
Elucidating the Prognostic and Therapeutic Implications of Insulin Resistance Genes in Breast Cancer: A Machine Learning-Powered Analysis

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
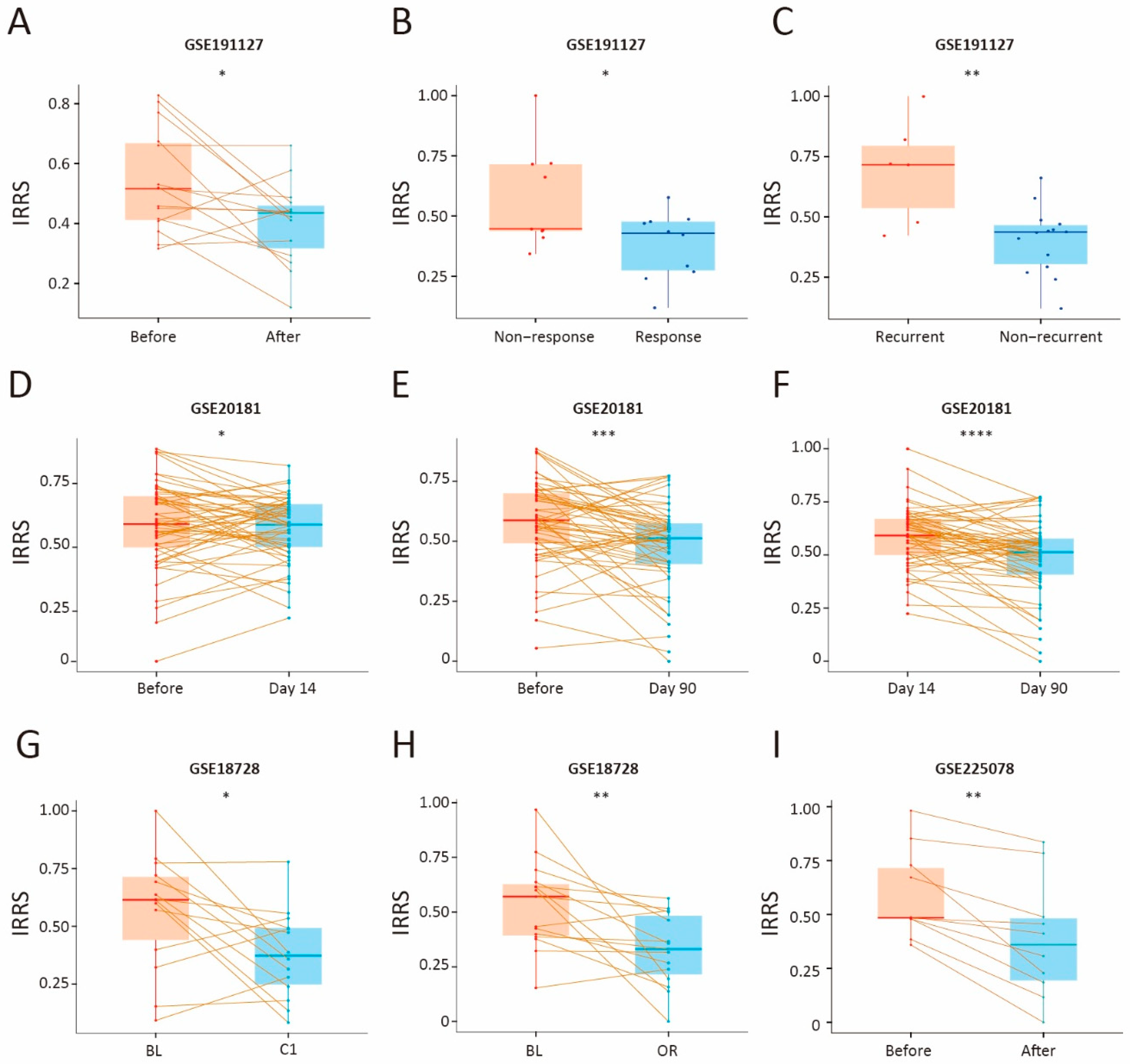
2.1. Data Collection and Processing
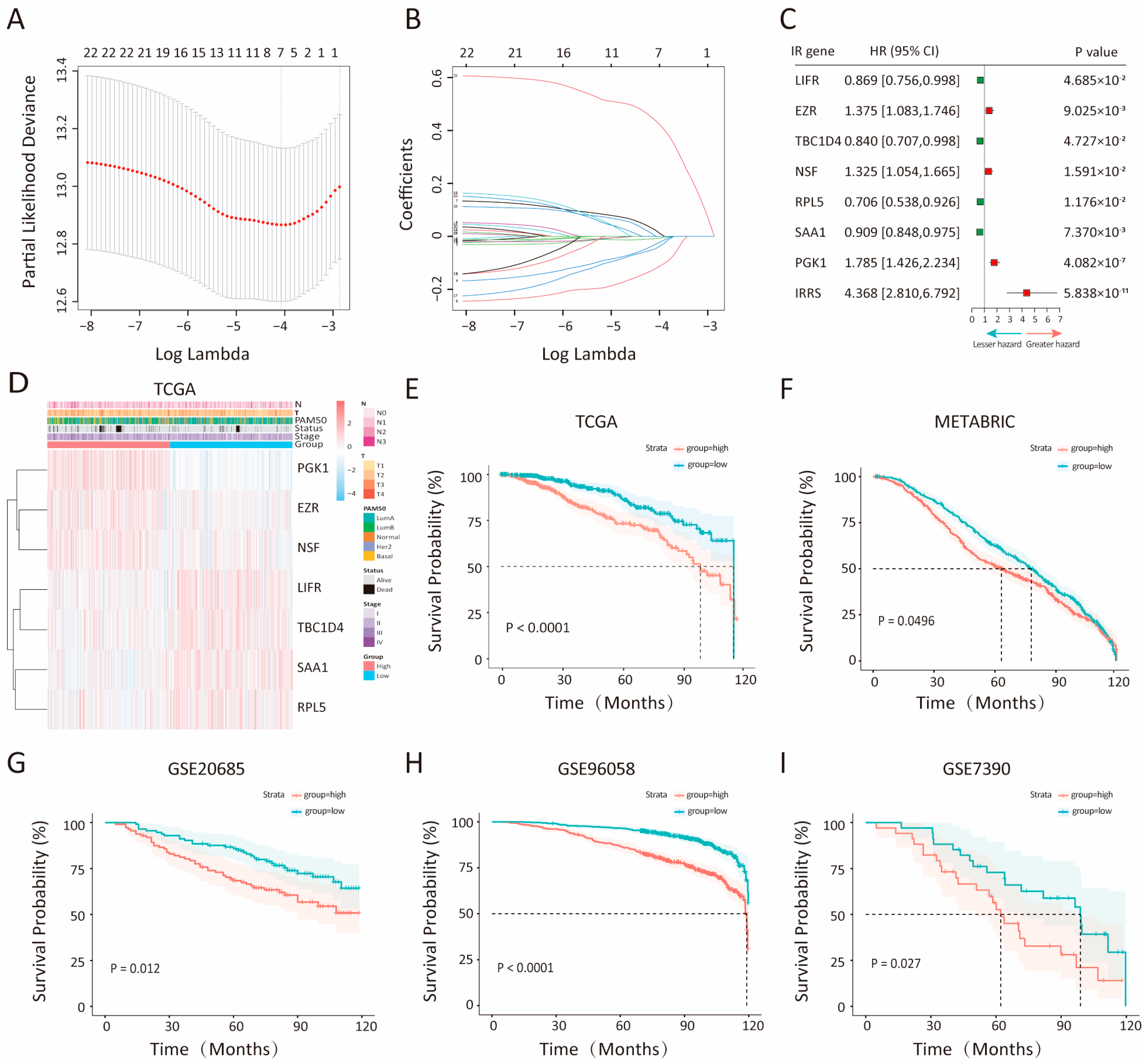
2.2. Construction and Validation of an IRG Prognostic Signature for Breast Cancer Patients
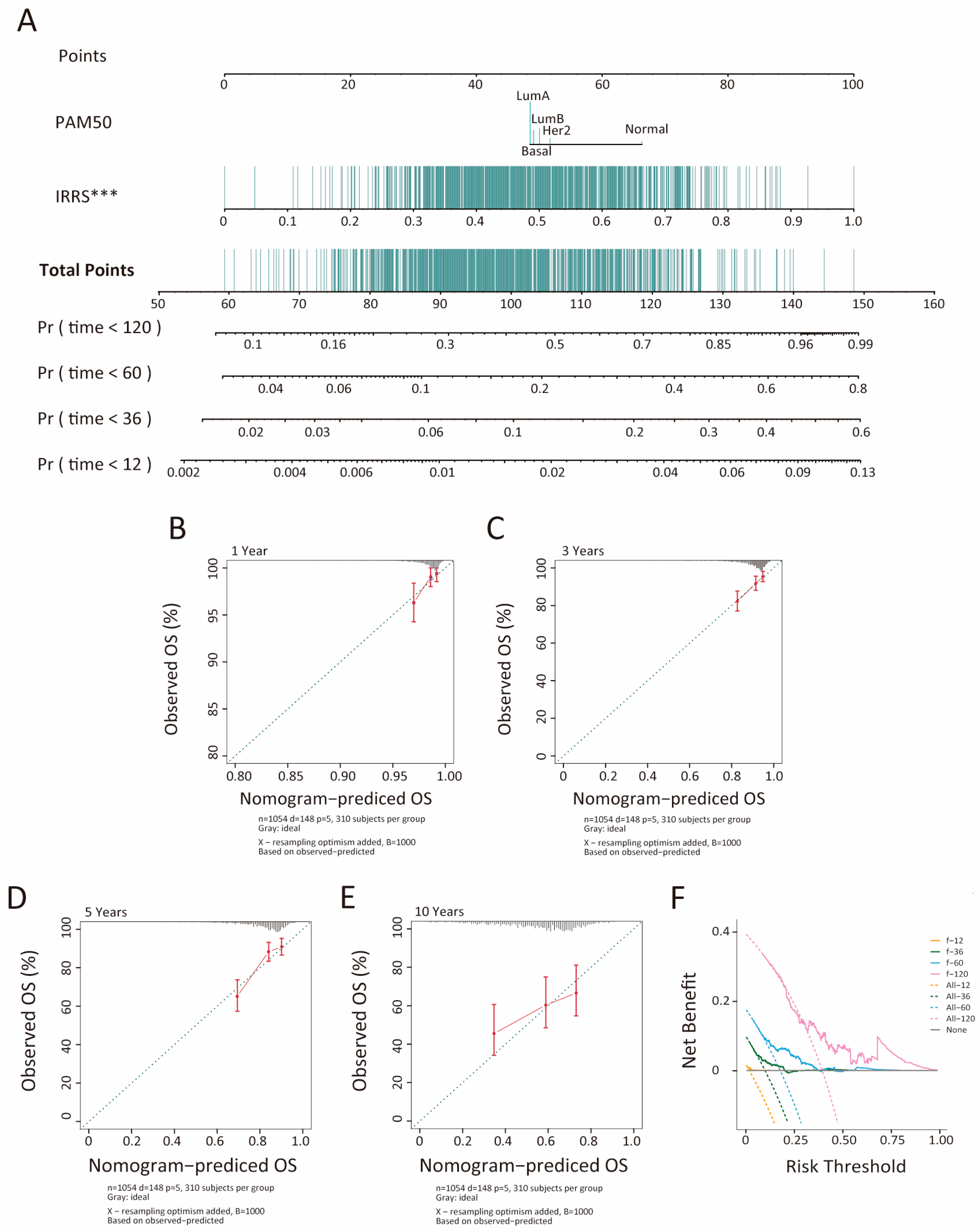
2.3. Development and Evaluation of an Insulin Resistance-Related Clinicopathological Nomogram
2.4. Tumor Microenvironment Characterization Analysis
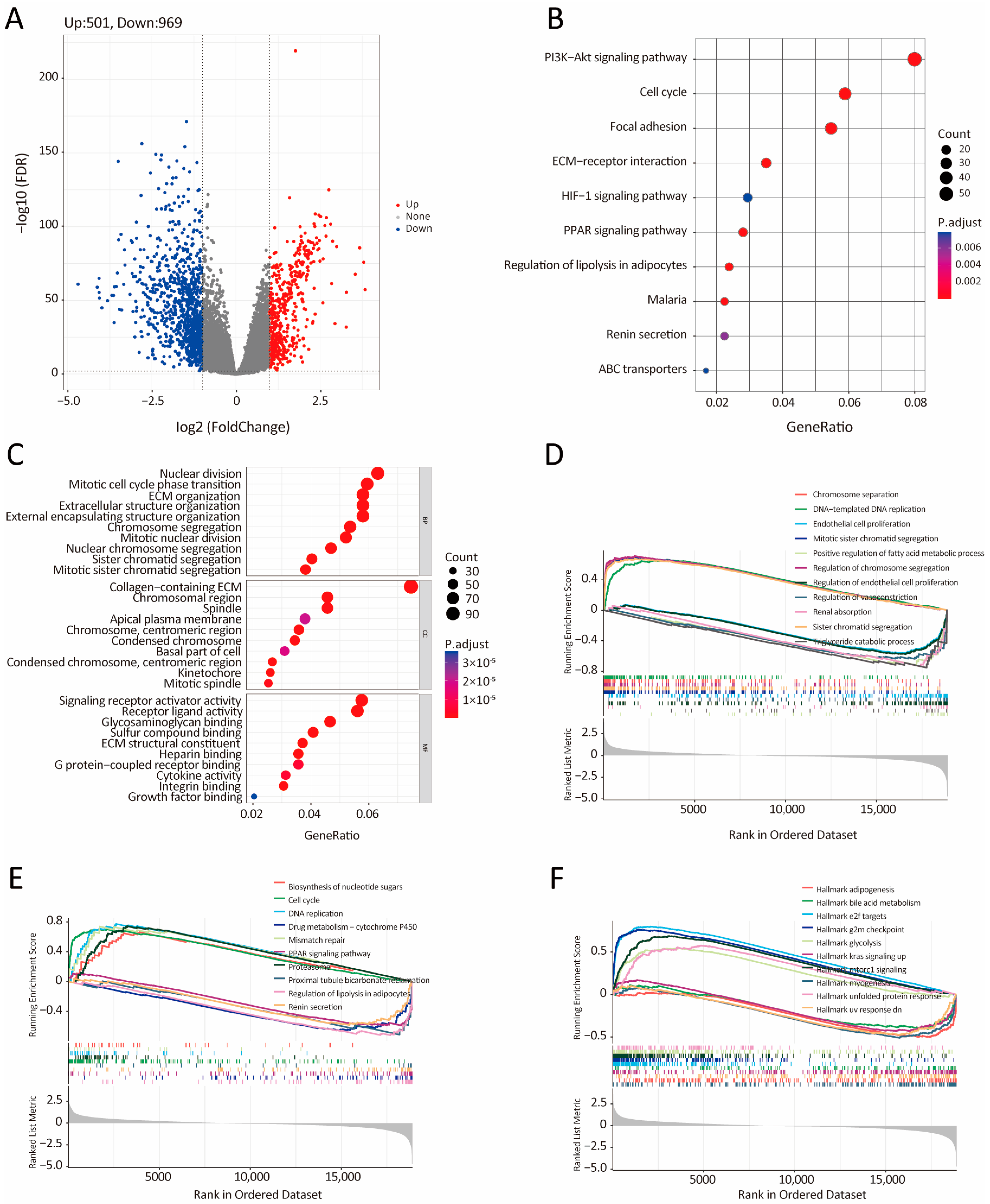
2.5. Differential Gene Expression of Low- and High-IRRS Groups and Functional Enrichment Analysis
2.6. Single-Cell RNA Sequencing Data Analysis
2.7. Ucell Analysis
2.8. Construction and Validation of the Machine Learning Model
2.9. Patients and Specimens
2.10. Immunohistochemistry
2.11. Statistical Analysis
3. Results
3.1. Construction and Validation of an Insulin Resistance-Relevant Prognostic Signature for Breast Cancer Patients
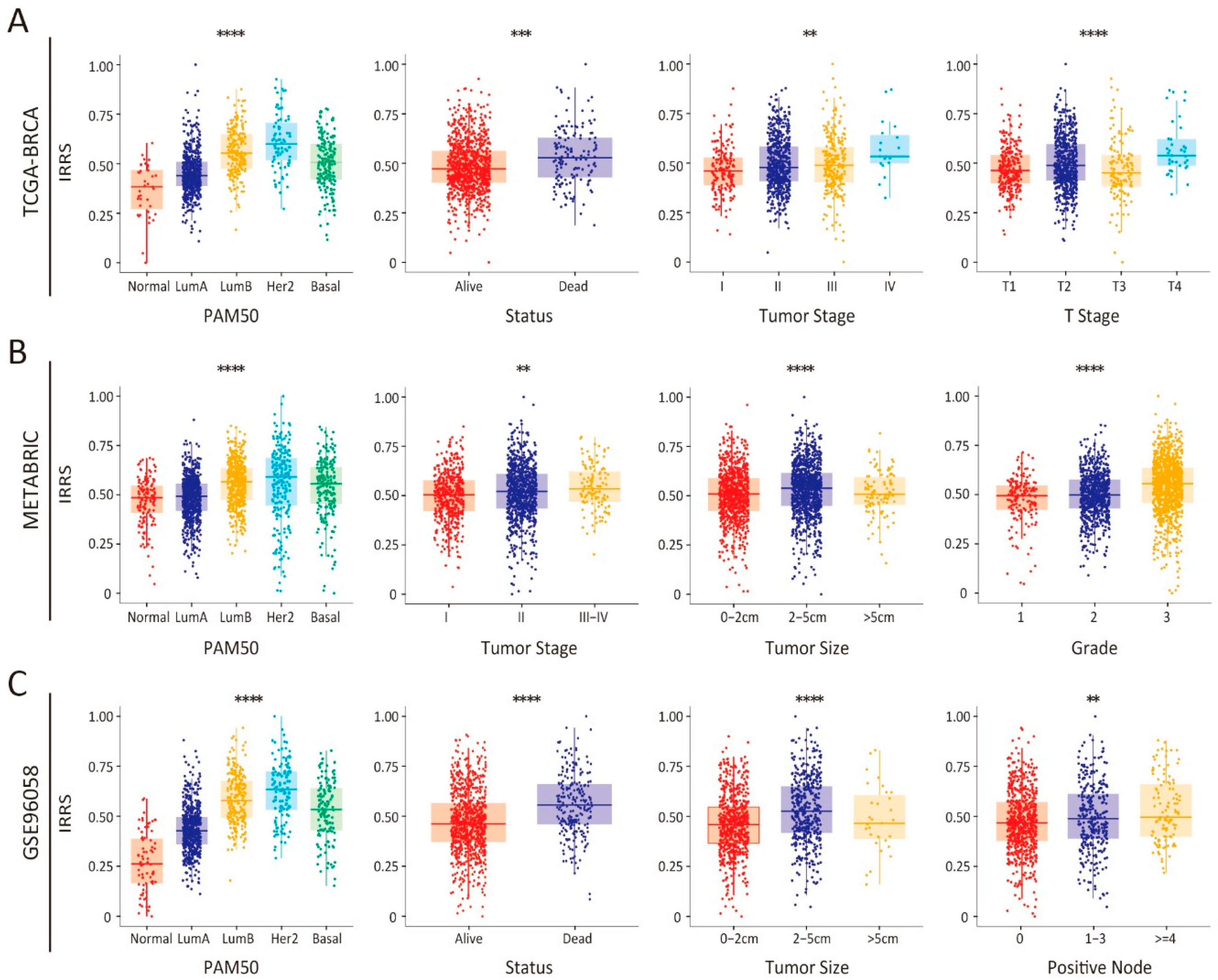
3.2. Integrated Assessment of the Prognostic Model and Clinical Parameters in Patients with Breast Cancer
3.3. Development and Evaluation of an Insulin Resistance-Related Clinicopathologic Nomogram
3.4. Association of TME Subcomponents with IRRS and Outcome in Patients with Breast Cancer
3.5. Bioinformatic Analysis of the Characteristics and Signaling Pathways Among Patients in Different Risk Groups
3.6. Therapeutic Benefit of the IRG Prognostic Signature
3.7. Single-Cell RNA Sequencing Data Analysis
3.8. Prediction of the Low- and High-IRRS Subtypes by the XGBoost Algorithm
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- Trapani, D.; Ginsburg, O.; Fadelu, T.; Lin, N.U.; Hassett, M.; Ilbawi, A.M.; Anderson, B.O.; Curigliano, G. Global challenges and policy solutions in breast cancer control. Cancer Treat. Rev. 2022, 104, 102339. [Google Scholar] [CrossRef] [PubMed]
