
Research on Plant RNA-Binding Protein Prediction Method Based on Improved Ensemble Learning



Simple Summary
Abstract
1. Introduction
2. Materials and Methods
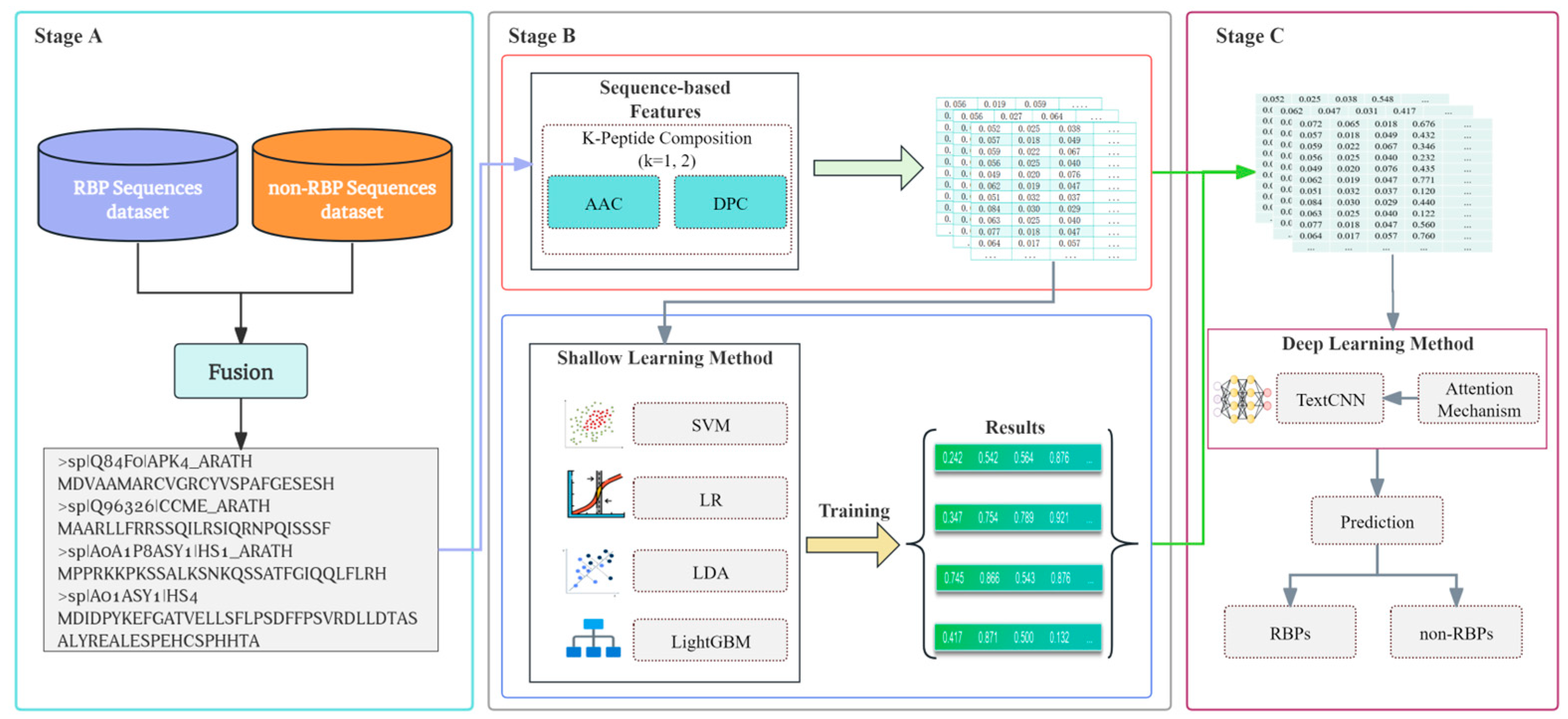
2.1. The Overall Framework of the Prediction Method
2.2. Dataset
2.3. Protein Sequence Encoding
2.4. Shallow Learning Method
2.5. Ensemble Learning Method
2.6. Deep Learning Method
2.7. Attention Mechanism
2.8. Evaluation Metrics
3. Results and Discussion
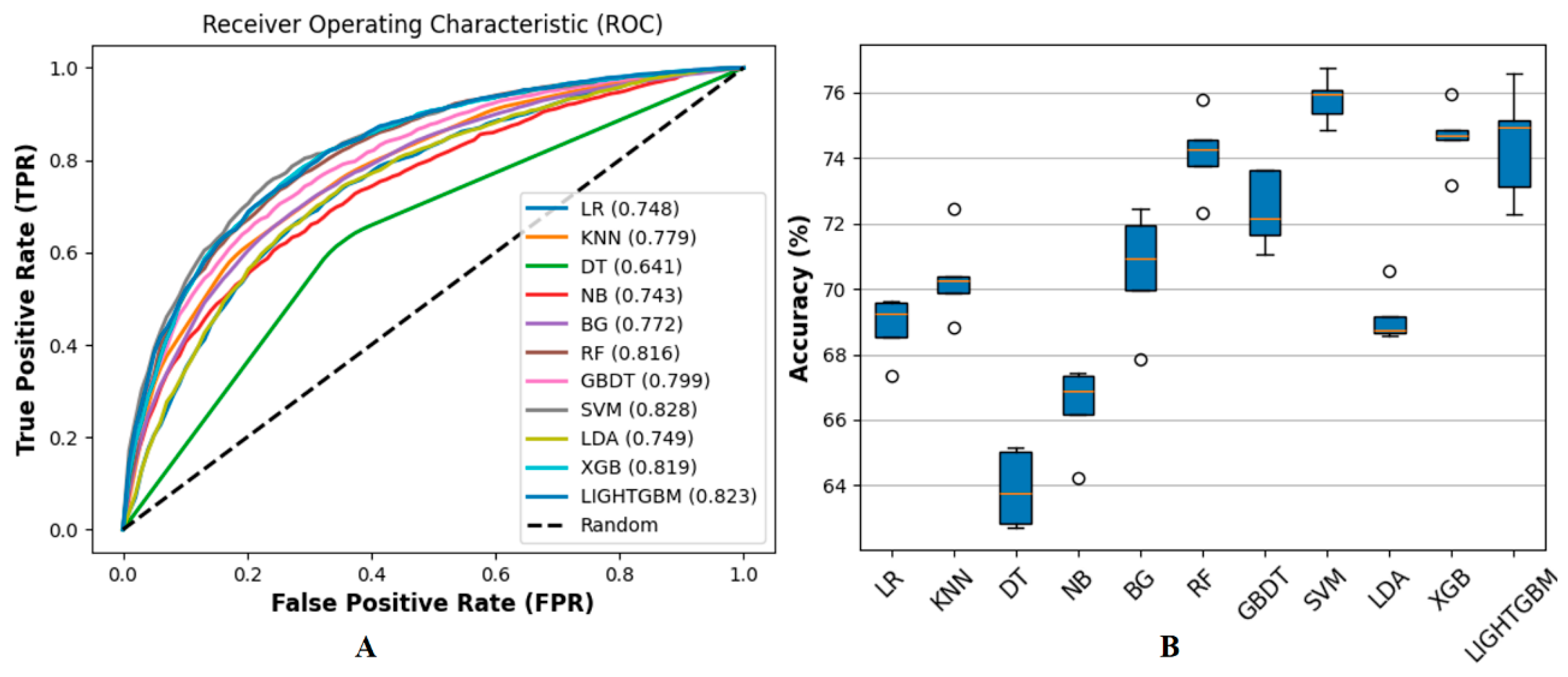
3.1. Performance on Benchmark Dataset
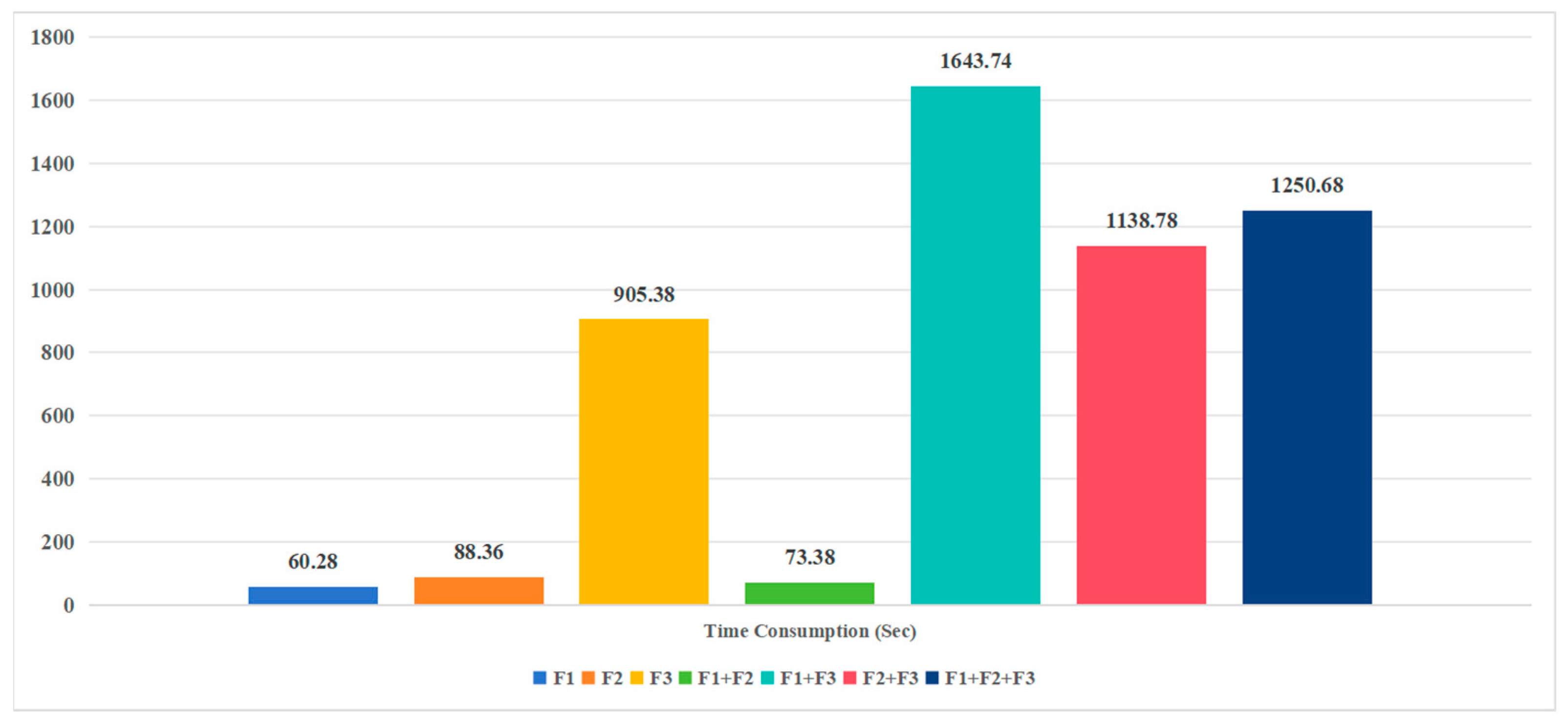
3.2. Performance on Feature Combinations
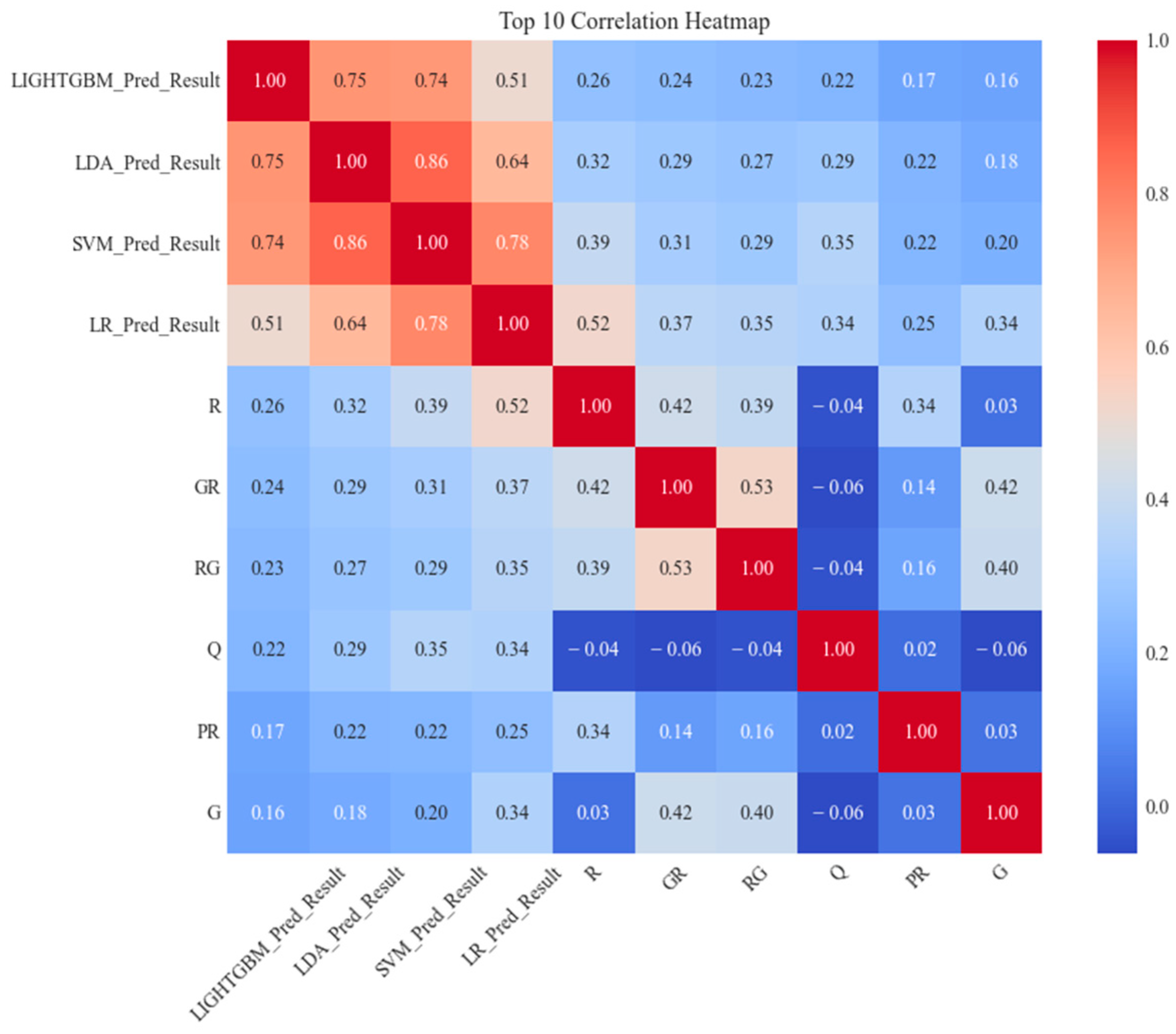
3.3. Performance on Ensemble Learning Framework
3.4. Performance on Independent Dataset
3.5. Comparison with State-of-the-Art Methods
3.6. Comparison with Original TextCNN
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| RBPs | RNA-Binding Proteins |
| GO | Gene Ontology |
| PSSM | Position-Specific Scoring Matrix |
| KPC | K-Peptide Composition |
| AAC | Amino Acid Composition |
| DPC | Di-Peptide Composition |
| CD-HIT | Cluster Database at High Identity with Tolerance |
| SVM | Support Vector Machine |
| LR | Logistic Regression |
| LDA | Linear Discriminant Analysis |
| LightGBM | Light Gradient Boosting Machine |
| TextCNN | Text Convolutional Neural Network |
| CNN | Convolutional Neural Network |
| RF | Random Forest |
| GBDT | Gradient Boosting Decision Tree |
| XGB | Extreme Gradient Boosting |
| KNN | K-Nearest Neighbors |
| DT | Decision Tree |
| NB | Naive Bayes |
| BG | Bagging |
| ACC | Accuracy |
| AUC | Area Under Curve |
| MCC | Matthews Correlation Coefficient |
| SN | Sensitivity |
| SP | Specificity |
| XGB-VIM | XGB Variable Importance Measures |
| LGBM-VIM | LightGBM Variable Importance Measure |
References
- Koletsou, E.; Huppertz, I. RNA-binding proteins as versatile metabolic regulators. Npj Metab. Health Disease 2025, 3, 1. [Google Scholar] [CrossRef]
