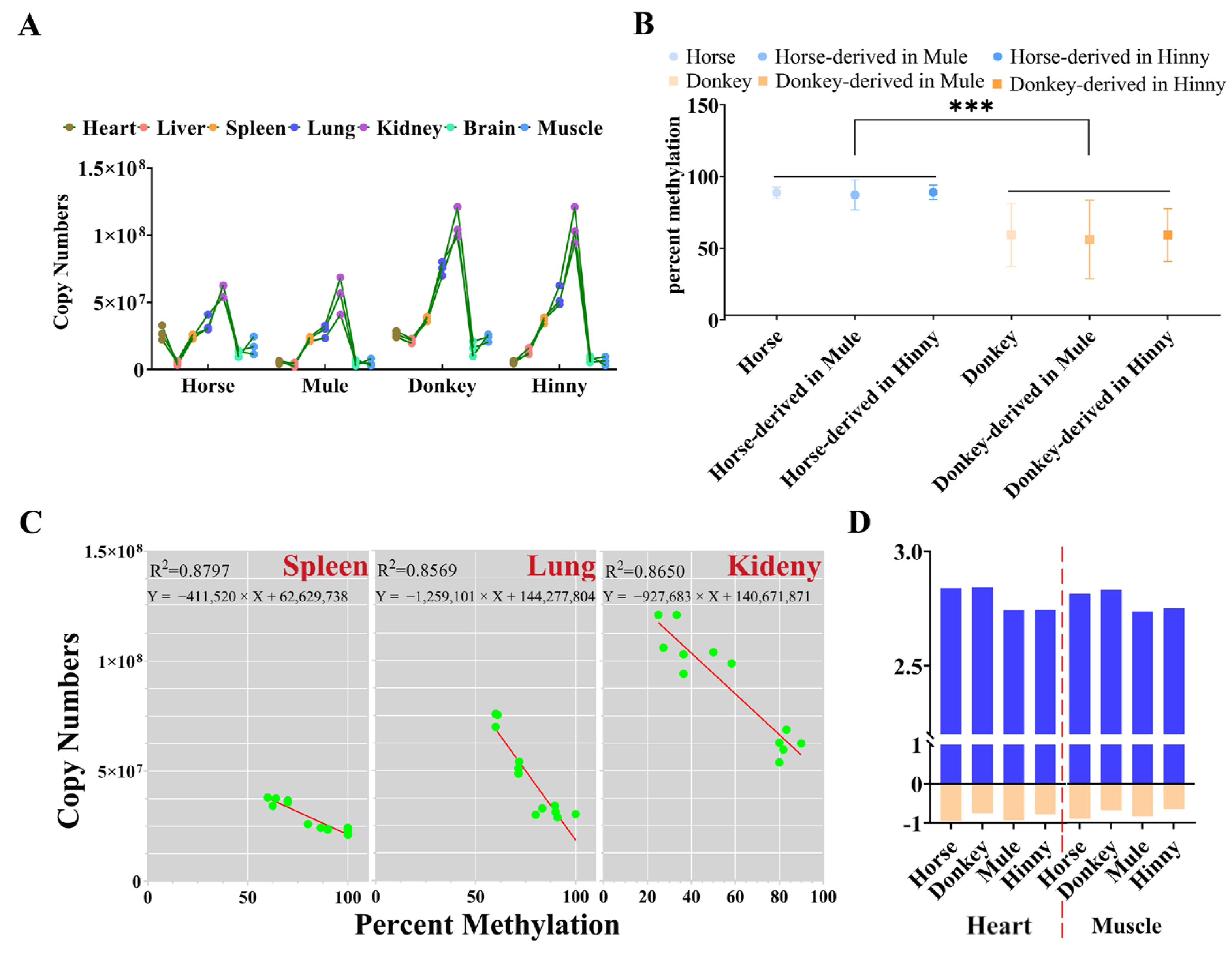
3.3. Cis-Acting Sequences Govern DNA Methylation Inheritance in Equines
3.4. Promoter CpGI2 DNA Methylation Drives Igf2r Expression Level in a Tissue-Dependent Manner
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schuff, M.; Strong, A.D.; Welborn, L.K.; Ziermann-Canabarro, J.M. Imprinting as Basis for Complex Evolutionary Novelties in Eutherians. Biology 2024, 13, 682. [Google Scholar] [CrossRef] [PubMed]
- Barlow, D.P.; Bartolomei, M.S. Genomic imprinting in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a018382. [Google Scholar] [CrossRef] [PubMed]
- Surani, M.A.; Barton, S.C.; Norris, M.L. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 1984, 308, 548–550. [Google Scholar] [CrossRef]
- Barton, S.C.; Surani, M.A.; Norris, M.L. Role of paternal and maternal genomes in mouse development. Nature 1984, 311, 374–376. [Google Scholar] [CrossRef]
- Qu, W.; Hashimoto, S.; Shimada, A.; Nakatani, Y.; Ichikawa, K.; Saito, T.L.; Ogoshi, K.; Matsushima, K.; Suzuki, Y.; Sugano, S.; et al. Genome-wide genetic variations are highly correlated with proximal DNA methylation patterns. Genome Res. 2012, 22, 1419–1425. [Google Scholar] [CrossRef]
- Yang, Y.L.; Fan, X.H.; Yan, J.Y.; Chen, M.Y.; Zhu, M.; Tang, Y.J.; Liu, S.Y.; Tang, Z.L. A comprehensive epigenome atlas reveals DNA methylation regulating skeletal muscle development. Nucleic Acids Res. 2021, 49, 1313–1329. [Google Scholar] [CrossRef]
- Vuu, Y.M.; Roberts, C.T.; Rastegar, M. MeCP2 Is an Epigenetic Factor That Links DNA Methylation with Brain Metabolism. Int. J. Mol. Sci. 2023, 24, 4218. [Google Scholar] [CrossRef]
- Hughes, J.; Surakhy, M.; Can, S.; Ducker, M.; Davies, N.; Szele, F.; Bühnemann, C.; Carter, E.; Trikin, R.; Crump, M.P.; et al. Maternal transmission of an Igf2r domain 11: IGF2 binding mutant allele (Igf2rI1565A) results in partial lethality, overgrowth and intestinal adenoma progression. Sci. Rep. 2019, 9, 11388. [Google Scholar] [CrossRef]
- Sandovici, I.; Georgopoulou, A.; Pérez-García, V.; Hufnagel, A.; López-Tello, J.; Lam, B.Y.H.; Schiefer, S.N.; Gaudreau, C.; Santos, F.; Hoelle, K.; et al. The imprinted Igf2-Igf2r axis is critical for matching placental microvasculature expansion to fetal growth. Dev. Cell 2022, 57, 63–79.e8. [Google Scholar] [CrossRef]