- Kerr, A.J.; Dodwell, D.; McGale, P.; Holt, F.; Duane, F.; Mannu, G.; Darby, S.C.; Taylor, C.W. Adjuvant and neoadjuvant breast cancer treatments: A systematic review of their effects on mortality. Cancer Treat. Rev. 2022, 105, 102375. [Google Scholar] [CrossRef]
- Jokar, N.; Velikyan, I.; Ahmadzadehfar, H.; Rekabpour, S.J.; Jafari, E.; Ting, H.H.; Biersack, H.J.; Assadi, M. Theranostic Approach in Breast Cancer: A Treasured Tailor for Future Oncology. Clin. Nucl. Med. 2021, 46, e410–e420. [Google Scholar] [CrossRef]
- Li, M.; Chi, X.; Wang, Y.; Setrerrahmane, S.; Xie, W.; Xu, H. Trends in insulin resistance: Insights into mechanisms and therapeutic strategy. Signal Transduct. Target Ther. 2022, 7, 216. [Google Scholar] [CrossRef] [PubMed]
- Dev, R.; Bruera, E.; Dalal, S. Insulin resistance and body composition in cancer patients. Ann. Oncol. 2018, 29, ii18–ii26. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Alex, J.M.; Bast, F. Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1R) signaling systems: Novel treatment strategies for cancer. Med. Oncol. 2014, 31, 805. [Google Scholar] [CrossRef]
- Chiefari, E.; Mirabelli, M.; La Vignera, S.; Tanyolaç, S.; Foti, D.P.; Aversa, A.; Brunetti, A. Insulin Resistance and Cancer: In Search for a Causal Link. Int. J. Mol. Sci. 2021, 22, 11137. [Google Scholar] [CrossRef]
- Chou, P.C.; Choi, H.H.; Huang, Y.; Fuentes-Mattei, E.; Velazquez-Torres, G.; Zhang, F.; Phan, L.; Lee, J.; Shi, Y.; Bankson, J.A.; et al. Impact of diabetes on promoting the growth of breast cancer. Cancer. Commun. 2021, 41, 414–431. [Google Scholar] [CrossRef]
- Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Kao, K.J.; Chang, K.M.; Hsu, H.C.; Huang, A.T. Correlation of microarray-based breast cancer molecular subtypes and clinical outcomes: Implications for treatment optimization. BMC Cancer 2011, 11, 143. [Google Scholar] [CrossRef] [PubMed]