- Hogan, D.J.; Riordan, D.P.; Gerber, A.P.; Herschlag, D.; Brown, P.O. Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLoS Biol. 2008, 6, e255. [Google Scholar] [CrossRef]
- Corley, M.; Burns, M.C.; Yeo, G.W. How RNA-binding proteins interact with RNA: Molecules and mechanisms. Mol. Cell 2020, 78, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, M.; Kim, J.H.; Kim, J.A.; Lee, S.I. Plant RNA binding proteins as critical modulators in drought, high salinity, heat, and cold stress responses: An updated overview. Int. J. Mol. Sci. 2021, 22, 6731. [Google Scholar] [CrossRef]
- Tao, Y.; Zhang, Q.; Wang, H.; Yang, X.; Mu, H. Alternative splicing and related RNA binding proteins in human health and disease. Signal Transduct. Target. Ther. 2024, 9, 26. [Google Scholar] [CrossRef]
- Gebauer, F.; Schwarzl, T.; Valcárcel, J.; Hentze, M.W. RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 2021, 22, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, E.L.; Freese, P.; Pratt, G.A.; Wang, X.; Wei, X.; Xiao, R.; Blue, S.M.; Chen, J.Y.; Cody, N.A.; Dominguez, D.; et al. A large-scale binding and functional map of human RNA-binding proteins. Nature 2020, 583, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Lorković, Z.J. Role of plant RNA-binding proteins in development, stress response and genome organization. Trends Plant Sci. 2009, 14, 229–236. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Y.; Skaggs, T.H.; Ferreira, J.F.; Chen, X.; Sandhu, D. Plant phase extraction: A method for enhanced discovery of the RNA-binding proteome and its dynamics in plants. Plant Cell 2023, 35, 2750–2772. [Google Scholar] [CrossRef]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Yan, Y.; Li, W.; Wang, S.; Huang, T. Seq-rbppred: Predicting rna-binding proteins from sequence. ACS Omega 2024, 9, 12734–12742. [Google Scholar] [CrossRef] [PubMed]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–888. [Google Scholar] [CrossRef]
