
Discovery of Genomic Targets and Therapeutic Candidates for Liver Cancer Using Single-Cell RNA Sequencing and Molecular Docking



Simple Summary
Abstract
1. Introduction
2. Methods and Materials
2.1. Data Description
2.2. Hybrid Model
2.2.1. scDEA
2.2.2. scHD4E
2.3. Individual Methods of Differential Expression Analysis
2.3.1. TPMM
2.3.2. ROSeq
2.3.3. Limma (Voom)
2.3.4. Seurat
2.4. Overview of the Analysis Process
2.5. Construction of PPI Network for DEGs
2.6. Gene Ontology and Pathway Enrichment Analysis
2.7. Validation of Differential Expression Analysis of hHub Genes
2.8. Liver Cancer Stage-Wise Relation to Hub Genes
2.9. Survival Analysis of hHub Genes for Liver Cancer
2.10. Molecular Docking
3. Results
3.1. Identification of Common Differentially Expressed Genes
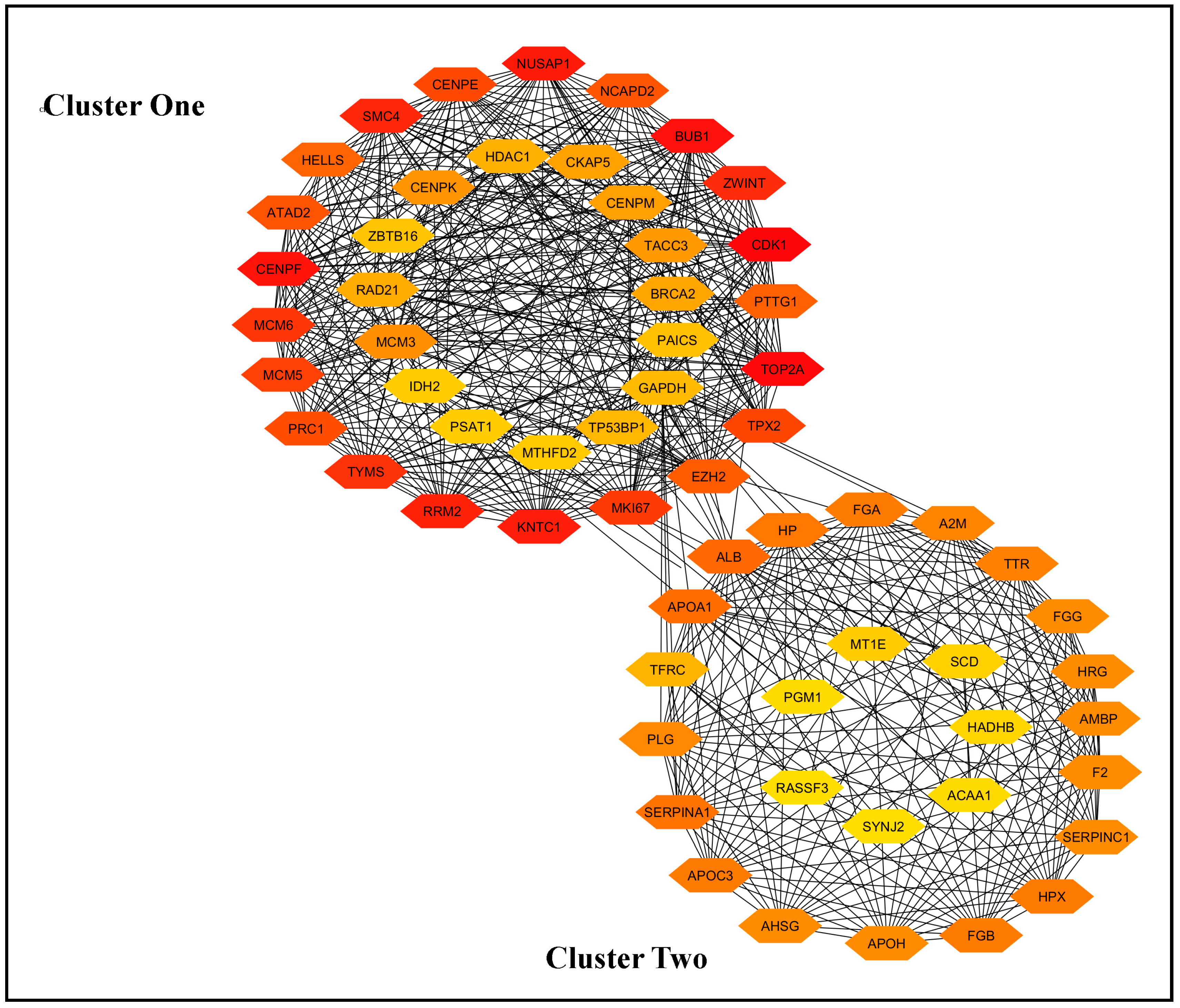
3.2. Identification of Hub Genes Using STRING and Cytoscape
3.3. Protein−Protein-Interaction (PPI) Network and Selection of the Most Important Genes
3.4. Checking Differentiability of hHub Genes Using GEPIA2
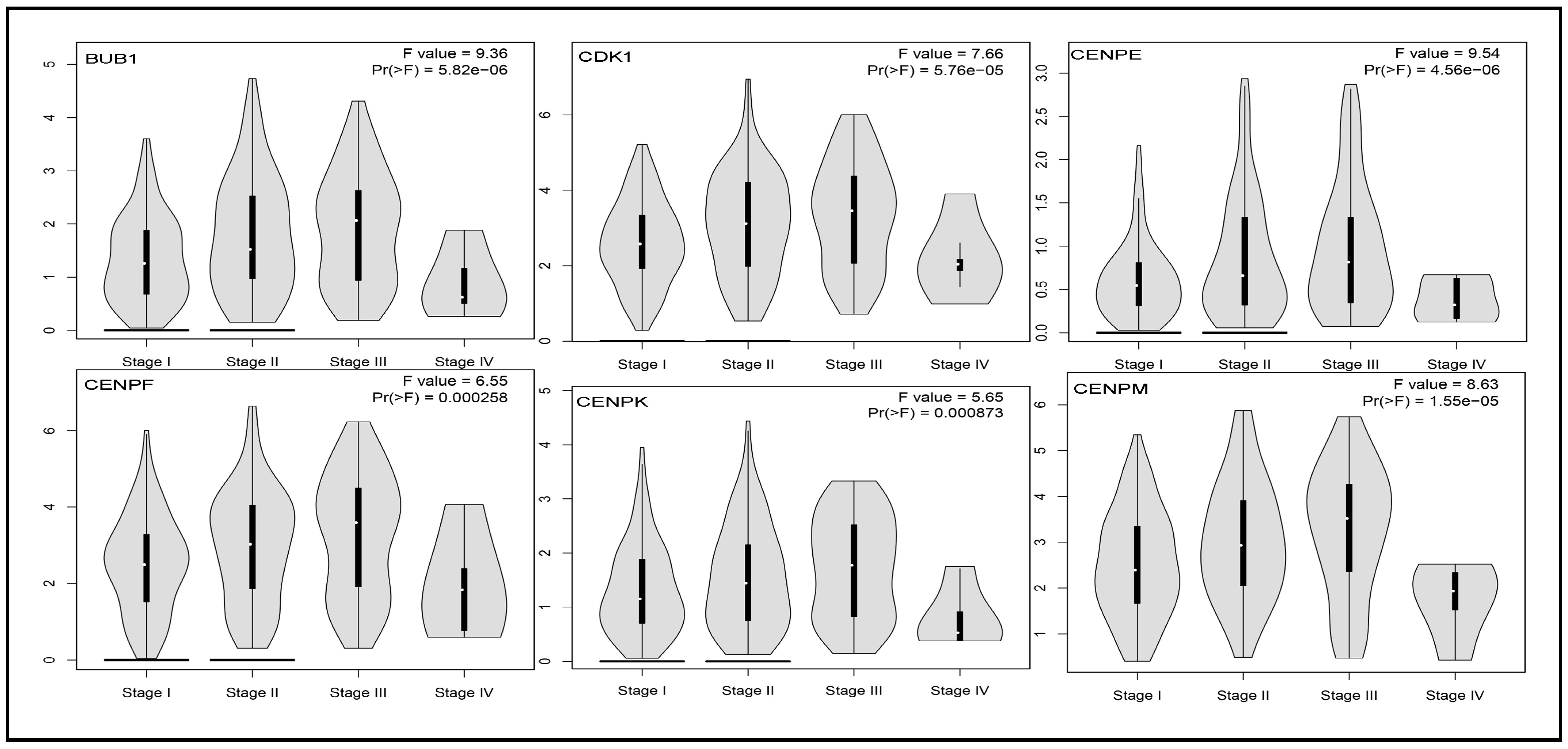
3.5. Impact of Hub Genes on the Stage of Liver Cancer
3.6. GO and Pathway Analysis of Hub Genes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | GO ID | Terms and References | Associated Gene Count | Common Associated Genes from at Least Two Databases | |||
|---|---|---|---|---|---|---|---|
| DAVID | STRING | Enrichr | WebGestalt | ||||
| Biological Process | GO:0051301 | cell division [66] | 14 | 16 | - | - | BUB1, PTTG1, TPX2, ZWINT, CENPE, CENPF, CDK1, HELLS, KNTC1, MCM5, NCAPD2, PRC1, SMC4, TACC3 |
| GO:0042730 | fibrinolysis [67] | 06 | 06 | 05 | 07 | F2, FGA, FGB, FGG, HRG, PLG | |
| GO:0000070 | mitotic sister chromatid segregation [68] | 05 | 08 | 09 | - | ZWINT, CENPK, KNTC1, NUSAP1, SMC4 | |
| GO:0031639 | plasminogen activation [69] | 04 | 04 | 03 | - | FGA, FGB, FGG, APOH | |
| GO:0030193 | regulation of blood coagulation [70] | 04 | 08 | 02 | - | APOH, F2, HRG, SERPINC1 | |
| GO:0007596 | blood coagulation [71] | 06 | 09 | - | - | F2, FGA, FGB, PLG, SERPINA1, SERPINC1 | |
| GO:0006953 | acute-phase response [72] | 06 | 06 | - | - | AHSG, A2M, F2, HP, SERPINA1, TFRC | |
| GO:0051918 | negative regulation of fibrinolysis | 04 | 04 | 02 | - | PLG, F2, HRG, APOH | |
| GO:0030168 | platelet activation [73] | 05 | 05 | - | - | F2, FGA, FGB, HRG, FGG | |
| GO:0051382 | kinetochore assembly [74] | 03 | 04 | 03 | - | CENPE, CENPF, CENPK, KNTC1 | |
| GO:0051383 | kinetochore organization | - | 05 | 02 | - | CENPE, CENPF | |
| GO:0007094 | mitotic spindle assembly checkpoint signaling [75] | 04 | 04 | 04 | - | BUB1, ZWINT, CENPF, KNTC1 | |
| GO:0072378 | blood coagulation, fibrin clot formation [76] | 03 | 05 | 03 | - | FGA, FGG, FGB | |
| GO:1900026 | positive regulation of substrate adhesion-dependent cell spreading | 04 | 04 | 04 | - | FGA, FGG, FGB, APOA1 | |
| GO:0090277 | positive regulation of peptide hormone secretion [77] | 03 | - | 03 | - | FGA, FGG, FGB, APOA1 | |
| GO:0034508 | centromere complex assembly [78] | - | 05 | 02 | - | CENPE, CENPF | |
| GO:0030195 | negative regulation of blood coagulation | - | 07 | 06 | - | FGA, FGB, FGG, PLG, F2, APOH | |
| GO:0140014 | mitotic nuclear division [79] | - | 09 | 05 | 13 | NCAPD2, CENPE, CENPK, PRC1, NUSAP1, TPX2, ZWINT, KNTC1, SMC4 | |
| GO:0000280 | nuclear division | - | 12 | - | 14 | ZWINT, BUB1, PRC1, PTTG1, TOP2A, SMC4, TPX2, CENPE, KNTC1, NUSAP1, NCAPD2 | |
| GO:1903047 | mitotic cell cycle process [55] | - | 16 | - | 21 | NCAPD2, KNTC1, MCM3, MCM6, PRC1, CENPF, SMC4, TACC3, TPX2, BUB1, CDK1, ZWINT, CENPE, NUSAP1, EZH2 | |
| Associated Gene Count | Associated Common Genes | ||||||
| Database | ID | Pathways | DAVID | STRING | Enrichr | WebGestalt | |
| KEGG | hsa04610 | complement and coagulation cascades [80] | 08 | 08 | 08 | 08 | A2M, F2, FGA, FGB, FGG, PLG, SERPINA1, SERPINC1 |
| hsa04110 | cell cycle [81] | 06 | 06 | 06 | 05 | BUB1, PTTG1, CDK1, MCM3, MCM5, MCM6 | |
| hsa03030 | DNA replication [82] | 03 | 03 | 03 | 03 | MCM3, MCM5, MCM6 | |
| hsa04611 | platelet activation [83] | 04 | 04 | 04 | 03 | F2, FGA, FGB, FGG | |
| hsa04979 | cholesterol metabolism [84] | 03 | 03 | 03 | 03 | APOA1, APOC3, APOH | |