- Brueffer, C.; Vallon-Christersson, J.; Grabau, D.; Ehinger, A.; Häkkinen, J.; Hegardt, C.; Malina, J.; Chen, Y.; Bendahl, P.O.; Manjer, J.; et al. Clinical Value of RNA Sequencing-Based Classifiers for Prediction of the Five Conventional Breast Cancer Biomarkers: A Report from the Population-Based Multicenter Sweden Cancerome Analysis Network-Breast Initiative. JCO Precis Oncol. 2018, 2, 1–18. [Google Scholar] [CrossRef]
- Desmedt, C.; Piette, F.; Loi, S.; Wang, Y.; Lallemand, F.o.; Haibe-Kains, B.; Viale, G.; Delorenzi, M.; Zhang, Y.; d’Assignies, M.S.; et al. Strong Time Dependence of the 76-Gene Prognostic Signature for Node-Negative Breast Cancer Patients in the TRANSBIG Multicenter Independent Validation Series. Clin. Cancer Res. 2007, 13, 3207–3214. [Google Scholar] [CrossRef] [PubMed]
- Hoogstraat, M.; Lips, E.H.; Mayayo-Peralta, I.; Mulder, L.; Kristel, P.; van der Heijden, I.; Annunziato, S.; van Seijen, M.; Nederlof, P.M.; Sonke, G.S.; et al. Comprehensive characterization of pre- and post-treatment samples of breast cancer reveal potential mechanisms of chemotherapy resistance. Npj Breast Cancer 2022, 8, 60. [Google Scholar] [CrossRef]
- Miller, W.R.; Larionov, A. Changes in expression of oestrogen regulated and proliferation genes with neoadjuvant treatment highlight heterogeneity of clinical resistance to the aromatase inhibitor, letrozole. Breast Cancer Res. 2010, 12, R52. [Google Scholar] [CrossRef]
- Korde, L.A.; Lusa, L.; McShane, L.; Lebowitz, P.F.; Lukes, L.; Camphausen, K.; Parker, J.S.; Swain, S.M.; Hunter, K.; Zujewski, J.A. Gene expression pathway analysis to predict response to neoadjuvant docetaxel and capecitabine for breast cancer. Breast Cancer Res. Treat. 2010, 119, 685–699. [Google Scholar] [CrossRef]