- Wang, X.S.; Asgenbaatar, N.; Shen, Y.C.; Yi, M.N.; Zhao, B.L.; Ren, H.; Davshilt, T.; Ulaangerel, T.; Wang, M.; Burenbaatar, A.; et al. Lower expression of the equine maternally imprinted gene IGF2R is related to the slow proliferation of hinny embryonic fibroblast. Mol. Biol. Rep. 2023, 50, 185–192. [Google Scholar] [CrossRef]
- Gicquel, C.; Weiss, J.; Amiel, J.; Gaston, V.; Le Bouc, Y.; Scott, C.D. Epigenetic abnormalities of the mannose-6-phosphate/ IGF2 receptor gene are uncommon in human overgrowth syndromes. J. Med. Genet. 2004, 41, e4. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.H.; Jirtle, R.L.; Hoffman, A.R. Cross-species clues of an epigenetic imprinting regulatory code for the IGF2R gene. Cytogenet. Genome Res. 2006, 113, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Wutz, A.; Smrzka, O.W.; Schweifer, N.; Schellander, K.; Wagner, E.F.; Barlow, D.P. Imprinted expression of the Igf2r gene depends on an intronic CpG island. Nature 1997, 389, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Sleutels, F.; Zwart, R.; Barlow, D.P. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 2002, 415, 810–813. [Google Scholar] [CrossRef]
- Wutz, A.; Smrzka, O.W.; Barlow, D.P. Making sense of imprinting the mouse and human IGF2R loci. Novartis Found. Symp. 1998, 214, 251–259; discussion 260–263. [Google Scholar] [CrossRef]
- Yotova, I.Y.; Vlatkovic, I.M.; Pauler, F.M.; Warczok, K.E.; Ambros, P.F.; Oshimura, M.; Theussl, H.C.; Gessler, M.; Wagner, E.F.; Barlow, D.P. Identification of the human homolog of the imprinted mouse Air non-coding RNA. Genomics 2008, 92, 464–473. [Google Scholar] [CrossRef]
- Dini, P.; Kalbfleisch, T.; Uribe-Salazar, J.M.; Carossino, M.; Ali, H.E.; Loux, S.C.; Esteller-Vico, A.; Norris, J.K.; Anand, L.; Scoggin, K.E.; et al. Parental bias in expression and interaction of genes in the equine placenta. Proc. Natl. Acad. Sci. USA 2021, 118, e2006474118. [Google Scholar] [CrossRef]
- Wang, X.; Miller, D.C.; Harman, R.; Antczak, D.F.; Clark, A.G. Paternally expressed genes predominate in the placenta. Proc. Natl. Acad. Sci. USA 2013, 110, 10705–10710. [Google Scholar] [CrossRef]
- Wang, X.S.; Bou, G.; Zhang, X.Z.; Tao, L.; Shen, Y.C.; Na, R.G.; Liu, G.Q.; Ren, H.; Ren, X.J.; Song, L.J.; et al. A Fast PCR Test for the Simultaneous Identification of Species and Gender in Horses, Donkeys, Mules and Hinnies. J. Equine Vet. Sci. 2021, 102, 103458. [Google Scholar] [CrossRef]
- Feinstein, S.I.; Miller, D.A.; Ehrlich, M.; Gehrke, C.W.; Eden, L.B.; Miller, O.J. DNA methylation is not increased in mouse-human somatic cell hybrids. Biochim. Biophys. Acta 1985, 824, 336–340. [Google Scholar] [CrossRef]