| hsa04114 | oocyte meiosis | 03 | - | 03 | 03 | BUB1, PTTG1, CDK1 | |
| coronavirus disease [85] | 04 | - | 04 | F2, FGA, FGB, FGG | |||
| hsa04115 | p53 signaling pathway [86] | - | - | 02 | 02 | CDK1, RRM2 | |
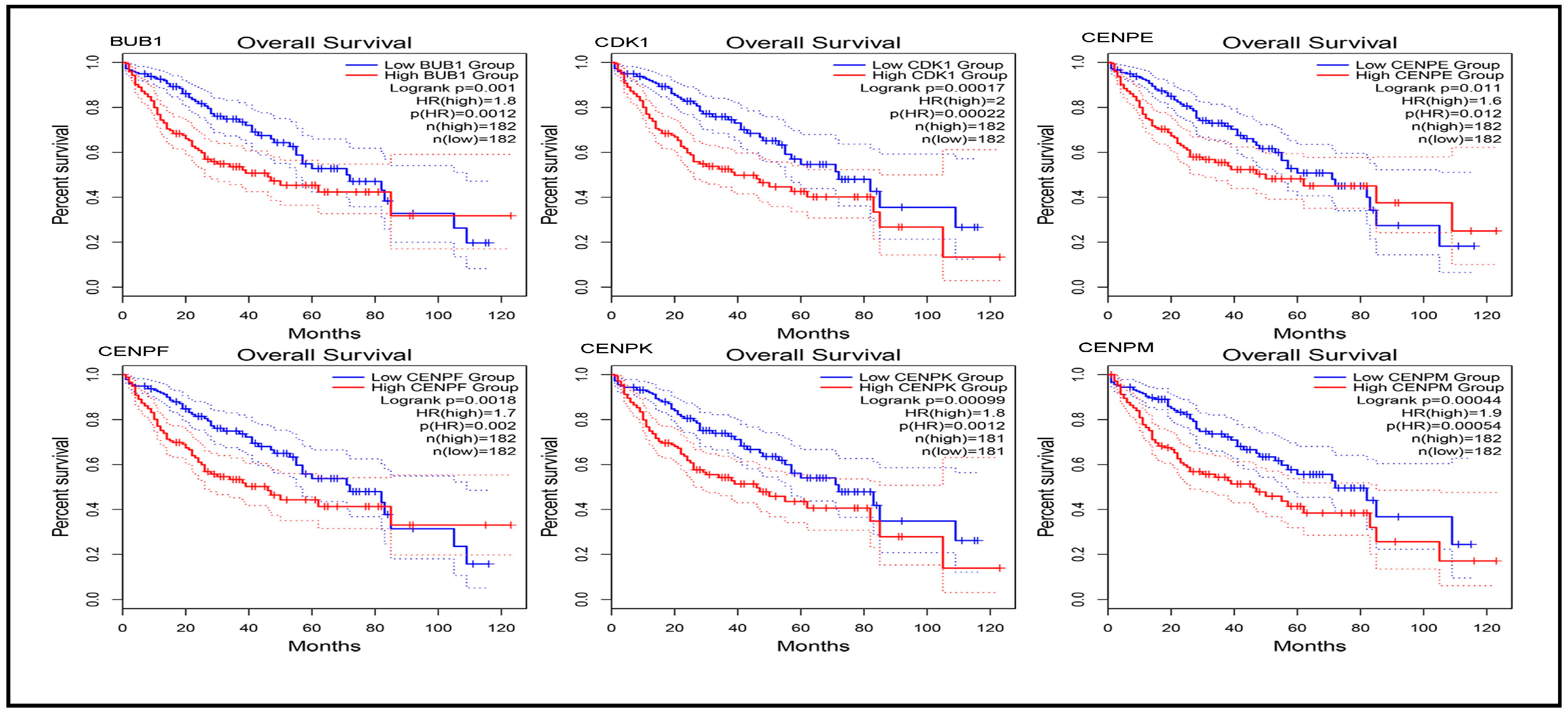
3.7. Prognostic Power of Hub Genes
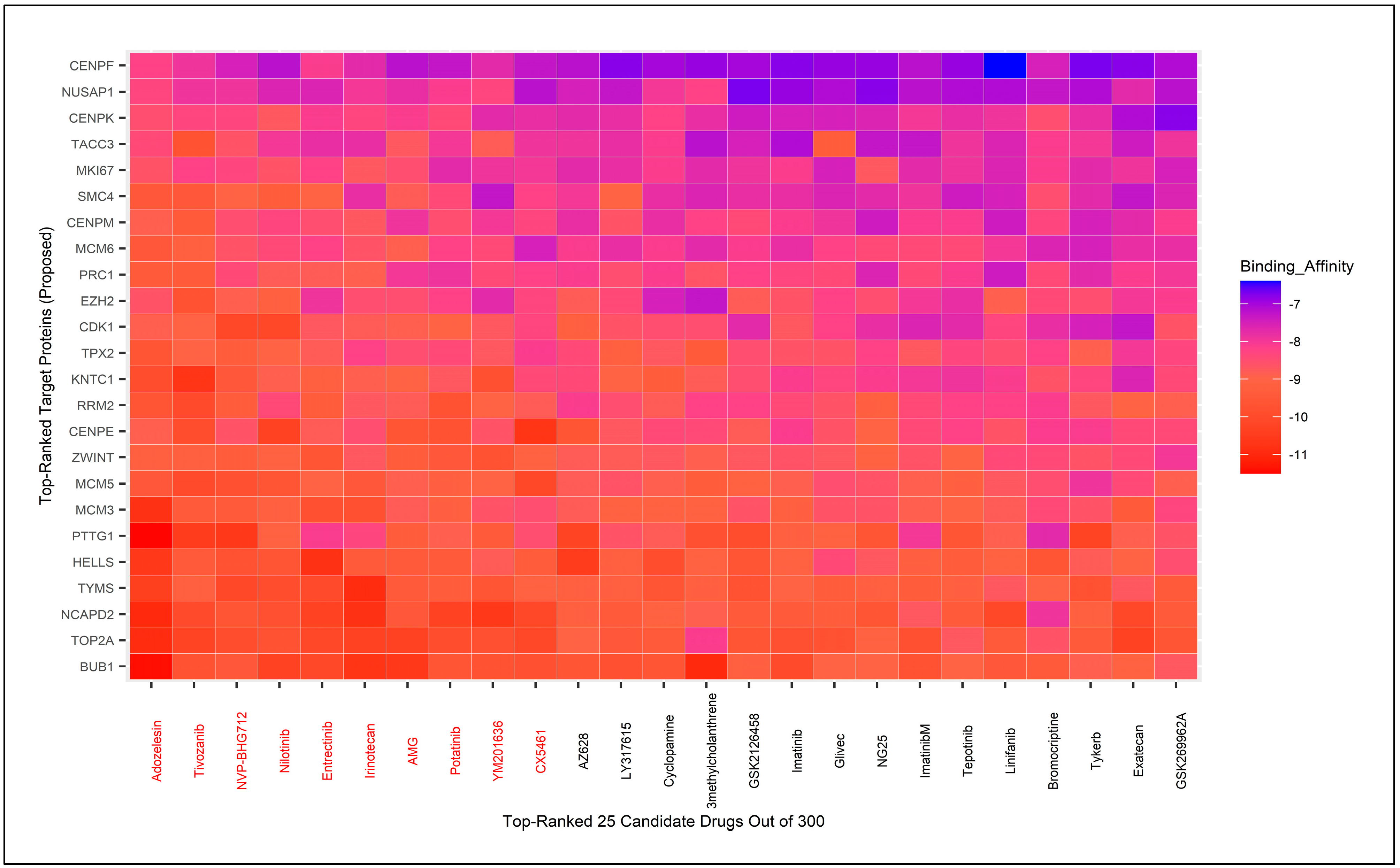
3.8. Molecular Docking for Drug Repurposing Guided by Biomarker Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ji, B.; Cai, H.; Jiao, Y. Identification of Core Genes Related with Trophinin-Associated Protein (TROAP) Expression in Liver Cancer. Res. Sq. 2020, 1–15. [Google Scholar] [CrossRef]
- Wu, M.; Liu, Z.; Zhang, A.; Li, N. Identification of key genes and pathways in hepatocellular carcinoma. Medicine 2019, 98, e14287. [Google Scholar] [CrossRef] [PubMed]
- Turdean, S.; Gurzu, S.; Turcu, M.; Voidazan, S.; Sin, A.; Marinescu, G.; Hospital, M. Current data in clinicopathological characteristics of primary hepatic tumors. Rom. J. Morphol. Embryol. 2012, 2012, 53. [Google Scholar]
- Severi, T.; Van Malenstein, H.; Verslype, C.; Van Pelt, J.F. Tumor initiation and progression in hepatocellular carcinoma: Risk factors, classification, and therapeutic targets. Acta Pharmacol. Sin. 2010, 31, 1409–1420. [Google Scholar] [CrossRef]
- Michielsen, P.; Ho, E. Viral hepatitis B and hepatocellular carcinoma. Acta Gastroenterol. Belg. 2011, 74, 4–8. [Google Scholar] [CrossRef]
- McGivern, D.R.; Lemon, S.M. Virus-specific mechanisms of carcinogenesis in hepatitis C virus associated liver cancer. Oncogene 2011, 30, 1969–1983. [Google Scholar] [CrossRef]
- Roman, S.; Fierro, N.A.; Moreno-Luna, L.E.; Panduro, A. Hepatitis B Virus Genotype H and Environmental Factors Associated to the Low Prevalence of Hepatocellular Carcinoma in Mexico. J. Cancer Ther. 2013, 4, 367–376. [Google Scholar] [CrossRef]
- Tang, R.; Liu, H.; Yuan, Y.; Xie, K.; Xu, P.; Liu, X.; Wen, J. Genetic factors associated with risk of metabolic syndrome and hepatocellular carcinoma. Oncotarget 2017, 8, 35403–35411. [Google Scholar] [CrossRef]
- Li, J.; Huang, Z.; Wei, L. Bioinformatics analysis of the gene expression profile of hepatocellular carcinoma: Preliminary results. Wspolczesna Onkol. 2016, 20, 20–27. [Google Scholar] [CrossRef]
- Wan, Z.; Zhang, X.; Luo, Y.; Zhao, B. Identification of Hepatocellular Carcinoma-Related Potential Genes and Pathways through Bioinformatic-Based Analyses. Genet. Test. Mol. Biomark. 2019, 23, 766–777. [Google Scholar] [CrossRef]
- Zhang, P.; Feng, J.; Wu, X.; Chu, W.; Zhang, Y.; Li, P. Bioinformatics Analysis of Candidate Genes and Pathways Related to Hepatocellular Carcinoma in China: A Study Based on Public Databases. Pathol. Oncol. Res. 2021, 27, 588532. [Google Scholar] [CrossRef] [PubMed]
- Identification of Critical Genes and Biological Signaling for Metformin Treated Liver Cancer. bioRxiv. 2021. Available online: https://www.biorxiv.org/content/10.1101/2021.12.29.474467v1?rss=1&utm_source=researcher_app&utm_medium=referral&utm_campaign=RESR_MRKT_Researcher_inbound (accessed on 10 December 2024).
- Das, S.; Rai, S.N. SwarnSeq: An improved statistical approach for differential expression analysis of single-cell RNA-seq data. Genomics 2021, 113, 1308–1324. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ning, B.; Shi, T. Single-cell RNA-seq technologies and related computational data analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef]
- Biswas, B.; Kumar, N.; Sugimoto, M.; Hoque, M.A. scHD4E: Novel ensemble learning-based differential expression analysis method for single-cell RNA-sequencing data. Comput. Biol. Med. 2024, 178, 108769. [Google Scholar] [CrossRef]
- Massalha, H.; Bahar Halpern, K.; Abu-Gazala, S.; Jana, T.; Massasa, E.E.; Moor, A.E.; Buchauer, L.; Rozenberg, M.; Pikarsky, E.; Amit, I.; et al. A single cell atlas of the human liver tumor microenvironment. Mol. Syst. Biol. 2020, 16, e9682. [Google Scholar] [CrossRef]
- Li, B.; Li, Y.; Zhou, H.; Xu, Y.; Cao, Y.; Cheng, C.; Peng, J.; Li, H.; Zhang, L.; Su, K.; et al. Multiomics identifies metabolic subtypes based on fatty acid degradation allocating personalized treatment in hepatocellular carcinoma. Hepatology 2024, 79, 289–306. [Google Scholar] [CrossRef]