- Page, D.B.; Pucilowska, J.; Chun, B.; Kim, I.; Sanchez, K.; Moxon, N.; Mellinger, S.; Wu, Y.; Koguchi, Y.; Conrad, V.; et al. A phase Ib trial of pembrolizumab plus paclitaxel or flat-dose capecitabine in 1st/2nd line metastatic triple-negative breast cancer. Npj Breast Cancer 2023, 9, 53. [Google Scholar] [CrossRef]
- Wu, S.Z.; Roden, D.L.; Wang, C.; Holliday, H.; Harvey, K.; Cazet, A.S.; Murphy, K.J.; Pereira, B.; Al-Eryani, G.; Bartonicek, N.; et al. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. EMBO J. 2020, 39, e104063. [Google Scholar] [CrossRef]
- Ginestet, C. ggplot2: Elegant Graphics for Data Analysis. J. R. Stat. Soc. Ser. A Stat. Soc. 2011, 174, 245–246. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e4. [Google Scholar] [CrossRef] [PubMed]
- Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L.; Hill, D.P.; et al. The Gene Ontology knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Haghverdi, L.; Lun, A.T.L.; Morgan, M.D.; Marioni, J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018, 36, 421–427. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e3529. [Google Scholar] [CrossRef]
- Wu, H.; Wang, W.; Zhang, Y.; Chen, Y.; Shan, C.; Li, J.; Jia, Y.; Li, C.; Du, C.; Cai, Y.; et al. Establishment of patient-derived organoids for guiding personalized therapies in breast cancer patients. Int. J. Cancer 2024, 155, 324–338. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Gonen, M.; Smith, J.J.; DeMatteo, R.P. Nomograms in oncology: More than meets the eye. Lancet Oncol. 2015, 16, e173–e180. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, H.L.; Liu, Y.J. A Novel Five-Gene Signature Related to Clinical Outcome and Immune Microenvironment in Breast Cancer. Front. Genet. 2022, 13, 912125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Fang, T.; Lv, Y. A novel lactate metabolism-related signature predicts prognosis and tumor immune microenvironment of breast cancer. Front. Genet. 