- Scott, E.Y.; Mansour, T.; Bellone, R.R.; Brown, C.T.; Mienaltowski, M.J.; Penedo, M.C.; Ross, P.J.; Valberg, S.J.; Murray, J.D.; Finno, C.J. Identification of long non-coding RNA in the horse transcriptome. BMC Genom. 2017, 18, 511. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Eichten, S.R.; Hermanson, P.J.; Springer, N.M. Inheritance Patterns and Stability of DNA Methylation Variation in Maize Near-Isogenic Lines. Genetics 2014, 196, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, Y.; Kayashima, T.; Soejima, H.; Kinoshita, A.; Yoshiura, K.; Matsumoto, N.; Ohta, T.; Urano, T.; Masuzaki, H.; Ishimaru, T.; et al. Neuron-specific relaxation of Igf2r imprinting is associated with neuron-specific histone modifications and lack of its antisense transcript Air. Hum. Mol. Genet. 2005, 14, 2511–2520. [Google Scholar] [CrossRef] [PubMed]
- Pervjakova, N.; Kasela, S.; Morris, A.P.; Kals, M.; Metspalu, A.; Lindgren, C.M.; Salumets, A.; Mägi, R. Imprinted genes and imprinting control regions show predominant intermediate methylation in adult somatic tissues. Epigenomics 2016, 8, 789–799. [Google Scholar] [CrossRef]
- Sharp, A.J.; Stathaki, E.; Migliavacca, E.; Brahmachary, M.; Montgomery, S.B.; Dupre, Y.; Antonarakis, S.E. DNA methylation profiles of human active and inactive X chromosomes. Genome Res. 2011, 21, 1592–1600. [Google Scholar] [CrossRef]
- Vilain, A.; Bernardino, J.; Gerbault-Seureau, M.; Vogt, N.; Niveleau, A.; Lefrançois, D.; Malfoy, B.; Dutrillaux, B. DNA methylation and chromosome instability in lymphoblastoid cell lines. Cytogenet. Cell Genet. 2000, 90, 93–101. [Google Scholar] [CrossRef]
- Santoro, F.; Mayer, D.; Klement, R.M.; Warczok, K.E.; Stukalov, A.; Barlow, D.P.; Pauler, F.M. Imprinted Igf2r silencing depends on continuous Airn lncRNA expression and is not restricted to a developmental window. Development 2013, 140, 1184–1195. [Google Scholar] [CrossRef]
- Springer, M.S.; Foley, N.M.; Brady, P.L.; Gatesy, J.; Murphy, W.J. Evolutionary Models for the Diversification of Placental Mammals Across the KPg Boundary. Front. Genet. 2019, 10, 1241. [Google Scholar] [CrossRef]
- Ge, D.Y.; Feijó, A.; Wen, Z.X.; Abramov, A.V.; Lu, L.; Cheng, J.L.; Pan, S.K.; Ye, S.C.; Xia, L.; Jiang, X.L.; et al. Demographic History and Genomic Response to Environmental Changes in a Rapid Radiation of Wild Rats. Mol. Biol. Evol. 2021, 38, 1905–1923. [Google Scholar] [CrossRef]