- Si, J.; Cui, J.; Cheng, J.; Wu, R. Computational prediction of RNA-binding proteins and binding sites. Int. J. Mol. Sci. 2015, 16, 26303–26317. [Google Scholar] [CrossRef] [PubMed]
- Avila-Lopez, P.; Lauberth, S.M. Exploring new roles for RNA-binding proteins in epigenetic and gene regulation. Curr. Opin. Genet. Dev. 2024, 84, 102136. [Google Scholar] [CrossRef]
- Goshisht, M.K. Machine learning and deep learning in synthetic biology: Key architectures, applications, and challenges. ACS Omega 2024, 9, 9921–9945. [Google Scholar] [CrossRef]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef]
- Ray, D.; Kazan, H.; Cook, K.B.; Weirauch, M.T.; Najafabadi, H.S.; Li, X.; Gueroussov, S.; Albu, M.; Zheng, H.; Yang, A.; et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature 2013, 499, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, S. RBPPred: Predicting RNA-binding proteins from sequence using SVM. Bioinformatics 2017, 33, 854–862. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Mishra, A.; Khanal, R.; Kabir, W.U.; Hoque, T. AIRBP: Accurate identification of RNA-binding proteins using machine learning techniques. Artif. Intell. Med. 2021, 113, 102034. [Google Scholar] [CrossRef]
- Niu, M.; Wu, J.; Zou, Q.; Liu, Z.; Xu, L. rBPDL: Predicting RNA-binding proteins using deep learning. IEEE J. Biomed. Health Inform. 2021, 25, 3668–3676. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Bottou, L.; Bengio, Y.; Haffner, P. Gradient-based learning applied to document recognition. Proc. IEEE 1998, 86, 2278–2324. [Google Scholar] [CrossRef]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Pradhan, U.K.; Meher, P.K.; Naha, S.; Pal, S.; Gupta, S.; Gupta, A.; Parsad, R. RBPLight: A computational tool for discovery of plant-specific RNA-binding proteins using light gradient boosting machine and ensemble of evolutionary features. Brief. Funct. Genom. 