2022, 13, 934830. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Bao, A.; Liu, S.; Dai, F.; Gong, Y.; Cheng, Y. A Pyroptosis-Related Gene Signature Predicts Prognosis and Immune Microenvironment for Breast Cancer Based on Computational Biology Techniques. Front. Genet. 2022, 13, 801056. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xiong, Y.; Zhang, Q.; Su, D.; Yu, C.; Cao, Y.; Pan, Y.; Lu, Q.; Zuo, Y.; Yang, L. Clinical significance and immunogenomic landscape analyses of the immune cell signature based prognostic model for patients with breast cancer. Brief Bioinform. 2021, 22, bbaa311. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Kohsaka, S.; Hinohara, K.; Wang, L.; Nishimura, T.; Urushido, M.; Yachi, K.; Tsuda, M.; Tanino, M.; Kimura, T.; Nishihara, H.; et al. Epiregulin enhances tumorigenicity by activating the ERK/MAPK pathway in glioblastoma. Neuro-Oncology 2014, 16, 960–970. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Gallagher, E.J.; Greco, G.; Lin, S.; Yagnik, R.; Feldman, S.M.; Port, E.; Friedman, N.B.; Boolbol, S.K.; Killelea, B.; Pilewskie, M.; et al. Insulin resistance and racial disparities in breast cancer prognosis: A multi-center cohort study. Endocr. Relat. Cancer 2022, 29, 693–701. [Google Scholar] [CrossRef]
- Dieli-Conwright, C.M.; Courneya, K.S.; Demark-Wahnefried, W.; Sami, N.; Lee, K.; Buchanan, T.A.; Spicer, D.V.; Tripathy, D.; Bernstein, L.; Mortimer, J.E. Effects of Aerobic and Resistance Exercise on Metabolic Syndrome, Sarcopenic Obesity, and Circulating Biomarkers in Overweight or Obese Survivors of Breast Cancer: A Randomized Controlled Trial. J. Clin. Oncol. 2018, 36, 875–883. [Google Scholar] [CrossRef]
