

Complete Mitochondrial Genome of Chlorogomphus papilio (Odonata: Anisoptera: Chlorogomphidae) and Phylogenetic Analyses

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Test Materials
2.2. Genomic DNA Extraction and High-Throughput Sequencing
2.3. Mitochondrial Genome Assembly and Annotation
2.4. Analysis of Mitochondrial Genome Characteristics
2.5. Phylogenetic Analyses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | GenBank Sequence Number | Reference | |
|---|---|---|---|
| 16S rRNA | cox1 | ||
| Chlorogomphus aritai | LC366454.1 | LC366751.1 | Futahashi, 2014 [50] |
| Chlorogomphus arooni | LC366488.1 | LC366785.1 | Futahashi, 2014 [50] |
| Chlorogomphus auratus | LC366510.1 | LC366807.1 | Futahashi, 2014 [50] |
| Chlorogomphus brevistigma | LC200915.1 | LC200926.1 | Futahashi, 2014 [50] |
| Chlorogomphus brunneus | LC366602.1 | LC366899.1 | Futahashi, 2014 [50] |
| Chlorogomphus caloptera | LC366445.1 | LC366742.1 | Futahashi, 2014 [50] |
| Chlorogomphus hiten | LC366512.1 | LC366809.1 | Futahashi, 2014 [50] |
| Chlorogomphus iriomotensis | LC200916.1 | LC200927.1 | Futahashi, 2014 [50] |
| Chlorogomphus kitawakii | LC366444.1 | LC366741.1 | Futahashi, 2014 [50] |
| Chlorogomphus magnificus | LC366469.1 | LC366766.1 | Futahashi, 2014 [50] |
| Chlorogomphus miyashitai | LC366385.1 | LC366682.1 | Futahashi, 2014 [50] |
| Chlorogomphus nakamurai | LC366441.1 | LC366738.1 | Futahashi, 2014 [50] |
| Chlorogomphus nasutus | LC366459.1 | LC366756.1 | Futahashi, 2014 [50] |
| Chlorogomphus okinawensis | LC200917.1 | LC200928.1 | Futahashi, 2014 [50] |
| Chlorogomphus piaoacensis | LC366386.1 | LC366683.1 | Futahashi, 2014 [50] |
| Chlorogomphus risi | LC200919.1 | LC200930.1 | Futahashi, 2014 [50] |
| Chlorogomphus shanicus | LC366451.1 | LC366748.1 | Futahashi, 2014 [50] |
| Chlorogomphus suzukii | LC366344.1 | LC366641.1 | Futahashi, 2014 [50] |
| Chlorogomphus tunti | LC366453.1 | LC366750.1 | Futahashi, 2014 [50] |
| Chlorogomphus vietnamensis | LC366468.1 | LC366765.1 | Futahashi, 2014 [50] |
| Chlorogomphus yokoii | LC200921.1 | LC200932.1 | Futahashi, 2014 [50] |
| Chloropetalia owadai | LC366460.1 | LC366757.1 | Futahashi, 2014 [50] |
| Watanabeopetalia uenoi | LC366456.1 | LC366753.1 | Futahashi, 2014 [50] |
| Watanabeopetalia usignata | LC366458.1 | LC366755.1 | Futahashi, 2014 [50] |
| Anotogaster klossi | AB707862.1 | AB708806.1 | Futahashi, 2011 [51] |