2023, 22, 401–410. [Google Scholar] [CrossRef]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.Y. Lightgbm: A highly efficient gradient boosting decision tree. In Proceedings of the 31st International Conference on Neural Information Processing Systems (NeurIPS 2017), Long Beach, CA, USA, 4–9 December 2017; Volume 30. [Google Scholar]
- Pradhan, U.K.; Naha, S.; Das, R.; Gupta, A.; Parsad, R.; Meher, P.K. RBProkCNN: Deep learning on appropriate contextual evolutionary information for RNA binding protein discovery in prokaryotes. Comput. Struct. Biotechnol. J. 2024, 23, 1631–1640. [Google Scholar] [CrossRef]
- Sandri, M.; Zuccolotto, P. A bias correction algorithm for the Gini variable importance measure in classification trees. J. Comput. Graph. Stat. 2008, 17, 611–628. [Google Scholar] [CrossRef]
- Gribskov, M.; McLachlan, A.D.; Eisenberg, D. Profile analysis: Detection of distantly related proteins. Proc. Natl. Acad. Sci. USA 1987, 84, 4355–4358. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liu, Y.; Shi, Y.; Zhang, W.; Yang, C.; Liu, H. Deep neural networks for inferring binding sites of RNA-binding proteins by using distributed representations of RNA primary sequence and secondary structure. BMC Genom. 2020, 21, 866. [Google Scholar] [CrossRef]
- Marchese, D.; de Groot, N.S.; Lorenzo Gotor, N.; Livi, C.M.; Tartaglia, G.G. Advances in the characterization of RNA-binding proteins. Wiley Interdiscip. Rev. RNA 2016, 7, 793–810. [Google Scholar] [CrossRef]
- LaValley, M.P. Logistic regression. Circulation 2008, 117, 2395–2399. [Google Scholar] [CrossRef]
- Ye, J.; Janardan, R.; Li, Q. Two-dimensional linear discriminant analysis. In Proceedings of the Advances in Neural Information Processing Systems 17 (NIPS 2004), Vancouver, BC, Canada, 13–18 December 2004; Volume 17. [Google Scholar]