- Yin, D.T.; He, H.; Yu, K.; Xie, J.; Lei, M.; Ma, R.; Li, H.; Wang, Y.; Liu, Z. The association between thyroid cancer and insulin resistance, metabolic syndrome and its components: A systematic review and meta-analysis. Int. J. Surg. 2018, 57, 66–75. [Google Scholar] [CrossRef]
- Argirion, I.; Weinstein, S.J.; Männistö, S.; Albanes, D.; Mondul, A.M. Serum Insulin, Glucose, Indices of Insulin Resistance, and Risk of Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2017, 26, 1519–1524. [Google Scholar] [CrossRef] [PubMed]
- Manousaki, D.; Kent, J.W., Jr.; Haack, K.; Zhou, S.; Xie, P.; Greenwood, C.M.; Brassard, P.; Newman, D.E.; Cole, S.; Umans, J.G.; et al. Toward Precision Medicine: TBC1D4 Disruption Is Common Among the Inuit and Leads to Underdiagnosis of Type 2 Diabetes. Diabetes Care 2016, 39, 1889–1895. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Ju, H.; Fan, J.; Shi, X.; Cheng, Y.; Cang, X.; Zheng, Z.; Duan, X.; Yi, W. O-GlcNAcylation of PGK1 coordinates glycolysis and TCA cycle to promote tumor growth. Nat. Commun. 2020, 11, 36. [Google Scholar] [CrossRef]
- Xiao, G.; Cheng, F.; Yuan, J.; Lu, W.; Wang, P.; Fan, H. Integrative multiomics analysis identifies a metastasis-related gene signature and the potential oncogenic role of EZR in breast cancer. Oncol. Res. 2022, 30, 35–51. [Google Scholar] [CrossRef]
- Chen, D.; Sun, Y.; Wei, Y.; Zhang, P.; Rezaeian, A.H.; Teruya-Feldstein, J.; Gupta, S.; Liang, H.; Lin, H.K.; Hung, M.C.; et al. LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat. Med. 2012, 18, 1511–1517. [Google Scholar] [CrossRef]
- Ma, X.; Li, Y.; Zhao, B. Ribosomal protein L5 (RPL5)/ E2F transcription factor 1 (E2F1) signaling suppresses breast cancer progression via regulating endoplasmic reticulum stress and autophagy. Bioengineered 2022, 13, 8076–8086. [Google Scholar] [CrossRef]