| Anotogaster kuchenbeiseri | AB707879.1 | AB708823.1 | Futahashi, 2011 [51] |
| Anotogaster chaoi | AB707859.1 | AB708803.1 | Futahashi, 2011 [51] |
| Anotogaster gregoryi | LC366511.1 | LC366808.1 | Futahashi, 2014 [50] |
| Anotogaster sakaii | LC366345.1 | LC366642.1 | Futahashi, 2014 [50] |
| Neallogaster pekinensis | AB707904.1 | AB708848.1 | Futahashi, 2011 [51] |
2.6. Divergence Time Estimation
3. Results
3.1. Mitochondrial Genome Structure of C. papilio
3.2. Mitochondrial Genome Nucleotide Composition
3.3. PCGs and Codon Usage
3.4. tRNA and rRNA
3.5. Control Region, Non-Coding, and Overlapping Areas
3.6. Phylogenetic Relationships
3.7. Divergence-Time Estimation Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DOAJ | Directory of open access journals |
| TLA | Three letter acronym |
| LD | Linear dichroism |
References
- Novelo-Gutiérrez, R.; Sites, R.W. The Dragonfly Nymphs of Thailand (Odonata: Anisoptera); Springer: New York, NY, USA, 2024; pp. 239–247. [Google Scholar]
- Ris, F. Ein neuer Chlorogomphus aus China (Odonata). Entomol. Mitteilungen 1927, 16, 103–105. [Google Scholar]
- Crampton-Platt, A.; Yu, D.W.; Zhou, X.; Vogler, A.P. Mitochondrial metagenomics: Letting the genes out of the bottle. GigaScience 2016, 5, 15. [Google Scholar] [CrossRef]
- Song, F.; Li, H.; Jiang, P.; Zhou, X.; Liu, J.; Sun, C.; Vogler, A.P.; Cai, W. Capturing the phylogeny of Holometabola with mitochondrial genome data and Bayesian site-heterogeneous mixture models. Genome Biol. Evol. 2016, 8, 1411–1426. [Google Scholar] [CrossRef]
- Sakai, M.; Suda, S.I.; Okeda, T.; Nomura, R.; Washitani, I. The importance of riparian subtropical lucidophyllous forest to odonate conservation. Aquat. Conserv. Mar. Freshw. Ecosyst. 2019, 29, 682–692. [Google Scholar] [CrossRef]
- Carle, F.L. Evolution, taxonomy and biogeography of ancient Gondwanian libelluloides, with comments on anisopteroid evolution and phylogenetic systematics (Anisoptera: Libelluloidea). Odonatologica 1995, 24, 383–424. [Google Scholar]
- Ishida, K. A new species of the genus Chlorogomphus from Hainan lsland of China. Jpn. J. Syst. Entomol. 1996, 2, 51–55. [Google Scholar]
- Carle, F.L.; Karl, M.; Kjer, K.M.; May, M.L. A molecular phylogeny and classification of Anisoptera (Odonata). Arthropod Syst. Phylogeny 2015, 73, 281–301. [Google Scholar] [CrossRef]