- Fan, J.; Ma, X.; Wu, L.; Zhang, F.; Yu, X.; Zeng, W. Light Gradient Boosting Machine: An efficient soft computing model for estimating daily reference evapotranspiration with local and external meteorological data. Agric. Water Manag. 2019, 225, 105758. [Google Scholar] [CrossRef]
- Lei, Z.; Dai, Y. An SVM-based system for predicting protein subnuclear localizations. BMC Bioinform. 2005, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, E.C.; Santana, K.; Josino, L.; Lima e Lima, A.H.; de Souza de Sales Júnior, C. Predicting cell-penetrating peptides using machine learning algorithms and navigating in their chemical space. Sci. Rep. 2021, 11, 7628. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, P.; Li, F.; Leier, A.; Marquez-Lago, T.T.; Wang, Y.; Webb, G.I.; Smith, A.I.; Daly, R.J.; Chou, K.C.; et al. iFeature: A python package and web server for features extraction and selection from protein and peptide sequences. Bioinformatics 2018, 34, 2499–2502. [Google Scholar] [CrossRef]
- Nakashima, H.; Nishikawa, K.; Ooi, T. The folding type of a protein is relevant to the amino acid composition. J. Biochem. 1986, 99, 153–162. [Google Scholar] [CrossRef]
- Reczko, M.; Bohr, H. The DEF data base of sequence-based protein fold class predictions. Nucleic Acids Res. 1994, 22, 3616. [Google Scholar]
- Shen, J.; Zhang, J.; Luo, X.; Zhu, W.; Yu, K.; Chen, K.; Li, Y.; Jiang, H. Predicting protein–protein interactions based only on sequences information. Proc. Natl. Acad. Sci. USA 2007, 104, 4337–4341. [Google Scholar] [CrossRef]
- Wei, L.; Xing, P.; Zeng, J.; Chen, J.; Su, R.; Guo, F. Improved prediction of protein–protein interactions using novel negative samples, features, and an ensemble classifier. Artif. Intell. Med. 2017, 83, 67–74. [Google Scholar] [CrossRef]