- Niu, X.; Yin, L.; Yang, X.; Yang, Y.; Gu, Y.; Sun, Y.; Yang, M.; Wang, Y.; Zhang, Q.; Ji, H. Serum amyloid A 1 induces suppressive neutrophils through the Toll-like receptor 2-mediated signaling pathway to promote progression of breast cancer. Cancer Sci. 2022, 113, 1140–1153. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, G.; Chu, H.; Wang, X.; Xiong, L.; Cai, G.; Liu, R.; Gao, H.; Tao, B.; Li, W.; et al. Macrophage-Associated PGK1 Phosphorylation Promotes Aerobic Glycolysis and Tumorigenesis. Mol. Cell. 2018, 71, 201–215.e7. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, Y.; Yu, H.; Zhang, T.; Guo, L.; Xu, J.; Wei, X.; Wang, N.; Wu, Y.; Wang, X.; et al. PGK1 represses autophagy-mediated cell death to promote the proliferation of liver cancer cells by phosphorylating PRAS40. Cell Death Dis. 2022, 13, 68. [Google Scholar] [CrossRef]
- Sun, S.; Liang, X.; Zhang, X.; Liu, T.; Shi, Q.; Song, Y.; Jiang, Y.; Wu, H.; Jiang, Y.; Lu, X.; et al. Phosphoglycerate kinase-1 is a predictor of poor survival and a novel prognostic biomarker of chemoresistance to paclitaxel treatment in breast cancer. Br. J. Cancer 2015, 112, 1332–1339. [Google Scholar] [CrossRef]
- Zeng, H.; Qu, J.; Jin, N.; Xu, J.; Lin, C.; Chen, Y.; Yang, X.; He, X.; Tang, S.; Lan, X.; et al. Feedback Activation of Leukemia Inhibitory Factor Receptor Limits Response to Histone Deacetylase Inhibitors in Breast Cancer. Cancer Cell 2016, 30, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.; Zhang, J.; Wang, X.; Li, Q.; Liu, L.Y.; Yang, J.; Guo, B.; Ni, L.; Zhao, L.; Huang, C. MeCP2 facilitates breast cancer growth via promoting ubiquitination-mediated P53 degradation by inhibiting RPL5/RPL11 transcription. Oncogenesis 2020, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Cancer stem cell in breast cancer therapeutic resistance. Cancer Treat. Rev. 2018, 69, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. The root cause of drug resistance in HER2-positive breast cancer and the therapeutic approaches to overcoming the resistance. Pharmacol. Ther. 2021, 218, 107677. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort | Data Type | Source | Sample Size | Reference |
