
Pathway-Specific Insights into Colorectal Cancer Through Comprehensive Multi-Omics Data Integration


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort and Experimental Design
2.2. Microbiome and Pathway Analysis
2.3. Transcriptome and Pathway Analysis
2.4. Data Standardization and Preprocessing
2.5. Data Integration Using sPLS Analysis
2.6. Data Integration Using N-Integration
2.7. Statistical Analysis
3. Results
3.1. Microbiome Analysis and Functional Pathway Prediction
3.2. Transcriptome and Pathway Enrichment Analysis
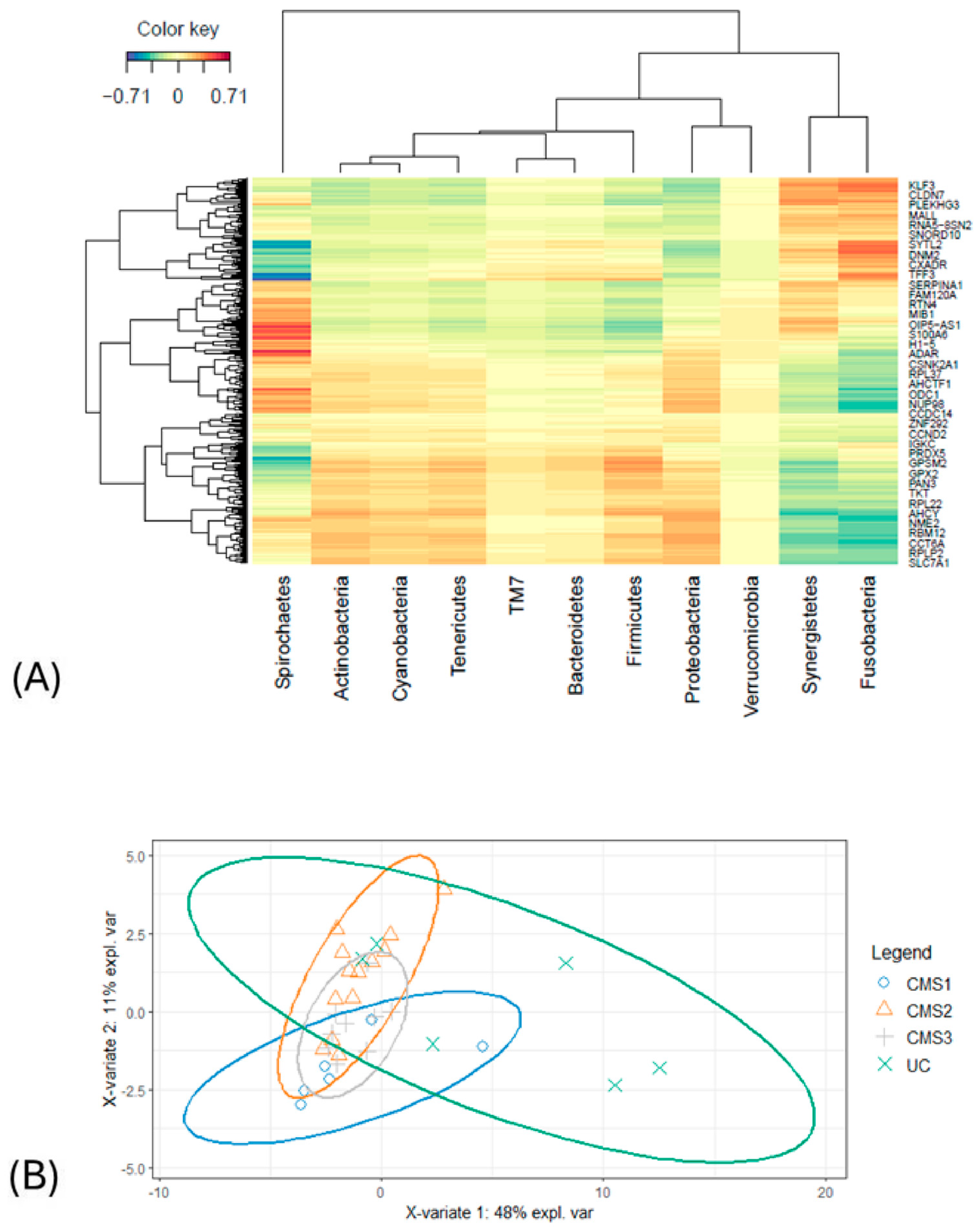
3.3. Integration of Microbiome and Transcriptome Datasets Using the sPLS Method
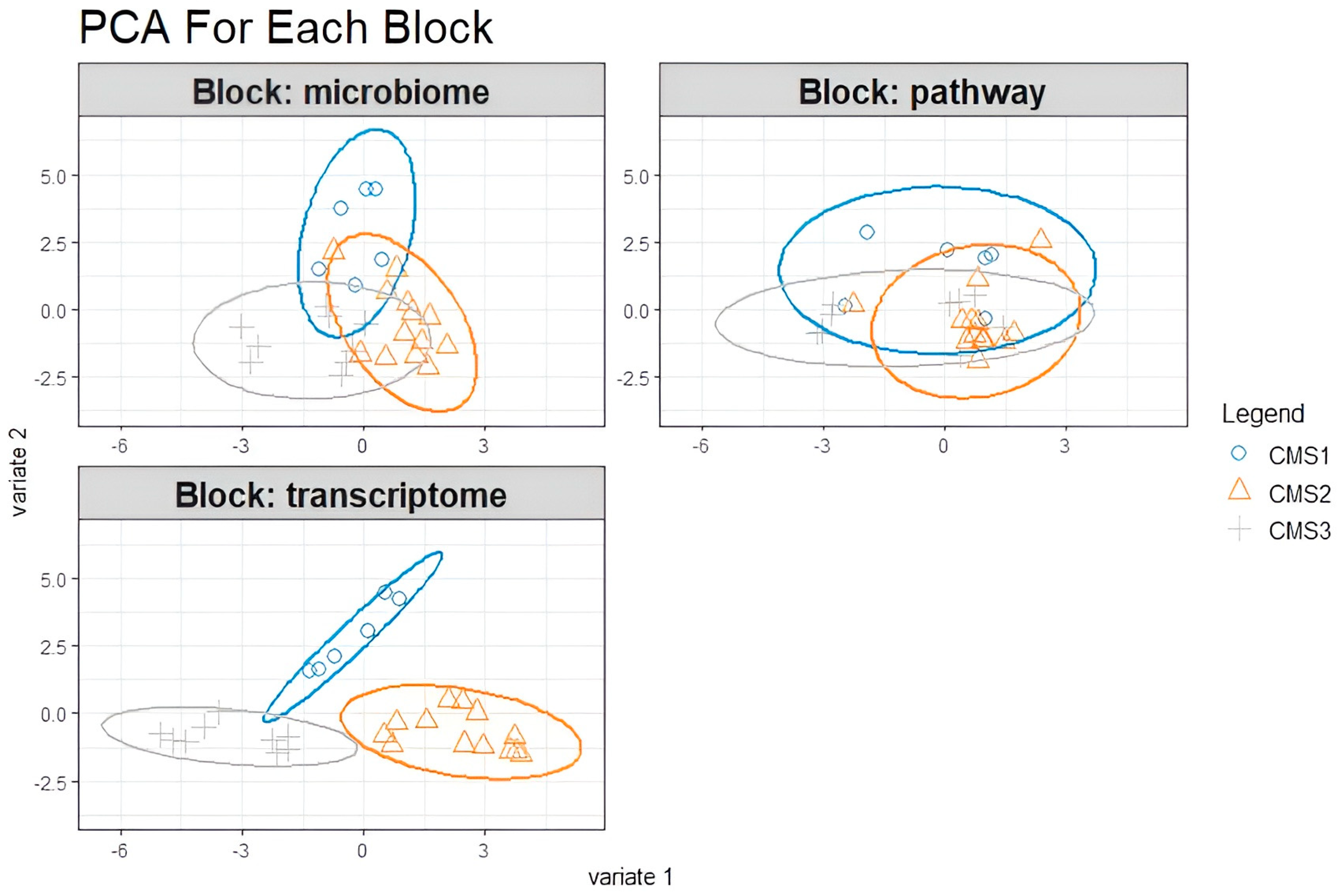
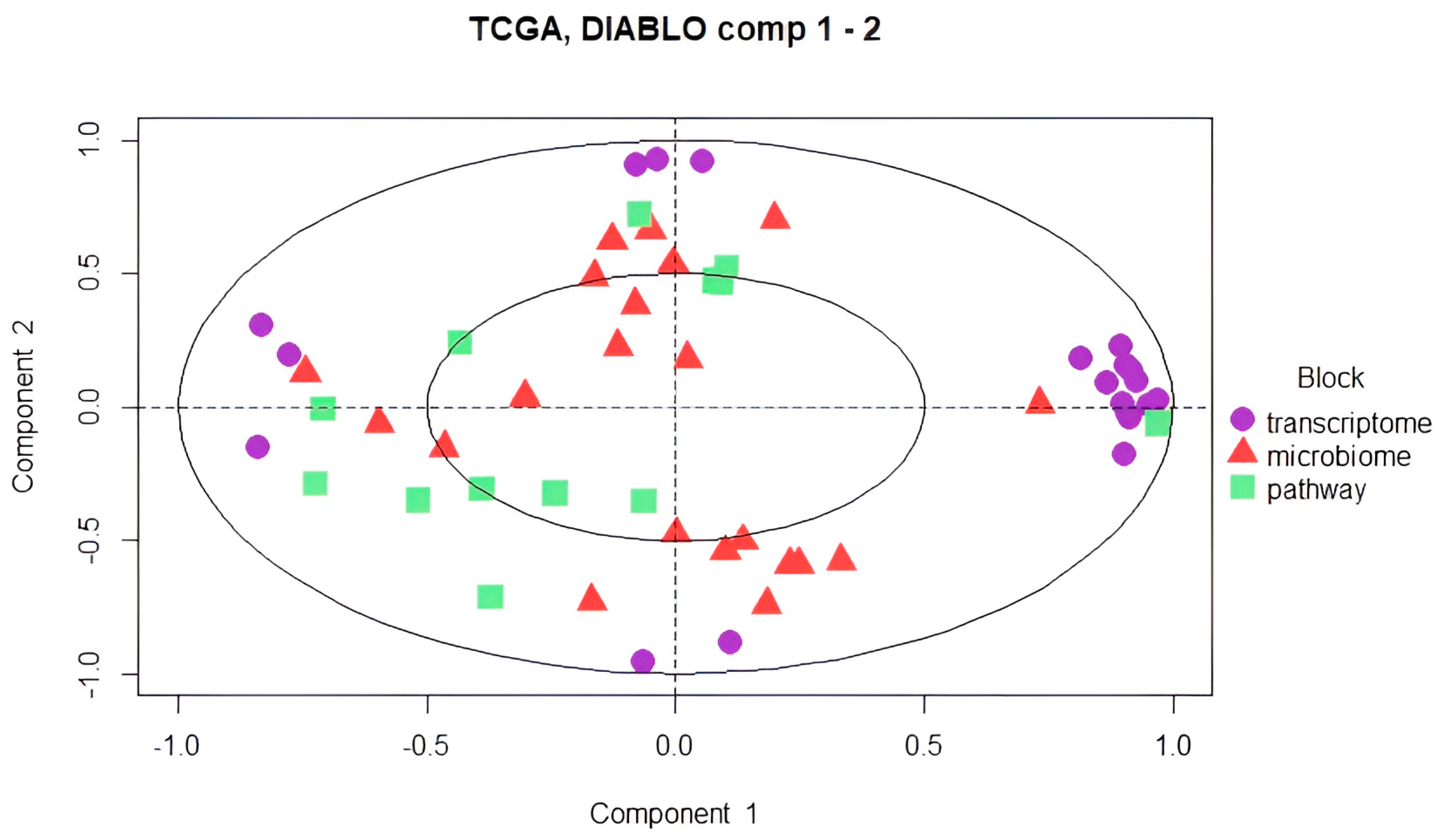
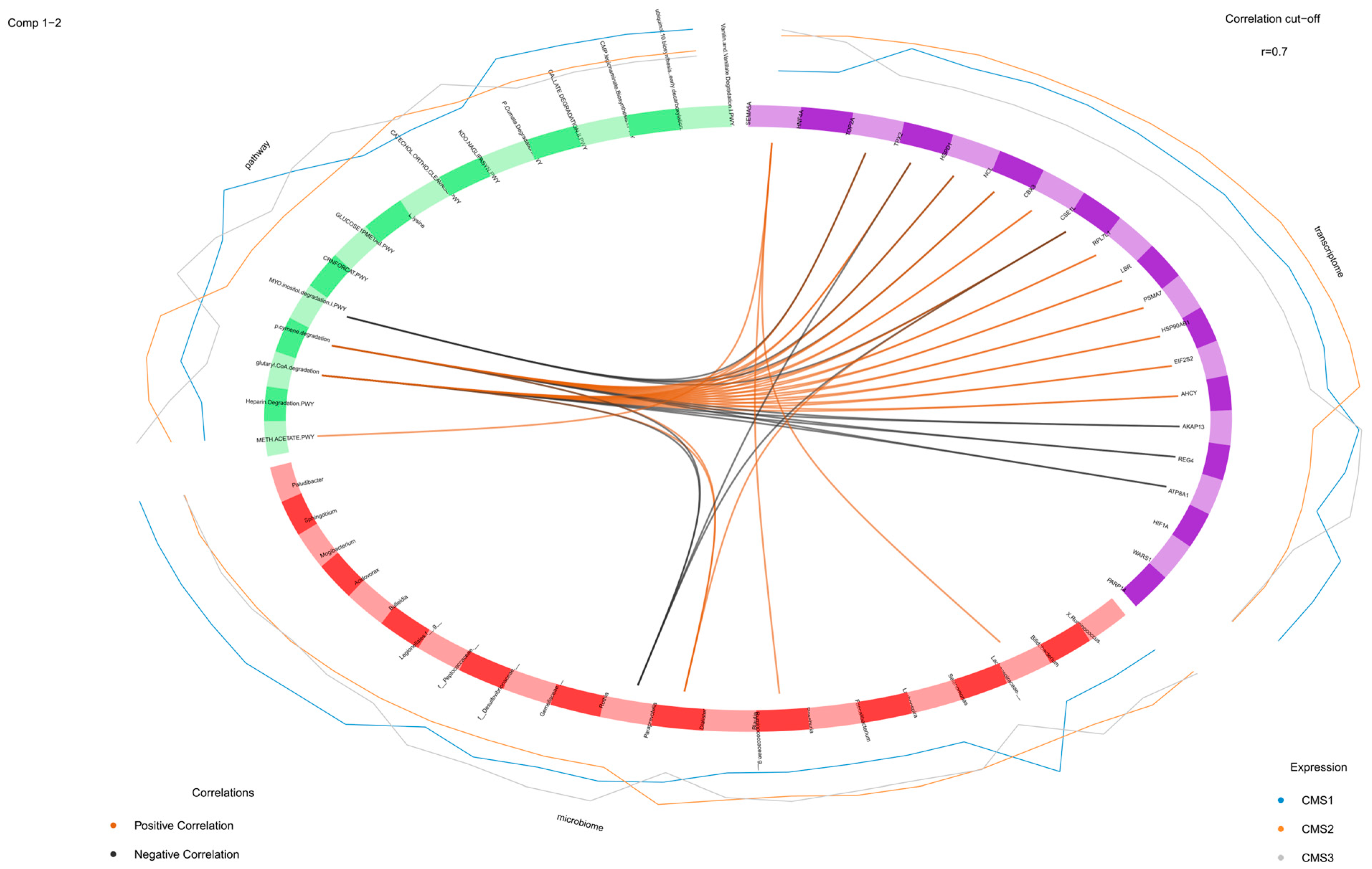
3.4. DIABLO Integration for the Preprocessed Microbiome, Transcriptome, and Microbial Pathway Datasets
4. Discussion
4.1. sPLS for Multi-Omics Data Integration
4.2. Impact of the Preprocessing Datasets
4.3. Multi-Block sPLS for Multi-Omics Data Integration for Preprocessed Datasets
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PCA | Principle Component Analysis |
| CCA | Canonical Correlation Analysis |
| CoIA | Co-Inertia Analysis |
| MFA | Multivariate Factor Analysis |