- Bybee, S.M.; Ogden, T.H.; Branham, M.A.; Whiting, M.F. Molecules, morphology and fossils: A comprehensive approach to odonate phylogeny and the evolution of the odonate wing. Cladistics 2008, 24, 477–514. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-W. A revision of the genus Chlorogomphus Selys of eastern China (Odonata: Cordulegastridae). Q. J. Taiwan Mus. 1950, 3, 137–150. [Google Scholar]
- Karube, H.; Phan, Q.T. Discovery of Chlorogomphus papilio Ris, 1927 from northern Vietnam. Tombo Fukui 2016, 58, 49–51. [Google Scholar]
- Curole, J.P.; Kocher, T.D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 1999, 14, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Nass, S.; Nass, M.M. Intramitochondrial fibers with DNA characteristics: II. Enzymatic and other hydrolytic treatments. J. Cell Biol. 1963, 19, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J.; Archibald, J.M. Organelle evolution: What’s in a name? Curr. Bology 2008, 18, 345–347. [Google Scholar] [CrossRef]
- Gong, L.; Jiang, H.; Zhu, K.; Lu, X.; Liu, L.; Liu, B.; Jiang, L.; Ye, Y.; Lü, Z. Large-scale mitochondrial gene rearrangements in the hermit crab Pagurus nigrofascia and phylogenetic analysis of the Anomura. Gene 2019, 695, 75–83. [Google Scholar] [CrossRef]
- Birky Jr, C.W. The inheritance of genes in mitochondria and chloroplasts: Laws, mechanisms, and models. Annu. Rev. Genet. 2001, 35, 125–148. [Google Scholar] [CrossRef]
- Sato, M.; Sato, K. Maternal inheritance of mitochondrial DNA by diverse mechanisms to eliminate paternal mitochondrial DNA. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 1979–1984. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.L.; Li, Q.; Gu, Y.; Shi, B.C.; van Achterberg, C.; Wei, S.J.; Chen, X.X. The complete mitochondrial genome of Taeniogonalos taihorina (Bischoff) (Hymenoptera: Trigonalyidae) reveals a novel gene rearrangement pattern in the Hymenoptera. Gene 2014, 543, 76–84. [Google Scholar] [CrossRef]
- Broughton, R.E.; Milam, J.E.; Roe, B.A. The complete sequence of the Zebrafish (Danio rerio) mitochondrial genome and evolutionary patterns in vertebrate mitochondrial DNA. Genome Res. 2001, 11, 1958–1967. [Google Scholar] [CrossRef]
- Yang, J.; Huang, Y. Strategy of high-throughput sequencing technology in the mitochondrial genome sequencing (in Chinese). Chin. Bull. Life Sci. 2016, 28, 112–117. [Google Scholar]