- Zheng, C.; Zheng, L.; Yoo, J.K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356. [Google Scholar] [CrossRef]
- Li, H.S.; Ou-Yang, L.; Zhu, Y.; Yan, H.; Zhang, X.F. ScDEA: Differential expression analysis in single-cell RNA-sequencing data via ensemble learning. Brief. Bioinform. 2022, 23, bbab402. [Google Scholar] [CrossRef]
- Srihari, S.; Leong, H.W. Temporal dynamics of protein complexes in PPI networks: A case study using yeast cell cycle dynamics. BMC Bioinform. 2012, 13 (Suppl. S1), S16. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Scardoni, G.; Lau, C. Centralities Based Analysis of Complex Networks. In New Frontiers in Graph Theory; InTech: Berlin, Germany, 2012. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models. Genome Res. 1971, 13, 426. [Google Scholar] [CrossRef]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Doms, A.; Schroeder, M. GoPubMed: Exploring PubMed with the gene ontology. Nucleic Acids Res. 2005, 33 (Suppl. S2), 783–786. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Győrffy, B. Integrated analysis of public datasets for the discovery and validation of survival-associated genes in solid tumors. Innovation 2024, 5, 100625. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank Helen. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Yasen, M.; Kajino, K.; Kano, S.; Tobita, H.; Yamamoto, J.; Uchiumi, T.; Kon, S.; Maeda, M.; Obulhasim, G.; Arii, S.; et al. The up-regulation of Y-box binding proteins (DNA binding protein A and Y-box binding protein-1) as prognostic markers of hepatocellular carcinoma. Clin. Cancer Res. 2005, 11, 7354–7361. [Google Scholar] [CrossRef] [PubMed]
- Malz, M.; Weber, A.; Singer, S.; Riehmer, V.; Bissinger, M.; Riener, M.O.; Longerich, T.; Soll, C.; Vogel, A.; Angel, P.; et al. Overexpression of far upstream element binding proteins: A mechanism regulating proliferation and migration in liver cancer cells. Hepatology 2009, 50, 1130–1139. [Google Scholar] [CrossRef]
- Raucci, R.; Colonna, G.; Guerriero, E.; Capone, F.; Accardo, M.; Castello, G.; Costantini, S. Structural and functional studies of the human selenium binding protein-1 and its involvement in hepatocellular carcinoma. Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 513–522. [Google Scholar] [CrossRef]
- Qi, Q.; Obianyo, O.; Du, Y.; Fu, H.; Li, S.; Ye, K. Blockade of Asparagine Endopeptidase Inhibits Cancer Metastasis. J. Med. Chem. 2017, 60, 7244–7255. [Google Scholar] [CrossRef]
- Qi, Y.; Qi, H.; Li, B. Expression and Significance of KLKB1 in Hepatocellular Carcinoma based on Multiple Gene Databases. Int. Core J. Eng. 2022, 8, 210–216. [Google Scholar]
- Kim, H.; Kim, Y.; Chung, Y.; Abdul, R.; Sim, J.; Ahn, H.; Shin, S.J.; Paik, S.S.; Kim, H.J.; Jang, K.; et al. Single-stranded DNA binding protein 2 expression is associated with patient survival in hepatocellular carcinoma. BMC Cancer 2018, 18, 1244. [Google Scholar] [CrossRef]
- Zhou, X.; Huang, J.M.; Li, T.M.; Liu, J.Q.; Wei, Z.L.; Lan, C.L.; Zhu, G.Z.; Liao, X.W.; Ye, X.P.; Peng, T. Clinical Significance and Potential Mechanisms of ATP Binding Cassette Subfamily C Genes in Hepatocellular Carcinoma. Front. Genet. 2022, 13, 805961. [Google Scholar] [CrossRef]
- Polireddy, K.; Chavan, H.; Abdulkarim, B.A.; Krishnamurthy, P. Functional significance of the ATP-binding cassette transporter B6 in hepatocellular carcinoma. Mol. Oncol. 2011, 5, 410–425. [Google Scholar] [CrossRef]
- Cai, C.; Song, X.; Yu, C. Identification of genes in hepatocellular carcinoma induced by non-alcoholic fatty liver disease. Cancer Biomark. 2020, 29, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Yu, G.; Lee, H.W.; Li, L.; Wang, L.; Yang, D.; Pan, Y.; Ding, C.; Qian, J.; Wu, L.; et al. The Nedd8-activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer Res. 2012, 72, 3360–3371. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.P.; He, Y.; Yang, J.; Wei, X.; Fan, Y.L.; Zhang, G.G.; Zhu, Y.D.; Li, Z.Q.; Liao, H.X.; Qin, D.J.; et al. Blockade of NMT1 enzymatic activity inhibits N-myristoylation of VILIP3 protein and suppresses liver cancer progression. Signal Transduct. Target. Ther. 2023, 8, 14. [Google Scholar] [CrossRef]