| PMA | Penalized Multivariate Analysis |
| PLD | Partial Least Squares |
| RFE | Recursive Feature Elimination |
| CRC | Colorectal Cancer |
| CMS | Consensus Molecular Subtype |
| NCBI | National Center for Biotechnology Information |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| UniProt | Universal Protein Knowledgebase |
| SPLS-DA | Sparse Partial Least Squares Discriminant Analysis |
| FDR | False Discovery Rate |
| TPM | Transcript per Million |
References
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-Generation Sequencing Technologies: An Overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Watts, G.S.; Hurwitz, B.L. Metagenomic Next-Generation Sequencing in Clinical Microbiology. Clin. Microbiol. Newsl. 2020, 42, 53–59. [Google Scholar] [CrossRef]
- Asrat, Z. Review on Mechanism of DNA Sequencing Technologies in Crop Improvement. Int. J. Res. Stud. Agric. Sci. 2021, 7, 27–36. [Google Scholar] [CrossRef]
- Reel, P.S.; Reel, S.; Pearson, E.; Trucco, E.; Jefferson, E. Using Machine Learning Approaches for Multi-Omics Data Analysis: A Review. Biotechnol. Adv. 2021, 49, 107739. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Miller, S.A. Clinical Metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef]
- Huo, T.; Canepa, R.; Sura, A.; Modave, F.; Gong, Y. Colorectal Cancer Stages Transcriptome Analysis. PLoS ONE 2017, 12, e0188697. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Yang, C.; Zhang, J.; Zhong, D.; Meng, M.; Zhang, L.; Chen, H.; Fang, L. Multi-Omic Profiling Reveals Associations between the Gut Microbiome, Host Genome and Transcriptome in Patients with Colorectal Cancer. J. Transl. Med. 2024, 22, 175. [Google Scholar] [CrossRef]
- Xie, Z.; Huang, J.; Sun, G.; He, S.; Luo, Z.; Zhang, L.; Li, L.; Yao, M.; Du, C.; Yu, W.; et al. Integrated Multi-Omics Analysis Reveals Gut Microbiota Dysbiosis and Systemic Disturbance in Major Depressive Disorder. Psychiatry Res. 2024, 334, 115804. [Google Scholar] [CrossRef]
- Creighton, C.J. Clinical Proteomics towards Multiomics in Cancer. Mass Spectrom. Rev. 2024, 43, 1255–1269. [Google Scholar] [CrossRef]
- Sankaran, K.; Holmes, S.P. Multitable Methods for Microbiome Data Integration. Front. Genet. 2019, 10, 627. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, A.; Mathé, E.; Merling, M.; Ma, Q.; Liu, B. Network Analyses in Microbiome Based on High-Throughput Multi-Omics Data. Brief. Bioinform. 2021, 22, 1639–1655. [Google Scholar] [CrossRef]