- Wang, L.; Brown, S.J. BindN: A web-based tool for efficient prediction of DNA and RNA binding sites in amino acid sequences. Nucleic Acids Res. 2006, 34 (Suppl. S2), W243–W248. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, Y.; Zhou, Y. Structure-based prediction of RNA-binding domains and RNA-binding sites and application to structural genomics targets. Nucleic Acids Res. 2011, 39, 3017–3025. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Friedman, J.H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd ACM Sigkdd International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13 August 2016; pp. 785–794. [Google Scholar]
- Yoon, K. Convolutional Neural Networks for Sentence Classification. In Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP), Doha, Qatar, 25–29 October 2014; Association for Computational Linguistics: Stroudsburg, PA, USA, 2024; pp. 1746–1751. [Google Scholar]
- Khurana, D.; Koli, A.; Khatter, K.; Singh, S. Natural language processing: State of the art, current trends and challenges. Multimed. Tools Appl. 2023, 82, 3713–3744. [Google Scholar] [CrossRef]
- Wei, J.; Chen, S.; Zong, L.; Gao, X.; Li, Y. Protein–RNA interaction prediction with deep learning: Structure matters. Brief. Bioinform. 2022, 23, bbab540. [Google Scholar] [CrossRef]
- Pan, X.; Shen, H.B. RNA-protein binding motifs mining with a new hybrid deep learning based cross-domain knowledge integration approach. BMC Bioinform. 2017, 18, 136. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. In Proceedings of the 31st International Conference on Neural Information Processing Systems (NeurIPS 2017), Long Beach, CA, USA, 4–9 December 2017; Volume 30. [Google Scholar]
- Zhang, S.; Zhou, J.; Hu, H.; Gong, H.; Chen, L.; Cheng, C.; Zeng, J. A deep learning framework for modeling structural features of RNA-binding protein targets. Nucleic Acids Res. 2016, 44, e32. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhu, M. A review about RNA–protein-binding sites prediction based on deep learning. IEEE Access 2020, 8, 150929–150944. [Google Scholar] [CrossRef]