- Nomura, S.; Niki, M.; Nisizawa, T.; Tamaki, T.; Shimizu, M. Microparticles as Biomarkers of Blood Coagulation in Cancer. Biomark. Cancer 2015, 7, 51–56. [Google Scholar] [CrossRef]
- Ren, Z.; Yue, Y.; Zhang, Y.; Dong, J.; Liu, Y.; Yang, X.; Lin, X.; Zhao, X.; Wei, Z.; Zheng, Y.; et al. Changes in the Peripheral Blood Treg Cell Proportion in Hepatocellular Carcinoma Patients After Transarterial Chemoembolization With Microparticles. Front. Immunol. 2021, 12, 624789. [Google Scholar] [CrossRef]
- Ramadori, P.; Klag, T.; Malek, N.P.; Heikenwalder, M. Platelets in chronic liver disease, from bench to bedside. JHEP Rep. 2019, 1, 448–459. [Google Scholar] [CrossRef]
- Liu, H.; Li, B. The functional role of exosome in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2018, 144, 2085–2095. [Google Scholar] [CrossRef]
- Coschi, C.H.; Martens, A.L.; Ritchie, K.; Francis, S.M.; Chakrabarti, S.; Berube, N.G.; Dick, F.A. Mitotic chromosome condensation mediated by the retinoblastoma protein is tumor-suppressive. Genes Dev. 2010, 24, 1351–1363. [Google Scholar] [CrossRef]
- Pavlović, N.; Heindryckx, F. Exploring the role of endoplasmic reticulum stress in hepatocellular carcinoma through the mining of the human protein atlas. Biology 2021, 10, 640. [Google Scholar] [CrossRef]
- Kakar, S.; Chen, X.; Ho, C.; Burgart, L.J.; Sahai, V.; Dachrut, S.; Yabes, A.; Jain, D.; Ferrell, L.D. Chromosomal changes in fibrolamellar hepatocellular carcinoma detected by array comparative genomic hybridization. Mod. Pathol. 2009, 22, 134–141. [Google Scholar] [CrossRef]
- Zhang, R.; Ma, M.; Lin, X.H.; Liu, H.H.; Chen, J.; Chen, J.; Gao, D.M.; Cui, J.F.; Ren, Z.G.; Chen, R.X. Extracellular matrix collagen i promotes the tumor progression of residual hepatocellular carcinoma after heat treatment. BMC Cancer 2018, 18, 901. [Google Scholar] [CrossRef]
- Ikehara, T.; Shimizu, T.; Hirano, S.; Fukushima, K.; Yoshizawa, J.; Nakamura, T.; Nakayama, A. Primary spindle cell tumor originating from the liver that was difficult to diagnose. Surg. Case Rep. 2022, 8, 171. [Google Scholar] [CrossRef] [PubMed]
- Bisteau, X.; Caldez, M.J.; Kaldis, P. The complex relationship between liver cancer and the cell cycle: A story of multiple regulations. Cancers 2014, 6, 79–111. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chen, X.; Wang, G. Platelets: A review of their function and effects in liver diseases. Liver Res. 2020, 4, 129–135. [Google Scholar] [CrossRef]
- Zanetto, A.; Senzolo, M.; Campello, E.; Bulato, C.; Gavasso, S.; Shalaby, S.; Gambato, M.; Vitale, A.; Cillo, U.; Farinati, F.; et al. Influence of hepatocellular carcinoma on platelet aggregation in cirrhosis. Cancers 2021, 13, 1150. [Google Scholar] [CrossRef]
- Adamek, A.; Kasprzak, A. Insulin-like growth factor (IGF) system in liver diseases. Int. J. Mol. Sci. 2018, 19, 1308. [Google Scholar] [CrossRef]
- Zeng, L.; Fan, X.; Wang, X.; Deng, H.; Zhang, X.; Zhang, K.; He, S.; Li, N.; Han, Q.; Liu, Z. Involvement of NEK2 and its interaction with NDC80 and CEP250 in hepatocellular carcinoma. BMC Med. Genomics 2020, 13, 158. [Google Scholar] [CrossRef]
- Greenbaum, L.E. Cell cycle regulation and hepatocarcinogenesis. Cancer Biol. Ther. 2004, 3, 1200–1207. [Google Scholar] [CrossRef]
- Quevedo-Ocampo, J.; Escobedo-Calvario, A.; Souza-Arroyo, V.; Miranda-Labra, R.U.; Bucio-Ortiz, L.; Gutiérrez-Ruiz, M.C.; Chávez-Rodríguez, L.; Gomez-Quiroz, L.E. Folate Metabolism in Hepatocellular Carcinoma. What Do We Know So Far? Technol. Cancer Res. Treat. 2022, 21, 15330338221144446. [Google Scholar] [CrossRef]
- Yuan, M.; Liu, L.; Wang, C.; Zhang, Y.; Zhang, J. The Complement System: A Potential Therapeutic Target in Liver Cancer. Life 2022, 12, 1532. [Google Scholar] [CrossRef]
- Li, D.; Zhang, X.; Jiang, L. Molecular mechanism and potential therapeutic targets of liver metastasis from gastric cancer. Front. Oncol. 2022, 12, 1000807. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Tan, H.Y.; Li, S.; Xu, Y.; Guo, W.; Feng, Y. Supplementation of micronutrient selenium in metabolic diseases: Its role as an antioxidant. Oxid. Med. Cell. Longev. 2017, 2017, 7478523. [Google Scholar] [CrossRef] [PubMed]