- Misra, B.B.; Langefeld, C.; Olivier, M.; Cox, L.A. Integrated Omics: Tools, Advances and Future Approaches. J. Mol. Endocrinol. 2019, 62, R21–R45. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.; Keleş, S. Sparse Partial Least Squares Regression for Simultaneous Dimension Reduction and Variable Selection. J. R. Stat. Soc. Ser. B Stat. Methodol. 2010, 72, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, K.; Wu, W.; Giannoulatou, E.; Ho, J.W.K.; Li, L. Host and Microbiome Multi-Omics Integration: Applications and Methodologies. Biophys. Rev. 2019, 11, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Holowatyj, A.N.; Gigic, B.; Herpel, E.; Scalbert, A.; Schneider, M.; Ulrich, C.M.; Achaintre, D.; Brezina, S.; Van Duijnhoven, F.J.B.; Gsur, A.; et al. Distinct Molecular Phenotype of Sporadic Colorectal Cancers Among Young Patients Based on Multiomics Analysis. Gastroenterology 2020, 158, 1155–1158.e2. [Google Scholar] [CrossRef]
- Daliri, E.B.-M.; Ofosu, F.K.; Chelliah, R.; Lee, B.H.; Oh, D.-H. Challenges and Perspective in Integrated Multi-Omics in Gut Microbiota Studies. Biomolecules 2021, 11, 300. [Google Scholar] [CrossRef]
- Louis, P.; Hold, G.L.; Flint, H.J. The Gut Microbiota, Bacterial Metabolites and Colorectal Cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Son, J.W.; Shoaie, S.; Lee, S. Systems Biology: A Multi-Omics Integration Approach to Metabolism and the Microbiome. Endocrinol. Metab. 2020, 35, 507–514. [Google Scholar] [CrossRef]