- Ghazikhani, H.; Butler, G. Journal of Integrative Bioinformatics: Ion Channel Classification Through Machine Learning and Protein Language Model Embeddings; Walter de Gruyter GmbH: Berlin, Germany, 2025. [Google Scholar]
- Abuelmakarem, H.S.; Majdy, A.; Maher, G.; Khaled, H.; Emad, M.; Asem Shaker, E. Precancer Detection Based on Mutations in Codons 248 and 249 Using Decision Tree (DT) and XGBoost Deep Learning Model. Int. J. Ind. Sustain. Dev. 2025, 6, 67–77. [Google Scholar] [CrossRef]
- Khan, S.; Noor, S.; Awan, H.H.; Iqbal, S.; AlQahtani, S.A.; Dilshad, N.; Ahmad, N. Deep-ProBind: Binding protein prediction with transformer-based deep learning model. BMC Bioinform. 2025, 26, 88. [Google Scholar] [CrossRef]
- Lakshmi, P.; Manikandan, P.; Ramyachitra, D. An Improved Bagging of Machine Learning Algorithms to Predict Motif Structures from Protein-Protein Interaction Networks. IEEE Access 2025, 13, 45077–45088. [Google Scholar] [CrossRef]
- Chen, Z.; Pang, M.; Zhao, Z.; Li, S.; Miao, R.; Zhang, Y.; Feng, X.; Feng, X.; Zhang, Y.; Duan, M.; et al. Feature selection may improve deep neural networks for the bioinformatics problems. Bioinformatics 2020, 36, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Mukaka, M.M. A guide to appropriate use of correlation coefficient in medical research. Malawi Med. J. 2012, 24, 69–71. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Folds | ACC (%) | AUC (%) | MCC (%) | (%) | SN (%) | SP (%) |
|---|---|---|---|---|---|---|
| Fold 1 | 96.10 | 98.89 | 92.27 | 96.10 | 94.27 | 98.11 |
| Fold 2 | 97.30 | 99.42 | 94.64 | 97.30 | 95.90 | 98.77 |
| Fold 3 | 97.60 | 99.38 | 95.19 | 97.59 | 96.68 | 98.45 |
| Fold 4 | 97.39 | 99.57 | 94.82 | 97.39 | 96.22 | 98.59 |
| Fold 5 | 97.60 | 99.19 | 95.23 | 97.59 | 95.59 | 99.43 |