- Chand, V.; Liao, X.; Guzman, G.; Benevolenskaya, E.; Raychaudhuri, P. Hepatocellular carcinoma evades RB1-induced senescence by activating the FOXM1–FOXO1 axis. Oncogene 2022, 41, 3778–3790. [Google Scholar] [CrossRef] [PubMed]
- Miyaoka, Y.; Ebato, K.; Kato, H.; Arakawa, S.; Shimizu, S.; Miyajima, A. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr. Biol. 2012, 22, 1166–1175. [Google Scholar] [CrossRef]
- Fan, M.; Lu, L.; Shang, H.; Lu, Y.; Yang, Y.; Wang, X.; Lu, H. Establishment and verification of a prognostic model based on coagulation and fibrinolysis-related genes in hepatocellular carcinoma. Aging 2024, 16, 7578–7595. [Google Scholar] [CrossRef]
- Kakar, M.U.; Mehboob, M.Z.; Akram, M.; Shah, M.; Shakir, Y.; Ijaz, H.W.; Aziz, U.; Ullah, Z.; Ahmad, S.; Ali, S.; et al. Identification of Differentially Expressed Genes Associated with the Prognosis and Diagnosis of Hepatocellular Carcinoma by Integrated Bioinformatics Analysis. BioMed Res. Int. 2022, 2022, 4237633. [Google Scholar] [CrossRef]
- Nam, D.; Seong, H.C.; Hahn, Y.S. Plasminogen Activator Inhibitor-1 and Oncogenesis in the Liver Disease. J. Cell. Signal. 2021, 2, 221–227. [Google Scholar] [CrossRef]
- Boccaccio, C.; Medico, E. Cancer and blood coagulation. Cell. Mol. Life Sci. 2006, 63, 1024–1027. [Google Scholar] [CrossRef]
- Pant, A.; Kopec, A.K.; Luyendyk, J.P. Role of the blood coagulation cascade in hepatic fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G171–G176. [Google Scholar] [CrossRef]
- Korbelik, M.; Cecic, I.; Merchant, S.; Sun, J. Acute phase response induction by cancer treatment with photodynamic therapy. Int. J. Cancer 2008, 122, 1411–1417. [Google Scholar] [CrossRef]
- Wang, B.; Zhu, J.; Ma, X.; Wang, H.; Qiu, S.; Pan, B.; Zhou, J.; Fan, J.; Yang, X.; Cheng, Y.; et al. Platelet activation status in the diagnosis and postoperative prognosis of hepatocellular carcinoma. Clin. Chim. Acta 2019, 495, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Jamasbi, E.; Hamelian, M.; Hossain, M.A.; Varmira, K. The cell cycle, cancer development and therapy. Mol. Biol. Rep. 2022, 49, 10875–10883. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.D.; Dai, Y.C.; Li, Z.Y.; Gan, M.F.; Zhang, S.R.; Pan, Y.; Lu, H.S.; Cao, X.Q.; Zheng, B.J.; Bao, L.F.; et al. Expression and function analysis of mitotic checkpoint genes identifies TTK as a potential therapeutic target for human hepatocellular carcinoma. PLoS ONE 2014, 9, e97739. [Google Scholar] [CrossRef]
- Lima, L.G.; Monteiro, R.Q. Activation of blood coagulation in cancer: Implications for tumour progression. Biosci. Rep. 2013, 33, 701–710. [Google Scholar] [CrossRef]
- Roskams, T.; Willems, M.; Campos, R.V.; Drucker, D.J.; Yap, S.H.; Desmet, V.J. Parathyroid hormone-related peptide expression in primary and metastatic liver tumours. Histopathology 1993, 23, 519–525. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, X.; Zheng, Z.; Chen, F.; Yang, N.; Zhou, Y. Centromere protein N may be a novel malignant prognostic biomarker for hepatocellular carcinoma. PeerJ 2021, 9, e11342. [Google Scholar] [CrossRef]
- Miyaoka, Y.; Miyajima, A. To divide or not to divide: Revisiting liver regeneration. Cell Div. 2013, 8, 8. [Google Scholar] [CrossRef]
- Shi, C.Y.; Li, Y.; Wei, R.Q.; Deng, J.; Zou, Q.L.; Yang, S.Q.; Piao, C.; Jin, M. Bioinformatics analysis reveals link between alternative complement cascade pathway and colorectal cancer liver metastasis. Res. Sq. 2023, 1–24. [Google Scholar] [CrossRef]
- Zheng, Y.; Shi, Y.; Yu, S.; Han, Y.; Kang, K.; Xu, H.; Gu, H.; Sang, X.; Chen, Y.; Wang, J. GTSE1, CDC20, PCNA, and MCM6 Synergistically Affect Regulations in Cell Cycle and Indicate Poor Prognosis in Liver Cancer. Anal. Cell. Pathol. 2019, 2019, 1038069. [Google Scholar] [CrossRef]
- Yang, S.F.; Chang, C.W.; Wei, R.J.; Shiue, Y.L.; Wang, S.N.; Yeh, Y.T. Involvement of DNA damage response pathways in hepatocellular carcinoma. BioMed Res. Int. 2014, 2014, 153867. [Google Scholar] [CrossRef]