- Mccormack, J.E.; Hird, S.M.; Zellmer, A.J.; Carstens, B.C.; Brumfield, R.T. Applications of next-generation sequencing to phylogeography and phylogenetics. Mol. Phylogenetics Evol. 2013, 66, 526–538. [Google Scholar] [CrossRef]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef] [PubMed]
- Ballard, J.W.; Whitlock, M.C. The incomplete natural history of mitochondria. Mol. Ecol. 2004, 13, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Et Biophys. Acta (BBA) Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Garesse, R. Drosophila melanogaster mitochondrial DNA: Gene organization and evolutionary considerations. Genetics 1988, 118, 649–663. [Google Scholar] [CrossRef]
- Chen, A.H. Complete mitochondrial genome of the double-striped cockroach Blattella bisignata (Insecta: Blattaria: Blaberoidea). Mitochondrial DNA 2013, 24, 14–16. [Google Scholar] [CrossRef]
- Xiao, B.; Feng, X.; Miao, W.J.; Jiang, G.F. The complete mitochondrial genome of grouse locust Tetrix japonica (Insecta: Orthoptera: Tetrigoidea). Mitochondrial DNA 2012, 23, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Huang, Y.; Shi, F. The mitochondrial genome of Ruspolia dubia (Orthoptera: Conocephalidae) contains a short A+ T-rich region of 70 bp in length. Genome 2007, 50, 855–866. [Google Scholar] [CrossRef]
- Dijkstra, K.D.B.; Bechly, G.; Bybee, S.M.; Dow, R.A.; Dumont, H.J.; Fleck, G.; Garrison, R.W.; Hämäläinen, M.; Kalkman, V.J.; Karube, H.; et al. The classification and diversity of dragonflies and damselflies (Odonata). Zootaxa 2013, 3703, 36–45. [Google Scholar]
- Martincová, I.; Aghová, T. Comparison of 12 DNA extraction kits for vertebrate samples. Anim. Biodivers. Conserv. 2020, 43, 67. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Li, H. and Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes de novo assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.D.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, 181–184. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Peden, J.F. Analysis of Codon Usage; University of Nottingham: Nottingham, UK, 1999. [Google Scholar]
- Lorenz, R.; Bernhart, S.H.; Honer Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Koichiro, T.; Glen, S.; Sudhir, K. Mega11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 7, 7. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar]
- Ronquist, F.; Huelsenbeck, J.; Teslenko, M. MrBayes Version 3.2 Manual: Tutorials and Model Summaries. 2011. Available online: https://gensoft.pasteur.fr/docs/mrbayes/3.2.7/Manual_MrBayes_v3.2.pdf (accessed on 19 October 2024).