- Bisht, V.; Nash, K.; Xu, Y.; Agarwal, P.; Bosch, S.; Gkoutos, G.V.; Acharjee, A. Integration of the Microbiome, Metabolome and Transcriptomics Data Identified Novel Metabolic Pathway Regulation in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 5763. [Google Scholar] [CrossRef]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; Van De Velde, C.J.H.; Watanabe, T. Colorectal Cancer. Nat. Rev. Dis. Primers 2015, 1, 15065. [Google Scholar] [CrossRef]
- Granados-Romero, J.J.; Valderrama-Treviño, A.I.; Contreras-Flores, E.H.; Barrera-Mera, B.; Herrera Enríquez, M.; Uriarte-Ruíz, K.; Ceballos-Villalba, J.C.; Estrada-Mata, A.G.; Alvarado Rodríguez, C.; Arauz-Peña, G. Colorectal Cancer: A Review. Int. J. Res. Med. Sci. 2017, 5, 4667. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Thanki, K.; Nicholls, M.E.; Gajjar, A.; Senagore, A.J.; Qiu, S.; Szabo, C.; Hellmich, M.R.; Chao, C. Consensus Molecular Subtypes of Colorectal Cancer and Their Clinical Implications. Int. Biol. Biomed. J. 2017, 3, 105. [Google Scholar]
- Purcell, R.V.; Visnovska, M.; Biggs, P.J.; Schmeier, S.; Frizelle, F.A. Distinct Gut Microbiome Patterns Associate with Consensus Molecular Subtypes of Colorectal Cancer. Sci. Rep. 2017, 7, 11590. [Google Scholar] [CrossRef]
- Rebhan, M.; Chalifa-Caspi, V.; Prilusky, J.; Lancet, D. GeneCards: A Novel Functional Genomics Compendium with Automated Data Mining and Query Reformulation Support. Bioinformatics 1998, 14, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Krieger, C.J. MetaCyc: A Multiorganism Database of Metabolic Pathways and Enzymes. Nucleic Acids Res. 2004, 32, D438–D442. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Narayan, N.R.; Weinmaier, T.; Laserna-Mendieta, E.J.; Claesson, M.J.; Shanahan, F.; Dabbagh, K.; Iwai, S.; DeSantis, T.Z. Piphillin Predicts Metagenomic Composition and Dynamics from DADA2-Corrected 16S rDNA Sequences. BMC Genom. 2020, 21, 56. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Ulgen, E.; Ozisik, O.; Sezerman, O.U. pathfindR: An R Package for Comprehensive Identification of Enriched Pathways in Omics Data Through Active Subnetworks. Front. Genet. 2019, 10, 858. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.-A. mixOmics: An R Package for ‘omics Feature Selection and Multiple Data Integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023; Available online: https://www.R-project.org/ (accessed on 1 April 2024).