- Pavlovic, N.; Rani, B.; Gerwins, P.; Heindryckx, F. Platelets as key factors in hepatocellular carcinoma. Cancers 2019, 11, 1022. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Sun, X. Cholesterol Metabolism: A Double-Edged Sword in Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2021, 9, 762828. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ding, Y.; Zhang, C.; Chen, R. Target and drug predictions for SARS-CoV-2 infection in hepatocellular carcinoma patients. PLoS ONE 2022, 17, e0269249. [Google Scholar] [CrossRef]
- Yu, S.; Ji, G.; Zhang, L. The role of p53 in liver fibrosis. Front. Pharmacol. 2022, 13, 1057829. [Google Scholar] [CrossRef]
- Alam, M.S.; Sultana, A.; Reza, M.S.; Amanullah, M.; Kabir, S.R.; Mollah, M.N.H. Integrated bioinformatics and statistical approaches to explore molecular biomarkers for breast cancer diagnosis, prognosis and therapies. PLoS ONE 2022, 17, e0268967. [Google Scholar] [CrossRef]
- Reza, M.S.; Hossen, M.A.; Harun-Or-Roshid, M.; Siddika, M.A.; Kabir, M.H.; Mollah, M.N.H. Metadata analysis to explore hub of the hub-genes highlighting their functions, pathways and regulators for cervical cancer diagnosis and therapies. Discov. Oncol. 2022, 13, 79. [Google Scholar] [CrossRef]
- Sarker, B.; Matiur Rahaman, M.; Alamin, M.H.; Ariful Islam, M.; Nurul Haque Mollah, M. Boosting edgeR (Robust) by dealing with missing observations and gene-specific outliers in RNA-Seq profiles and its application to explore biomarker genes for diagnosis and therapies of ovarian cancer. Genomics 2024, 116, 110834. [Google Scholar] [CrossRef]
- Chen, Q.F.; Xia, J.G.; Li, W.; Shen, L.J.; Huang, T.; Wu, P. Examining the key genes and pathways in hepatocellular carcinoma development from hepatitis B virus-positive cirrhosis. Mol. Med. Rep. 2018, 18, 4940–4950. [Google Scholar] [CrossRef]
- Qiang, R.; Zhao, Z.; Tang, L.; Wang, Q.; Wang, Y.; Huang, Q. Identification of 5 Hub Genes Related to the Early Diagnosis, Tumour Stage, and Poor Outcomes of Hepatitis B Virus-Related Hepatocellular Carcinoma by Bioinformatics Analysis. Comput. Math. Methods Med. 2021, 2021, 9991255. [Google Scholar] [CrossRef]
- Li, N.; Li, L.; Chen, Y. The identification of core gene expression signature in hepatocellular carcinoma. Oxid. Med. Cell. Longev. 2018, 2018, 3478305. [Google Scholar] [CrossRef]
- Shi, Z.; Xiao, Z.; Hu, L.; Gao, Y.; Zhao, J.; Liu, Y.; Shen, G.; Xu, Q.; Huang, D. The genetic association between type 2 diabetic and hepatocellular carcinomas. Ann. Transl. Med. 2020, 8, 380. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Wang, W.; Du, G.; Huang, G.Z.; Han, L.T.; Tang, Z.Y.; Fan, D.G.; Li, J.; Zhang, S.Z. Identifying hub genes and dysregulated pathways in hepatocellular carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 592–601. [Google Scholar] [PubMed]
- Li, C.; Xu, J. Identification of Potentially Therapeutic Target Genes of Hepatocellular Carcinoma. In Proceedings of the 2021 IEEE 9th International Conference on Bioinformatics and Computational Biology (ICBCB), Taiyuan, China, 25–27 May 2021; pp. 96–101. [Google Scholar] [CrossRef]
- Nie, H.; Wang, Y.; Yang, X.; Liao, Z.; He, X.; Zhou, J.; Ou, C. Clinical Significance and Integrative Analysis of the SMC Family in Hepatocellular Carcinoma. Front. Med. 2021, 8, 727965. [Google Scholar] [CrossRef]
- Li, S.; Zhao, J.; Lv, L.; Dong, D. Identification and Validation of TYMS as a Potential Biomarker for Risk of Metastasis Development in Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 762821. [Google Scholar] [CrossRef]
- Huang, J.L.; Cao, S.W.; Ou, Q.S.; Yang, B.; Zheng, S.H.; Tang, J.; Chen, J.; Hu, Y.W.; Zheng, L.; Wang, Q. The long non-coding RNA PTTG3P promotes cell growth and metastasis via up-regulating PTTG1 and activating PI3K/AKT signaling in hepatocellular carcinoma. Mol. Cancer 2018, 17, 93. [Google Scholar] [CrossRef]
- Schuller, S.; Sieker, J.; Riemenschneider, P.; Köhler, B.; Drucker, E.; Weiler, S.M.E.; Dauch, D.; Sticht, C.; Goeppert, B.; Roessler, S.; et al. HELLS Is Negatively Regulated by Wild-Type P53 in Liver Cancer by a Mechanism Involving P21 and FOXM1. Cancers 2022, 14, 459. [Google Scholar] [CrossRef]
- Li, H.T.; Wei, B.; Li, Z.Q.; Wang, X.; Jia, W.X.; Xu, Y.Z.; Liu, J.Y.; Shao, M.N.; Chen, S.X.; Mo, N.F.; et al. Diagnostic and prognostic value of MCM3 and its interacting proteins in hepatocellular carcinoma. Oncol. Lett. 2020, 20, 308. [Google Scholar] [CrossRef]