|---|---|---|---|---|
| TCGA-BRCA | RNASeq | TCGA | 1095 | [10] |
| METABRIC | microarray | cBioPortal | 1906 | [11] |
| GSE20685 | microarray | GEO | 327 | [12] |
| GSE96058 | RNASeq | GEO | 3069 | [13] |
| GSE7390 | microarray | GEO | 198 | [14] |
| GSE191127 | RNAseq | GEO | 40 | [15] |
| GSE20181 | microarray | GEO | 176 | [16] |
| GSE18728 | microarray | GEO | 61 | [17] |
| GSE225078 | RNAseq | GEO | 30 | [18] |
| SCP1106 | scRNA-seq | Single-cell portal | 5 | [19] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, L.; Li, D.; Chen, H.; Pu, Y.; Wang, Q.; Li, J.; Zhou, M.; Liu, C.; Long, P. Elucidating the Prognostic and Therapeutic Implications of Insulin Resistance Genes in Breast Cancer: A Machine Learning-Powered Analysis. Biology 2025, 14, 539. https://doi.org/10.3390/biology14050539
Wei L, Li D, Chen H, Pu Y, Wang Q, Li J, Zhou M, Liu C, Long P. Elucidating the Prognostic and Therapeutic Implications of Insulin Resistance Genes in Breast Cancer: A Machine Learning-Powered Analysis. Biology. 2025; 14(5):539. https://doi.org/10.3390/biology14050539
Chicago/Turabian StyleWei, Lengyun, Dashuai Li, Hongjin Chen, Yajing Pu, Qun Wang, Jintao Li, Meng Zhou, Chenfeng Liu, and Pengpeng Long. 2025. "Elucidating the Prognostic and Therapeutic Implications of Insulin Resistance Genes in Breast Cancer: A Machine Learning-Powered Analysis" Biology 14, no. 5: 539. https://doi.org/10.3390/biology14050539
APA StyleWei, L., Li, D., Chen, H., Pu, Y., Wang, Q., Li, J., Zhou, M., Liu, C., & Long, P. (2025). Elucidating the Prognostic and Therapeutic Implications of Insulin Resistance Genes in Breast Cancer: A Machine Learning-Powered Analysis. Biology, 14(5), 539. https://doi.org/10.3390/biology14050539