- Wang, K.; Xu, C.; Li, W.; Ding, L. Emerging Clinical Significance of Claudin-7 in Colorectal Cancer: A Review. Cancer Manag. Res. 2018, 10, 3741–3752. [Google Scholar] [CrossRef] [PubMed]
- Karahashi, Y.; Cueno, M.E.; Kamio, N.; Takahashi, Y.; Takeshita, I.; Soda, K.; Maruoka, S.; Gon, Y.; Sato, S.; Imai, K. Fusobacterium Nucleatum Putatively Affects the Alveoli by Disrupting the Alveolar Epithelial Cell Tight Junction, Enlarging the Alveolar Space, and Increasing Paracellular Permeability. Biochem. Biophys. Res. Commun. 2023, 682, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.L.; Yang, M.; Wang, Y.C.F.; Song, C.C.; Du, G.H.; Tang, G.Y. Effects of Fusobacterium nucleus derived outer membrane vesicles on claudin-4 expression in oral epithelial cells. Shanghai Kou Qiang Yi Xue 2023, 32, 126–131. [Google Scholar] [PubMed]
- Shen, W.; Yuan, L.; Hao, B.; Xiang, J.; Cheng, F.; Wu, Z.; Li, X. KLF3 Promotes Colorectal Cancer Growth by Activating WNT1. Aging 2024, 16, 2475. [Google Scholar] [CrossRef]
- Li, X.; Huang, J.; Yu, T.; Fang, X.; Lou, L.; Xin, S.; Ji, L.; Jiang, F.; Lou, Y. Fusobacterium Nucleatum Promotes the Progression of Colorectal Cancer Through Cdk5-Activated Wnt/β-Catenin Signaling. Front. Microbiol. 2021, 11, 545251. [Google Scholar] [CrossRef]
- Li, J.; Liu, C.; Sato, T. Novel Antitumor Invasive Actions of p-Cymene by Decreasing MMP-9/TIMP-1 Expression Ratio in Human Fibrosarcoma HT-1080 Cells. Biol. Pharm. Bull. 2016, 39, 1247–1253. [Google Scholar] [CrossRef]
- Bhatti, M.Z.; Ali, A.; Duong, H.-Q.; Chen, J.; Rahman, F.-U. Anticancer Activity and Mechanism of Bis-Pyrimidine Based Dimetallic Ru(II)(H6-p-Cymene) Complex in Human Non-Small Cell Lung Cancer via P53-Dependent Pathway. J. Inorg. Biochem. 2019, 194, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Leng, Q.; Zhang, C.; Zhu, Y.; Wang, J. P-Cymene Prevent High-Fat Diet-Associated Colorectal Cancer by Improving the Structure of Intestinal Flora. J. Cancer 2021, 12, 4355–4361. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.P.; Mills, C.A.; Anderson, K.A.; Henriques, B.J.; Lucas, T.G.; Francisco, S.; Liu, J.; Ilkayeva, O.R.; Adams, A.E.; Kulkarni, S.R.; et al. Deglutarylation of Glutaryl-CoA Dehydrogenase by Deacylating Enzyme SIRT5 Promotes Lysine Oxidation in Mice. J. Biol. Chem. 2022, 298, 101723. [Google Scholar] [CrossRef]
- Xie, L.; Xiao, Y.; Meng, F.; Li, Y.; Shi, Z.; Qian, K. Functions and Mechanisms of Lysine Glutarylation in Eukaryotes. Front. Cell Dev. Biol. 2021, 9, 667684. [Google Scholar] [CrossRef]
- Minogue, E.; Cunha, P.P.; Wadsworth, B.J.; Grice, G.L.; Sah-Teli, S.K.; Hughes, R.; Bargiela, D.; Quaranta, A.; Zurita, J.; Antrobus, R.; et al. Glutarate Regulates T Cell Metabolism and Anti-Tumour Immunity. Nat. Metab. 2023, 5, 1747–1764. [Google Scholar] [CrossRef]
- Wachoski-Dark, E.; Zhao, T.; Khan, A.; Shutt, T.E.; Greenway, S.C. Mitochondrial Protein Homeostasis and Cardiomyopathy. Int. J. Mol. Sci. 2022, 23, 3353. [Google Scholar] [CrossRef]
- Duan, Y.; Yu, J.; Chen, M.; Lu, Q.; Ning, F.; Gan, X.; Liu, H.; Ye, Y.; Lu, S.; Lash, G.E. Knockdown of Heat Shock Protein Family D Member 1 (HSPD1) Promotes Proliferation and Migration of Ovarian Cancer Cells via Disrupting the Stability of Mitochondrial 3-Oxoacyl-ACP Synthase (OXSM). J. Ovarian Res. 2023, 16, 81. [Google Scholar] [CrossRef]