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Futahashi, R. A revisional study of Japanese dragonflies based on DNA analysis (2). Tombo 2014, 56, 57–59. [Google Scholar]
- Futahashi, R. A revisional study of Japanese dragonflies based on DNA analysis (1). Tombo 2011, 53, 67–74. [Google Scholar]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De, M.N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef]
- Osozawa, S.; Wakabayashi, J. Odonata timetree; exponentially increased base substitution rate toward the recent and within the Carboniferous. Available online: https://www.researchsquare.com/article/rs-116942/v1 (accessed on 19 October 2024).
- Osozawa, S.; Su, Z.H.; Oba, Y.; Yagi, T.; Watanabe, Y.; Wakabayashi, J. Vicariant speciation due to 1.55 Ma isolation of the Ryukyu islands, Japan, based on geological and Gen Bank data. Entomol. Sci. 2013, 16, 267–277. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- O’Reilly, J.E.; Dos Reis, M.; Donoghue, P.C. Dating tips for divergence-time estimation. Trends Genet. 2015, 31, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zheng, Y.; Zhao, X.; Xu, X.; Xu, Z.; Cui, C. Molecular phylogeny and evolution of the Tuerkayana (Decapoda: Brachyura: Gecarcinidae) genus based on whole mitochondrial genome sequences. Biology 2023, 12, 974. [Google Scholar] [CrossRef]
- Wang, Y.; Du, Y.; Song, X.; Huang, A. Complete mitochondrial genome sequence of Anax parthenope (Odonata: Anisoptera: Aeshnidae) and phylogenetic analysis. Mitochondrial DNA Part B 2021, 6, 122–123. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Zhu, J.C.; Zheng, B.Y.; Wei, S.J.; Sharkey, M.; Chen, X.X.; Vogler, A.P. Mitochondrial phylogenomics of the Hymenoptera. Mol. Phylogenetics Evol. 2019, 131, 8–18. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Liao, J.; Han, B.P. The complete mitochondrial genome of Chlorogomphus shanicus Wilson, 2002 (Anisoptera: Chlorogomphidae), an endemic species in South China. Mitochondrial DNA Part B 2023, 8, 1192–1195. [Google Scholar] [CrossRef]
- Liu, J.; Yang, Y.; Yan, Z.; Wang, H.; Bai, M.; Shi, C.; Li, J. Analysis of the mitogenomes of two helotid species provides new insights into the phylogenetic relationship of the Basal Cucujoidea (Insecta: Coleoptera). Biology 2023, 12, 135. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.J.; Chen, X.X. Progress in research on the comparative mitogenomics of insects. Chin. J. Appl. Entomol. 2011, 48, 1573–1585. [Google Scholar]