- Marra, N.J.; Richards, V.P.; Early, A.; Bogdanowicz, S.M.; Bitar, P.D.P.; Stanhope, M.J.; Shivji, M.S. Comparative transcriptomics of elasmobranchs and teleosts highlight important processes in adaptive immunity and regional endothermy. BMC Genom. 2017, 18, 87. [Google Scholar] [CrossRef]
- Vrana, P.B.; Guan, X.J.; Ingram, R.S.; Tilghman, S.M. Genomic imprinting is disrupted in interspecific Peromyscus hybrids. Nat. Genet. 1998, 20, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.P.; Parameswaran, S.; Cai, L.; Elston, S.; Pham, C.; Barski, A.; Weirauch, M.T.; Ji, H. TET1 regulates responses to house dust mite by altering chromatin accessibility, DNA methylation, and gene expression in airway epithelial cells. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Cao, S.; Chen, K.; Lu, K.; Chen, S.; Zhang, X.; Shen, C.; Zhu, S.; Niu, Y.; Fan, L.; Chen, Z.J.; et al. Asymmetric variation in DNA methylation during domestication and de-domestication of rice. Plant Cell 2023, 35, 3429–3443. [Google Scholar] [CrossRef]
- Cusack, M.; King, H.W.; Spingardi, P.; Kessler, B.M.; Klose, R.J.; Kriaucionis, S. Distinct contributions of DNA methylation and histone acetylation to the genomic occupancy of transcription factors. Genome Res. 2020, 30, 1393–1406. [Google Scholar] [CrossRef]
- Kalscheuer, V.M.; Mariman, E.C.; Schepens, M.T.; Rehder, H.; Ropers, H.H. The insulin-like growth factor type-2 receptor gene is imprinted in the mouse but not in humans. Nat. Genet. 1993, 5, 74–78. [Google Scholar] [CrossRef]
- Wang, X.Y.; Moazed, D. DNA sequence-dependent epigenetic inheritance of gene silencing and histone H3K9 methylation. Science 2017, 356, 88–91. [Google Scholar] [CrossRef]
- Xiong, X.; Geden, C.J.; Tan, Y.J.; Zhang, Y.; Zhang, D.P.; Werren, J.H.; Wang, X. Genome Structure, Evolution, and Host Shift of. Biol. Basel 2024, 13, 952. [Google Scholar] [CrossRef]
- Lee, J.; Inoue, K.; Ono, R.; Ogonuki, N.; Kohda, T.; Kaneko-Ishino, T.; Ogura, A.; Ishino, F. Erasing genomic imprinting memory in mouse clone embryos produced from day 11.5 primordial germ cells. Development 2002, 129, 1807–1817. [Google Scholar] [CrossRef]
- Castora, F.J.; Arnheim, N.; Simpson, M.V. Mitochondrial DNA polymorphism: Evidence that variants detected by restriction enzymes differ in nucleotide sequence rather than in methylation. Proc. Natl. Acad. Sci. USA 1980, 77, 6415–6419. [Google Scholar] [CrossRef]