- Nahm, J.H.; Kim, H.; Lee, H.; Cho, J.Y.; Choi, Y.R.; Yoon, Y.S.; Han, H.S.; Park, Y.N. Transforming acidic coiled-coil-containing protein 3 (TACC3) overexpression in hepatocellular carcinomas is associated with “stemness” and epithelial-mesenchymal transition-related marker expression and a poor prognosis. Tumor Biol. 2016, 37, 393–403. [Google Scholar] [CrossRef]
- Xiao, Y.; Najeeb, R.M.; Ma, D.; Yang, K.; Zhong, Q.; Liu, Q. Upregulation of CENPM promotes hepatocarcinogenesis through mutiple mechanisms. J. Exp. Clin. Cancer Res. 2019, 38, 458. [Google Scholar] [CrossRef]
- Liu, M.; Hu, Q.; Tu, M.; Wang, X.; Yang, Z.; Yang, G.; Luo, R. MCM6 promotes metastasis of hepatocellular carcinoma via MEK/ERK pathway and serves as a novel serum biomarker for early recurrence. J. Exp. Clin. Cancer Res. 2018, 37, 10. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Liao, P.; Yan, L.-Y.; Zhao, Q.-Y.; Xie, Z.-Y.; Dong, J.; Sun, H.-T. Correlation of MKI67 with prognosis, immune infiltration, and T cell exhaustion in hepatocellular carcinoma. BMC Gastroenterol. 2021, 21, 416. [Google Scholar] [CrossRef]
- Fernandes, A.D.; Reid, J.N.; Macklaim, J.M.; McMurrough, T.A.; Edgell, D.R.; Gloor, G.B. Unifying the analysis of high-throughput sequencing datasets: Characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2014, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Yi, S.J.; Wang, L.X.; Zhao, J.X.; Xiao, J.; Xie, N.; Zeng, Z.; Han, Q.; Tang, H.O.; Li, Y.K.; et al. Identification of the DNA Replication Regulator MCM Complex Expression and Prognostic Significance in Hepatic Carcinoma. BioMed Res. Int. 2020, 2020, 3574261. [Google Scholar] [CrossRef]
- He, P.; Hu, P.; Yang, C.; He, X.; Shao, M.; Lin, Y. Reduced expression of CENP-E contributes to the development of hepatocellular carcinoma and is associated with adverse clinical features. Biomed. Pharmacother. 2020, 123, 109795. [Google Scholar] [CrossRef]
- Zhang, W.; Dong, J.; Wu, Y.; Liang, X.; Suo, L.; Wang, L. Integrated Bioinformatic Analysis Reveals the Oncogenic, Survival, and Prognostic Characteristics of TPX2 in Hepatocellular Carcinoma. Biochem. Genet. 2024. [Google Scholar] [CrossRef]
- Gu, J.X.; Huang, K.; Zhao, W.L.; Zheng, X.M.; Wu, Y.Q.; Yan, S.R.; Huang, Y.G.; Hu, P. NCAPD2 augments the tumorigenesis and progression of human liver cancer via the PI3K-Akt-mTOR signaling pathway. Int. J. Mol. Med. 2024, 54, 84. [Google Scholar] [CrossRef]
- Au, S.L.K.; Wong, C.C.L.; Lee, J.M.F.; Fan, D.N.Y.; Tsang, F.H.; Ng, I.O.L.; Wong, C.M. Enhancer of zeste homolog 2 epigenetically silences multiple tumor suppressor microRNAs to promote liver cancer metastasis. Hepatology 2012, 56, 622–631. [Google Scholar] [CrossRef]
- Wang, B.; Qiang, L.; Zhang, G.; Chen, W.; Sheng, Y.; Wu, G.; Deng, C.; Zeng, S.; Zhang, Q. APOC3 as a potential prognostic factor for hepatitis B virus-related acute-on-chronic liver failure. Medicine 2025, 104, e41503. [Google Scholar] [CrossRef]
- Han, X.; Liu, Z.; Cui, M.; Lin, J.; Li, Y.; Qin, H.; Sheng, J.; Zhang, X. FGA influences invasion and metastasis of hepatocellular carcinoma through the PI3K/AKT pathway. Aging 2024, 16, 12806–12819. [Google Scholar] [CrossRef]
- Yang, W.; Shi, J.; Zhou, Y.; Liu, T.; Li, J.; Hong, F.; Zhang, K.; Liu, N. Co-expression network analysis identified key proteins in association with hepatic metastatic colorectal cancer. Proteom. Clin. Appl. 2019, 13, 1900017. [Google Scholar] [CrossRef]
- Gong, Y.; Zou, B.; Peng, S.; Li, P.; Zhu, G.; Chen, J.; Chen, J.; Liu, X.; Zhou, W.; Ding, L.; et al. Nuclear GAPDH is vital for hypoxia-induced hepatic stellate cell apoptosis and is indicative of aggressive hepatocellular carcinoma behaviour. Cancer Manag. Res. 2019, 11, 4947–4956. [Google Scholar] [CrossRef] [PubMed]
- Duarte, T.L.; Viveiros, N.; Godinho, C.; Duarte, D. Heme (dys)homeostasis and liver disease. Front. Physiol. 2024, 15, 1436897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, F.; Huang, Y.; Ke, K.; Zhao, B.; Chen, L.; Liao, N.; Wang, L.; Li, Q.; Liu, X.; et al. FGG promotes migration and invasion in hepatocellular carcinoma cells through activating epithelial to mesenchymal transition. Cancer Manag. Res. 2019, 11, 1653–1665. [Google Scholar] [CrossRef]