- Yokobori, S.I.; Paabo, S. Polyadenylation creates the discriminator nucleotide of chicken mitochondrial tRNA Tyr. J. Mol. Biol. 1997, 265, 95–99. [Google Scholar] [CrossRef]
- Passmore, L.A.; Coller, J. Roles of mRNA poly (A) tails in regulation of eukaryotic gene expression. Nat. Rev. Mol. Cell Biol. 2022, 23, 93–106. [Google Scholar] [CrossRef]
- Attardi, G. Animal Mitochondrial DNA: An extreme example of genetic economy. Int. Rev. Cytol. 1985, 93, 93–145. [Google Scholar] [PubMed]
- Varani, G.; McClain, W.H. The G· U wobble base pair. Eur. Mol. Biol. Organ. Rep. 2000, 1, 18–23. [Google Scholar] [CrossRef]
- Masta, S.E.; Boore, J.L. Parallel evolution of truncated transfer RNA genes in arachnid mitochondrial genomes. Mol. Biol. Evol. 2008, 25, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.M.; Tong, X.L. Chlorogomphid dragonflies of Guizhou Province, China with first descriptions of Chlorogomphus tunti and Watanabeopetalia usignata larvae (Anisoptera: Cordulegastridae). Odonatologica 2010, 39, 341–352. [Google Scholar]
- Fleck, G.N.A.; MartínezDelclòs, X. The oldest record of the Libellulidae from the Upper Cretaceous of Kazakhstan (Odonata, Anisoptera). Cretac. Res. 1999, 20, 655–658. [Google Scholar] [CrossRef]
- Nel, A.; Ribeiro, G.C. New fossil wings shed light on Lower Cretaceous Araripechlorogomphidae and minimum age of the Chlorogomphoidea (Odonata: Anisoptera): Crato formation, Araripe Basin, NE Brazil. Cretac. Res. 2024, 156. [Google Scholar] [CrossRef]
- Gupta, S.; Kumar, K. Precursors of the Paleocene–Eocene Thermal Maximum (PETM) in the Subathu Group, NW sub-Himalaya, India. J. Asian Earth Sci. 2019, 169, 21–46. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, S. Extinction vs. rapid radiation: The juxtaposed evolutionary histories of coelotine spiders support the Eocene–Oligocene orogenesis of the Tibetan Plateau. Syst. Biol. 2017, 66, 988–1006. [Google Scholar] [CrossRef]
- Hofmann, C.C.; Zhao, W.Y. Unravelling the palaeobiogeographical history of the living fossil genus Rehderodendron (Styracaceae) with fossil and extant pollen and fruit data. BMC Ecol. Evol. 2022, 22, 145. [Google Scholar] [CrossRef]
- Currano, E.D.; Wilf, P.; Wing, S.L.; Labandeira, C.C.; Lovelock, E.C.; Royer, D.L. Sharply increased insect herbivory during the Paleocene–Eocene Thermal Maximum. Proc. Natl. Acad. Sci. USA 2008, 105, 1960–1964. [Google Scholar] [CrossRef]