| Average | 97.20 ± 0.56 | 99.29 ± 0.23 | 94.43 ± 1.10 | 97.19 ± 0.56 | 95.73 ± 0.82 | 98.67 ± 0.44 |
| (a) | ||||||
| Feature Set | ACC (%) | AUC (%) | MCC (%) | (%) | SN (%) | SP (%) |
| D0 | 64.24 ± 2.26 | 70.42 ± 3.14 | 28.02 ± 4.94 | 64.02 ± 2.53 | 66.75 ± 5.43 | 61.02 ± 8.97 |
| D1 | 76.28 ± 1.35 | 82.61 ± 1.78 | 53.36 ± 2.82 | 76.08 ± 1.34 | 67.08 ± 2.04 | 85.38 ± 2.11 |
| D2 | 76.68 ± 1.28 | 83.74 ± 1.53 | 53.55 ± 2.57 | 76.62 ± 1.29 | 71.42 ± 2.21 | 81.87 ± 1.55 |
| D3 | 79.41 ± 1.79 | 86.40 ± 1.73 | 58.89 ± 3.58 | 79.39 ± 1.80 | 76.08 ± 2.05 | 82.72 ± 2.36 |
| D4 | 97.48 ± 0.83 | 99.39 ± 0.23 | 95.01 ± 1.62 | 97.48 ± 0.83 | 95.66 ± 1.34 | 99.31 ± 0.57 |
| (b) | ||||||
| Feature Set Comparison | p-Value (ACC) | p-Value (MCC) | ||||
| D1 vs. D0 | <0.00001 | <0.00001 | ||||
| D2 vs. D1 | 0.16304 | 0.3848 | ||||
| D3 vs. D2 | 0.00034 | 0.00022 | ||||
| D4 vs. D3 | <0.00001 | <0.00001 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Shi, Y.; Wang, Y.; Yang, X.; Li, K.; Im, S.-K.; Han, Y. Research on Plant RNA-Binding Protein Prediction Method Based on Improved Ensemble Learning. Biology 2025, 14, 672. https://doi.org/10.3390/biology14060672
Zhang H, Shi Y, Wang Y, Yang X, Li K, Im S-K, Han Y. Research on Plant RNA-Binding Protein Prediction Method Based on Improved Ensemble Learning. Biology. 2025; 14(6):672. https://doi.org/10.3390/biology14060672
Chicago/Turabian StyleZhang, Hongwei, Yan Shi, Yapeng Wang, Xu Yang, Kefeng Li, Sio-Kei Im, and Yu Han. 2025. "Research on Plant RNA-Binding Protein Prediction Method Based on Improved Ensemble Learning" Biology 14, no. 6: 672. https://doi.org/10.3390/biology14060672
APA StyleZhang, H., Shi, Y., Wang, Y., Yang, X., Li, K., Im, S.-K., & Han, Y. (2025). Research on Plant RNA-Binding Protein Prediction Method Based on Improved Ensemble Learning. Biology, 14(6), 672. https://doi.org/10.3390/biology14060672