- Parma, B.; Ramesh, V.; Gollavilli, P.N.; Siddiqui, A.; Pinna, L.; Schwab, A.; Marschall, S.; Zhang, S.; Pilarsky, C.; Napoli, F.; et al. Metabolic Impairment of Non-Small Cell Lung Cancers by Mitochondrial HSPD1 Targeting. J. Exp. Clin. Cancer Res. 2021, 40, 248. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Zhao, X.; Xia, L.; Lian, J.; You, J. Clinicopathological and Prognostic Significance of CBX3 Expression in Human Cancer: A Systematic Review and Meta-Analysis. Dis. Markers 2020, 2020, 2412741. [Google Scholar] [CrossRef]
- Niu, H.; Chen, P.; Fan, L.; Sun, B. Comprehensive Pan-Cancer Analysis on CBX3 as a Prognostic and Immunological Biomarker. BMC Med. Genom. 2022, 15, 29. [Google Scholar] [CrossRef]
- Li, H.; Wang, L.; Ruan, Z.; Li, X.; Yang, Y.; Fang, J.; Wang, R. CSE1L as a prognostic biomarker associated with pan cancer immune infiltration and drug sensitivity. Expert Rev. Clin. Immunol. 2024, 20, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Shaath, H.; Vishnubalaji, R.; Elango, R.; Velayutham, D.; Jithesh, P.V.; Alajez, N.M. Therapeutic Targeting of the TPX2/TTK Network in Colorectal Cancer. Cell Commun. Signal 2023, 21, 265. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.; Deshpande, S.; Sengupta, K. Nuclear Envelope Protein Lamin B Receptor Protects the Genome from Chromosomal Instability and Tumorigenesis. Hum. Mol. Genet. 2023, 32, 745–763. [Google Scholar] [CrossRef] [PubMed]
- Loftus, M.; Hassouneh, S.A.-D.; Yooseph, S. Bacterial Community Structure Alterations within the Colorectal Cancer Gut Microbiome. BMC Microbiol. 2021, 21, 98. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group 1 | Group 2 | H | p-Value | q-Value |
|---|---|---|---|---|
| CMS1 | CMS2 | 0.769231 | 0.380455 | 0.570683 |
| CMS3 | 0.222222 | 0.637352 | 0.764822 | |
| UC | 3.692308 | 0.054664 | 0.231300 | |
| CMS2 | CMS3 | 0.054627 | 0.8152 | 0.8152 |
| UC | 1.3 | 0.254213 | 0.508426 | |
| CMS3 | UC | 3.125 | 0.0771 | 0.231300 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karaman, T.; Oktem Okullu, S.; Bayram Akçapınar, G.; Sezerman, O.U. Pathway-Specific Insights into Colorectal Cancer Through Comprehensive Multi-Omics Data Integration. Biology 2025, 14, 468. https://doi.org/10.3390/biology14050468
Karaman T, Oktem Okullu S, Bayram Akçapınar G, Sezerman OU. Pathway-Specific Insights into Colorectal Cancer Through Comprehensive Multi-Omics Data Integration. Biology. 2025; 14(5):468. https://doi.org/10.3390/biology14050468
Chicago/Turabian StyleKaraman, Tayyip, Sinem Oktem Okullu, Günseli Bayram Akçapınar, and Osman Ugur Sezerman. 2025. "Pathway-Specific Insights into Colorectal Cancer Through Comprehensive Multi-Omics Data Integration" Biology 14, no. 5: 468. https://doi.org/10.3390/biology14050468
APA StyleKaraman, T., Oktem Okullu, S., Bayram Akçapınar, G., & Sezerman, O. U. (2025). Pathway-Specific Insights into Colorectal Cancer Through Comprehensive Multi-Omics Data Integration. Biology, 14(5), 468. https://doi.org/10.3390/biology14050468