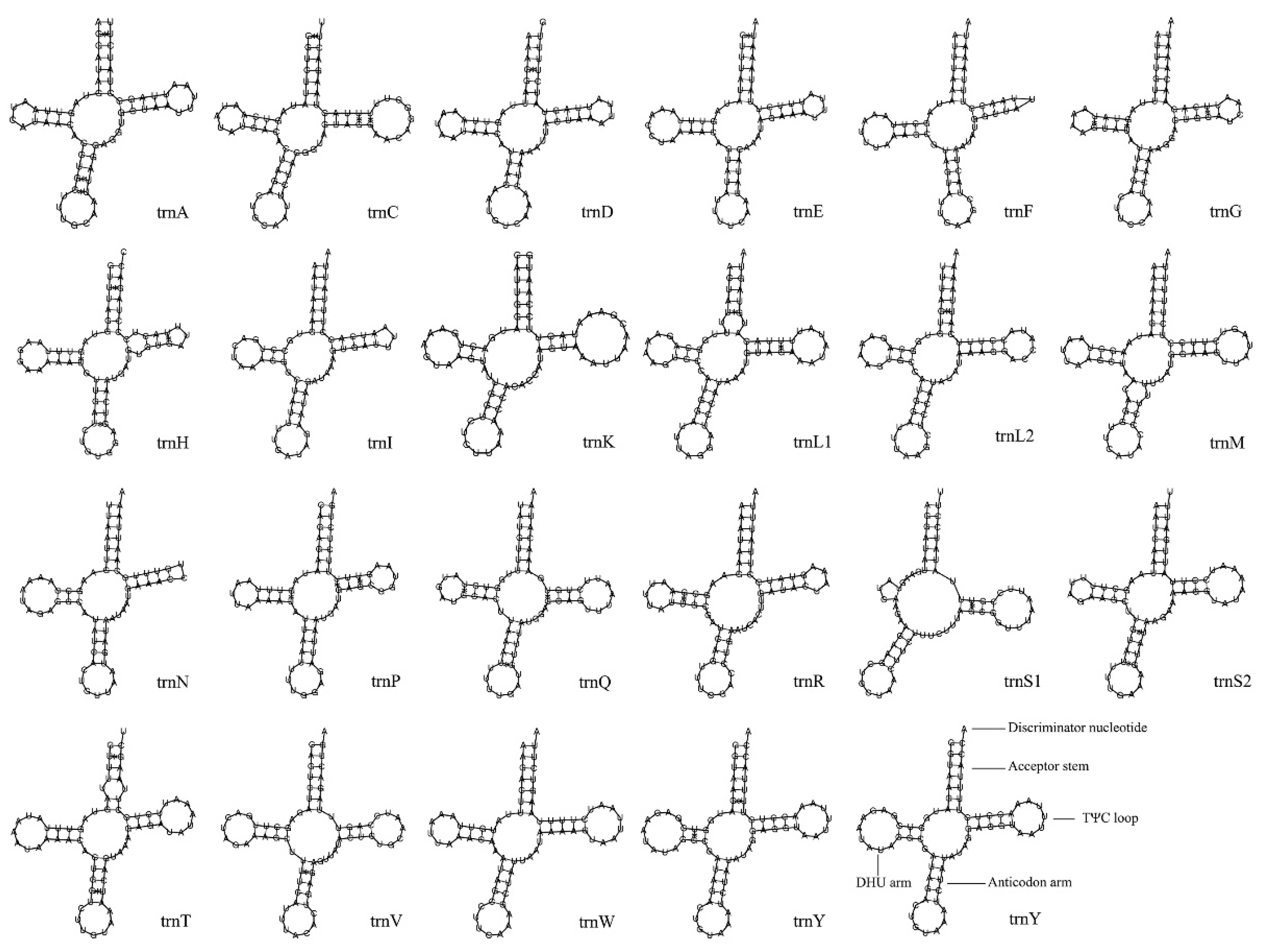
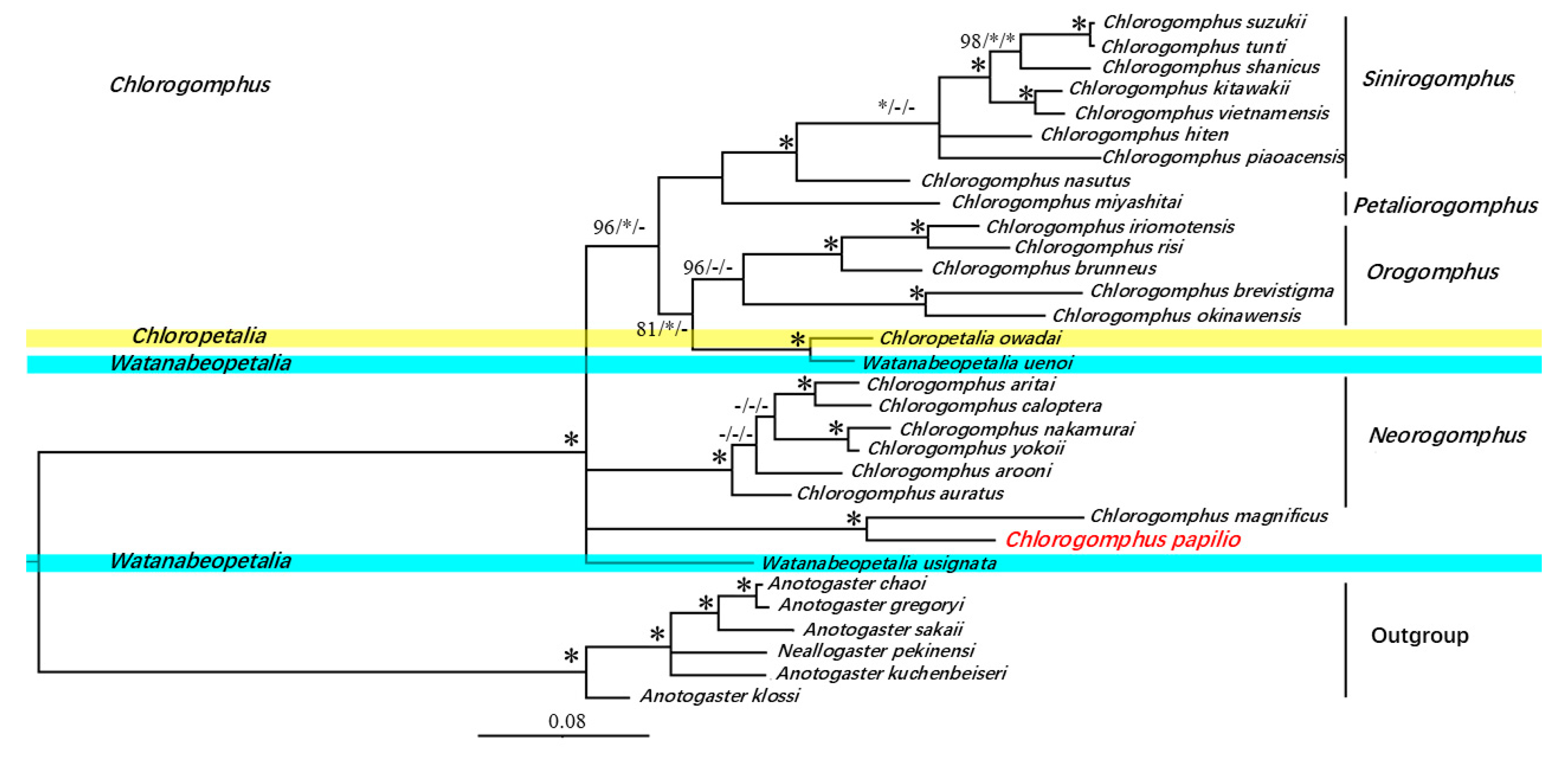
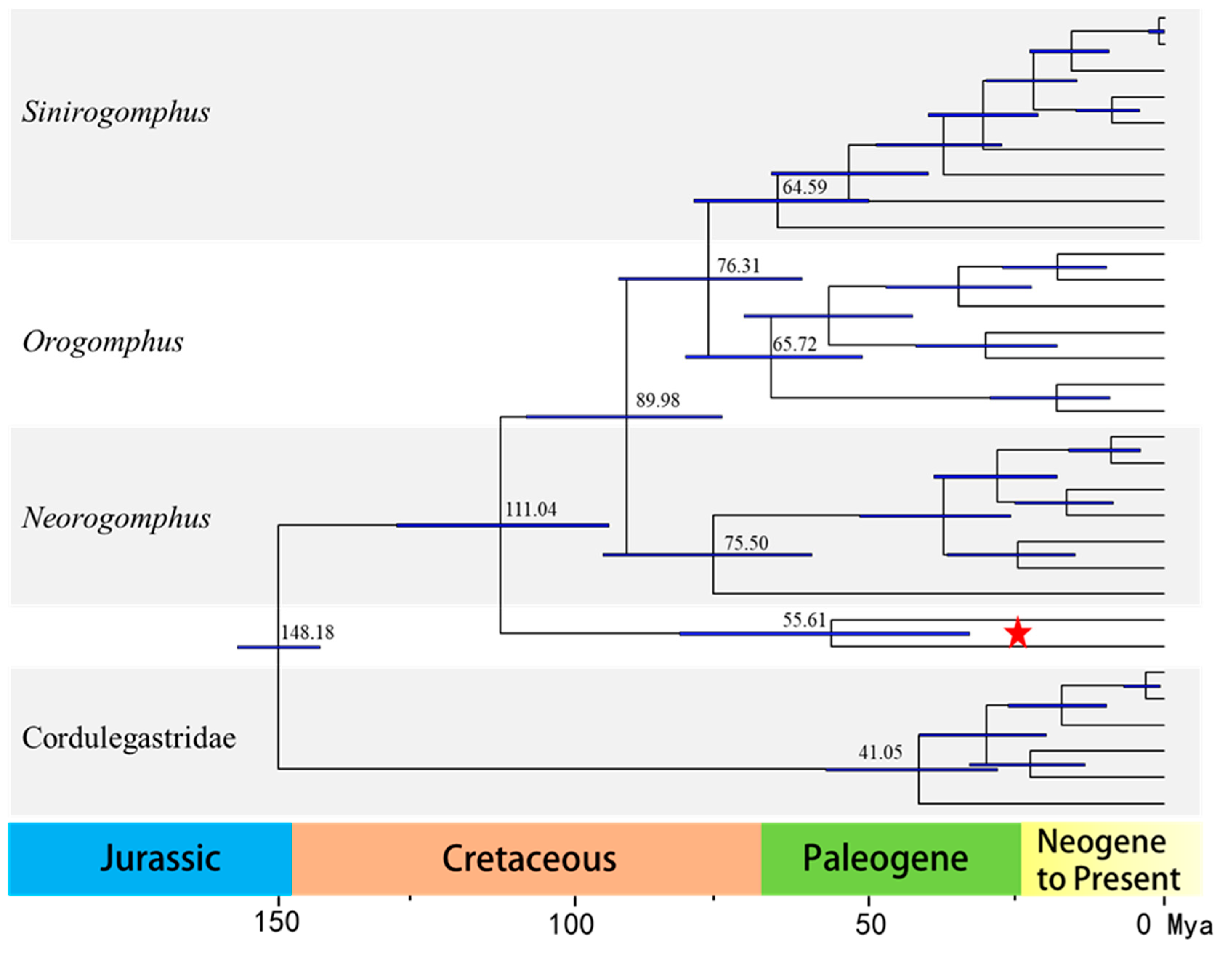
| Gene | Strand | Start Position | Stop Position | Gene Length (bp) | Intergenic Nucleotide (bp) | Overlapping Nucleotide (bp) | Start Codon | Stop Codon |
|---|---|---|---|---|---|---|---|---|
| trnI | H | 1 | 65 | 65 | - | - | - | - |
| trnQ | L | 63 | 130 | 68 | - | 3 | - | - |
| trnM | H | 130 | 198 | 69 | - | 1 | - | - |
| nad2 | H | 199 | 1200 | 1002 | - | - | ATT | TAA |
| trnW | H | 1199 | 1269 | 71 | - | 2 | - | - |
| trnC | L | 1262 | 1332 | 71 | - | 8 | - | - |
| trnY | L | 1333 | 1403 | 71 | - | - | - | - |
| cox1 | H | 1447 | 2983 | 1537 | 43 | - | ATA | T |
| trnL | H | 2984 | 3050 | 67 | - | - | - | - |
| cox2 | H | 3051 | 3738 | 688 | - | - | ATG | T |
| trnK | H | 3739 | 3809 | 71 | - | - | - | - |
| trnD | H | 3810 | 3875 | 66 | - | - | - | - |
| atp8 | H | 3876 | 4034 | 159 | - | - | ATC | TAA |
| atp6 | H | 4031 | 4705 | 675 | - | 4 | ATA | TAA |
| cox3 | H | 4705 | 5491 | 787 | - | 1 | ATG | T |
| trnG | H | 5492 | 5556 | 65 | - | - | - | - |
| nad3 | H | 5566 | 5910 | 345 | 9 | - | ATA | TAG |
| trnA | H | 5909 | 5973 | 65 | - | 2 | - | - |
| trnR | H | 5974 | 6041 | 68 | - | - | - | - |
| trnN | H | 6042 | 6107 | 66 | - | - | - | - |
| trnS1 | H | 6108 | 6175 | 68 | - | - | - | - |
| trnE | H | 6177 | 6244 | 68 | 1 | - | - | - |
| trnF | L | 6243 | 6307 | 65 | - | 2 | - | - |
| nad5 | L | 6308 | 8036 | 1729 | - | - | ATT | T |
| trnH | L | 8037 | 8102 | 66 | - | - | - | - |
| nad4 | L | 8102 | 9433 | 1332 | - | 1 | ATT | TAG |
| nad4l | L | 9439 | 9732 | 294 | 5 | - | ATG | TAA |
| trnT | H | 9735 | 9803 | 69 | 2 | - | - | - |
| trnP | L | 9814 | 9878 | 65 | 10 | - | - | - |
| nad6 | H | 9880 | 10,398 | 519 | 1 | - | ATC | TAA |
| cob | H | 10,398 | 11,531 | 1134 | - | 1 | ATG | TAA |
| trnS2 | H | 11,530 | 11,596 | 67 | - | 2 | - | - |
| nad1 | L | 11,614 | 12,564 | 951 | 17 | - | ATG | TAA |
| trnL1 | L | 12,567 | 12,634 | 68 | 2 | - | - | - |
| rrnL | L | 12,634 | 13,949 | 1316 | - | 1 | - | - |
| trnV | L | 13,927 | 13,997 | 71 | - | 23 | - | - |
| rrnS | L | 14,001 | 14,774 | 774 | 3 | - | - | - |
| Regions | T (%) | C (%) | A (%) | G (%) | A + T (%) | AT Skew | GC Skew |
|---|---|---|---|---|---|---|---|
| Complete sequence | 31.18 | 18.04 | 38.95 | 11.82 | 70.14 | 0.111 | −0.208 |