- de Oliveira, N.F.P.; Persuhn, D.C.; Dos Santos, M. Can Global DNA Methylation Be Influenced by Polymorphisms in Genes Involved in Epigenetic Mechanisms? A Review. Genes 2024, 15, 1504. [Google Scholar] [CrossRef]
- Kieffer-Kwon, K.R.; Tang, Z.H.; Mathe, E.; Qian, J.S.; Sung, M.H.; Li, G.L.; Resch, W.; Baek, S.; Pruett, N.; Grontved, L.; et al. Interactome Maps of Mouse Gene Regulatory Domains Reveal Basic Principles of Transcriptional Regulation. Cell 2013, 155, 1507–1520. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.H.; Zhang, C.R.; Bi, X.M.; Xu, J.K.; Guo, S.N.; Li, P.H.; Shen, Y.T.; Cai, J.L.; Zhang, N.H.; Tian, G.H.; et al. Mapping enhancer and chromatin accessibility landscapes charts the regulatory network of Alzheimer’s disease. Comput. Biol. Med. 2024, 168, 107802. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Liu, S.X. CRISPR/dCas9-Tet1-Mediated DNA Methylation Editing. Bio-Protocol 2024, 14, e4976. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Primer Sequence (5′-3′ Orientation) | Annealing Temperature (°C) | Product Size (bp) |
|---|---|---|---|
| CpGI1 | F: GGGGGAGGGTTTTTAAGG | 57 | 321 |
| R: AATAATAACAAATTTCAAAACTAACC | |||
| CpGI2 | F: AGATAATTTGTGTAAGAGGGAATATTTT | 60 | 259 |
| R: CCAATCTACCCCTAATCTATCCTTCCTA | |||
| CpGI3 | F: GTGGTTTAGGGGTGGAGATTA | 54 | 362 |
| R: TATTCTCCAAACTACCCCCTTCC | |||
| Intron 2 | F: GGTTATTTTGTTTTTGGGTTTGTATAG | 58 | 341 |
| R: TCTCTTCTTAAAAATCAAATCACAT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Shen, Y.; Ren, H.; Yi, M.; Bou, G. DNA Methylation of Igf2r Promoter CpG Island 2 Governs Cis-Acting Inheritance and Gene Dosage in Equine Hybrids. Biology 2025, 14, 678. https://doi.org/10.3390/biology14060678
Wang X, Shen Y, Ren H, Yi M, Bou G. DNA Methylation of Igf2r Promoter CpG Island 2 Governs Cis-Acting Inheritance and Gene Dosage in Equine Hybrids. Biology. 2025; 14(6):678. https://doi.org/10.3390/biology14060678
Chicago/Turabian StyleWang, Xisheng, Yingchao Shen, Hong Ren, Minna Yi, and Gerelchimeg Bou. 2025. "DNA Methylation of Igf2r Promoter CpG Island 2 Governs Cis-Acting Inheritance and Gene Dosage in Equine Hybrids" Biology 14, no. 6: 678. https://doi.org/10.3390/biology14060678
APA StyleWang, X., Shen, Y., Ren, H., Yi, M., & Bou, G. (2025). DNA Methylation of Igf2r Promoter CpG Island 2 Governs Cis-Acting Inheritance and Gene Dosage in Equine Hybrids. Biology, 14(6), 678. https://doi.org/10.3390/biology14060678