- Zou, X.; Zhang, D.; Song, Y.; Liu, S.; Long, Q.; Yao, L.; Li, W.; Duan, Z.; Wu, D.; Liu, L. HRG switches TNFR1-mediated cell survival to apoptosis in Hepatocellular Carcinoma. Theranostics 2020, 10, 10434–10447. [Google Scholar] [CrossRef]
- Hu, Z.G.; Chen, Y.B.; Huang, M.; Tu, J.B.; Tu, S.J.; Pan, Y.J.; Chen, X.L.; He, S.Q. PLG inhibits Hippo signaling pathway through SRC in the hepatitis B virus-induced hepatocellular-carcinoma progression. Am. J. Transl. Res. 2021, 13, 515–531. [Google Scholar]
- Xu, D.; Wu, J.; Dong, L.; Luo, W.; Li, L.; Tang, D.; Liu, J. Serpinc1 Acts as a Tumor Suppressor in Hepatocellular Carcinoma Through Inducing Apoptosis and Blocking Macrophage Polarization in an Ubiquitin-Proteasome Manner. Front. Oncol. 2021, 11, 738607. [Google Scholar] [CrossRef]
- Guo, S.; Chen, Y.; Xue, X.; Yang, Y.; Wang, Y.; Qiu, S.; Cui, J.; Zhang, X.; Ma, L.; Qiao, Y.; et al. TRIB2 desensitizes ferroptosis via βTrCP-mediated TFRC ubiquitiantion in liver cancer cells. Cell Death Discov. 2021, 7, 196. [Google Scholar] [CrossRef]
- Cao, P.R.; McHugh, M.M.; Melendy, T.; Beerman, T. The DNA minor groove-alkylating cyclopropylpyrroloindole drugs adozelesin and bizelesin induce different DNA damage response pathways in human colon carcinoma HCT116 cells. Mol. Cancer Ther. 2003, 2, 651–659. [Google Scholar]
- Fountzilas, C.; Gupta, M.; Lee, S.; Krishnamurthi, S.; Estfan, B.; Wang, K.; Attwood, K.; Wilton, J.; Bies, R.; Bshara, W.; et al. A multicentre phase 1b/2 study of tivozanib in patients with advanced inoperable hepatocellular carcinoma. Br. J. Cancer 2020, 122, 963–970. [Google Scholar] [CrossRef]
- Yu, H.C.; Lin, C.S.; Tai, W.T.; Liu, C.Y.; Shiau, C.W.; Chen, K.F. Nilotinib induces autophagy in hepatocellular carcinoma through AMPK activation. J. Biol. Chem. 2013, 288, 18249–18259. [Google Scholar] [CrossRef]
- Liu, C.; Mu, X.; Wang, X.; Zhang, C.; Zhang, L.; Yu, B.; Sun, G. Ponatinib inhibits proliferation and induces apoptosis of liver cancer cells, but its efficacy is compromised by its activation on PDK1/Akt/mTOR signaling. Molecules 2019, 24, 1363. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.Z.; Xi, Z.Q.; Niu, J.; Li, W.; Wang, X.; Liang, C.; Sun, H.; Fang, D.; Xie, S.Q. Inhibition of PIKfyve using YM201636 suppresses the growth of liver cancer via the induction of autophagy. Oncol. Rep. 2019, 41, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Li, M.; Li, H.; Chen, L. Entrectinib, a new multi-target inhibitor for cancer therapy. Biomed. Pharmacother. 2022, 150, 112974. [Google Scholar] [CrossRef]




| Accession No. | Compared Cell Subset | Number of Samples per Group | Number of Features | Organism | Data Source |
|---|---|---|---|---|---|
| GSE98638 | CD8+ T cells from adjacent normal liver tissues (NTC) vs. CD8+ T cells from liver tumor (TTC) | 412 vs. 777 | 9288 | Human | GEO |
| GSE146409 | Non-malignant liver tumor vs. malignant liver tumor | 12 vs. 63 | 15061 | Human | GEO |
| GSE202069 | Non-tumor vs. tumor patient | 25 vs. 41 | 24492 | Human | GEO |
| GSE189935 | Adjacent normal tissues vs. tumor tissues | 987 vs. 2199 | 15782 | Human | GEO |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biswas, B.; Sugimoto, M.; Hoque, M.A. Discovery of Genomic Targets and Therapeutic Candidates for Liver Cancer Using Single-Cell RNA Sequencing and Molecular Docking. Biology 2025, 14, 431. https://doi.org/10.3390/biology14040431
Biswas B, Sugimoto M, Hoque MA. Discovery of Genomic Targets and Therapeutic Candidates for Liver Cancer Using Single-Cell RNA Sequencing and Molecular Docking. Biology. 2025; 14(4):431. https://doi.org/10.3390/biology14040431
Chicago/Turabian StyleBiswas, Biplab, Masahiro Sugimoto, and Md. Aminul Hoque. 2025. "Discovery of Genomic Targets and Therapeutic Candidates for Liver Cancer Using Single-Cell RNA Sequencing and Molecular Docking" Biology 14, no. 4: 431. https://doi.org/10.3390/biology14040431
APA StyleBiswas, B., Sugimoto, M., & Hoque, M. A. (2025). Discovery of Genomic Targets and Therapeutic Candidates for Liver Cancer Using Single-Cell RNA Sequencing and Molecular Docking. Biology, 14(4), 431. https://doi.org/10.3390/biology14040431