| tRNAs | 35.03 | 12.35 | 36.24 | 16.38 | 71.28 | 0.017 | 0.14 |
| rRNAs | 39.43 | 10.1 | 33.44 | 17.03 | 72.87 | −0.082 | 0.256 |
| Control region | 44.03 | 7.76 | 41.51 | 6.71 | 85.54 | −0.029 | −0.073 |
| atp6 | 34.67 | 20 | 34.67 | 10.67 | 69.34 | 0 | −0.304 |
| atp8 | 32.7 | 23.27 | 35.85 | 8.18 | 68.55 | 0.046 | −0.48 |
| cob | 34.39 | 19.75 | 31.48 | 14.37 | 65.87 | −0.044 | −0.158 |
| cox1 | 32.08 | 20.95 | 29.93 | 17.05 | 62.01 | −0.035 | −0.103 |
| cox2 | 29.36 | 19.48 | 36.34 | 14.83 | 65.7 | 0.106 | −0.136 |
| cox3 | 32.53 | 18.68 | 32.53 | 16.26 | 65.06 | 0 | −0.069 |
| nad1 | 48.05 | 10.94 | 22.4 | 18.61 | 70.45 | −0.364 | 0.26 |
| nad2 | 37.23 | 16.27 | 35.43 | 11.08 | 72.66 | −0.025 | −0.19 |
| nad3 | 33.62 | 19.42 | 34.78 | 12.17 | 68.4 | 0.017 | −0.23 |
| nad4 | 48.5 | 9.68 | 22.9 | 18.92 | 71.4 | −0.359 | 0.323 |
| nad4l | 52.72 | 7.14 | 22.45 | 17.69 | 75.17 | −0.403 | 0.425 |
| nad5 | 46.73 | 9.72 | 24.99 | 18.57 | 71.72 | −0.303 | 0.313 |
| nad6 | 36.61 | 16.76 | 37.19 | 9.44 | 73.8 | 0.008 | −0.279 |
| rrnL | 39.59 | 9.35 | 34.8 | 16.26 | 74.39 | −0.064 | 0.27 |
| rrnS | 39.15 | 11.37 | 31.14 | 18.35 | 70.29 | −0.114 | 0.235 |
| First position | 38.5 | 13.83 | 31.42 | 16.25 | 69.92 | −0.101 | 0.08 |
| Second position | 38.15 | 17.03 | 28.3 | 16.52 | 66.45 | −0.148 | −0.015 |
| Third position | 40.97 | 15.9 | 28.97 | 14.15 | 69.94 | −0.172 | −0.058 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, X.; Lin, X.; Wang, S.; Fang, J. Complete Mitochondrial Genome of Chlorogomphus papilio (Odonata: Anisoptera: Chlorogomphidae) and Phylogenetic Analyses. Biology 2025, 14, 493. https://doi.org/10.3390/biology14050493
Jin X, Lin X, Wang S, Fang J. Complete Mitochondrial Genome of Chlorogomphus papilio (Odonata: Anisoptera: Chlorogomphidae) and Phylogenetic Analyses. Biology. 2025; 14(5):493. https://doi.org/10.3390/biology14050493
Chicago/Turabian StyleJin, Xiaoxiao, Xiaojia Lin, Simeng Wang, and Jie Fang. 2025. "Complete Mitochondrial Genome of Chlorogomphus papilio (Odonata: Anisoptera: Chlorogomphidae) and Phylogenetic Analyses" Biology 14, no. 5: 493. https://doi.org/10.3390/biology14050493
APA StyleJin, X., Lin, X., Wang, S., & Fang, J. (2025). Complete Mitochondrial Genome of Chlorogomphus papilio (Odonata: Anisoptera: Chlorogomphidae) and Phylogenetic Analyses. Biology, 14(5), 493. https://doi.org/10.3390/biology14050493