
Establishment of Novel Simple Sequence Repeat Markers in Phragmites australis and Application in Wetlands of Nanhui Dongtan, Shanghai

 ,
,
Simple Summary
Abstract
1. Introduction
- (1)
- Development and screening of reed SSR primers: To utilize simple sequence repeats derived from the entire reed genome in order to develop and screen SSR primers that provide a robust tool for investigating reed genetic diversity.
- (2)
- Validation of primer stability and reliability: To use the reed community in Shanghai Nanhui Dongtan as a case study to validate the stability and reliability of the newly developed primers, thereby confirming their feasibility.
- (3)
- Assessment of genetic diversity and population differentiation: To employ the developed primers in exploring the genetic diversity and population genetic differentiation of four reed communities in Nanhui Dongtan, thus establishing an important foundation for future selective breeding for salt tolerance and ecological restoration initiatives.
2. Materials and Methods
2.1. Common Reed Samples
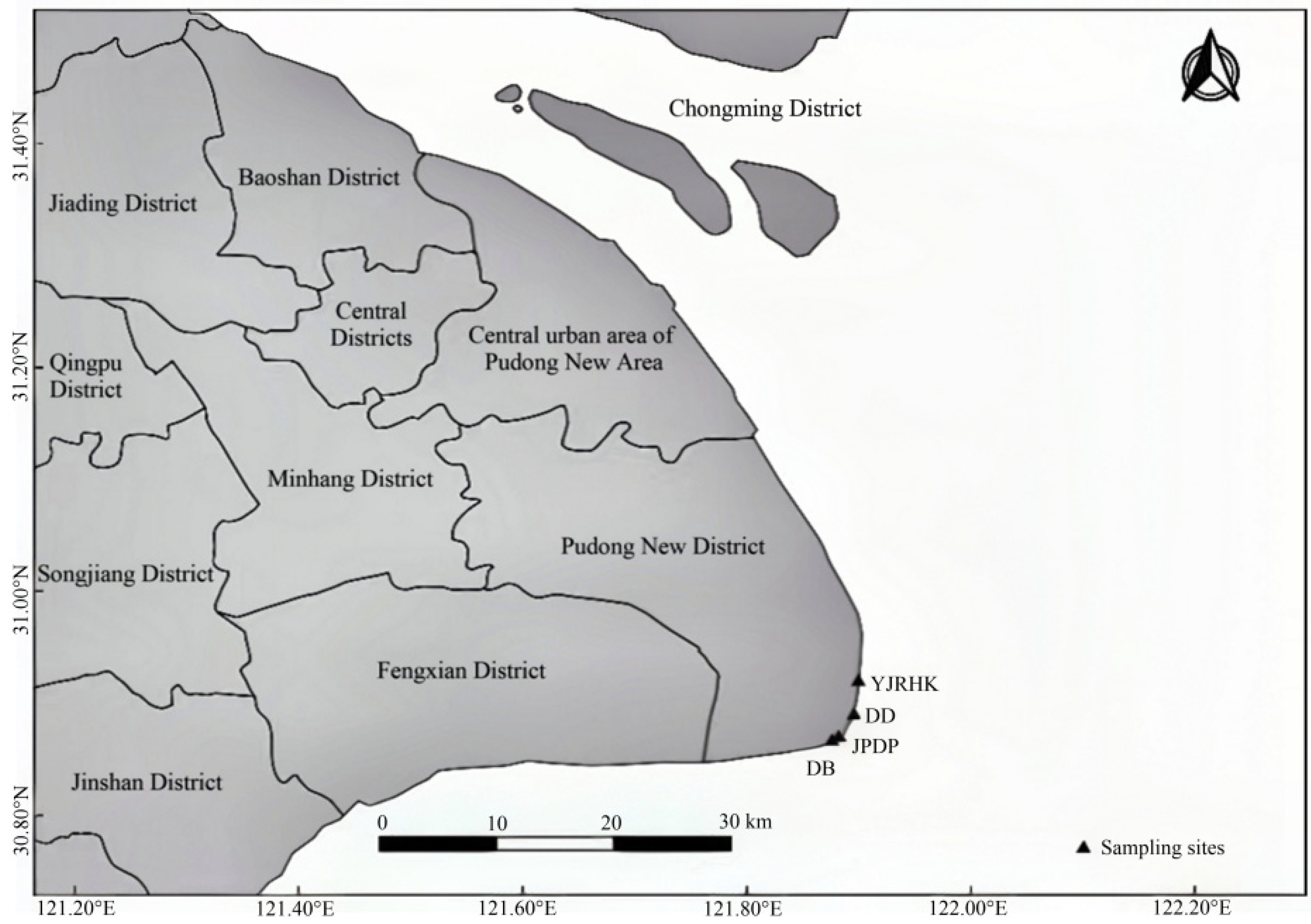
2.2. Sampling Sites
2.3. DNA Extraction
2.4. Development of SSR Molecular Markers
2.5. Analysis of Genetic Diversity
2.5.1. Genetic Diversity and Variation Analysis in Natural Populations
2.5.2. Analysis of Strain Genetic Structure
2.5.3. Analysis of Group Structure
2.6. Statistical Analysis of Data
3. Results
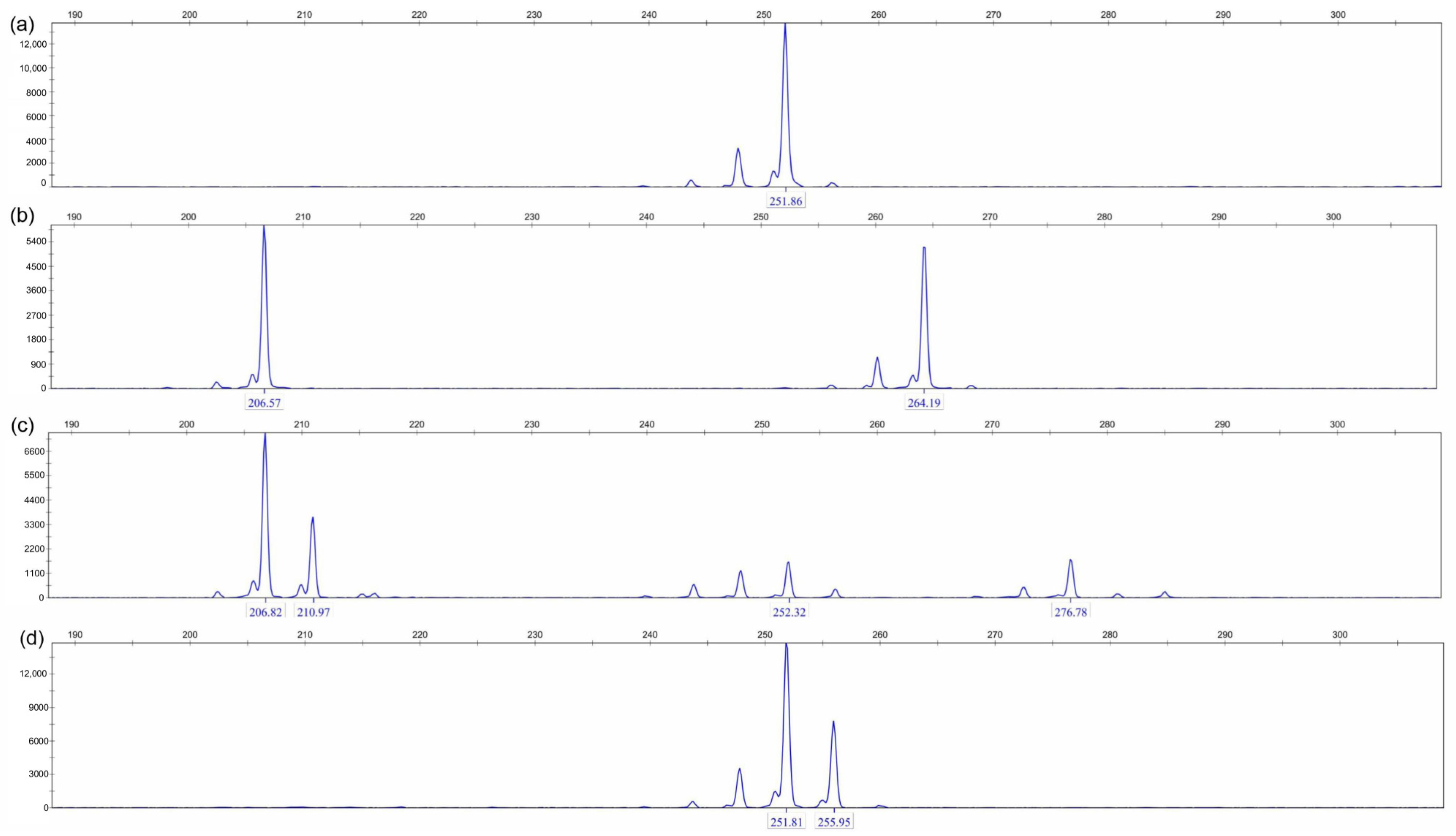
3.1. Development of SSR Molecular Markers in P. australis Populations
3.2. SSR Molecular Marker Polymorphism Analysis
3.3. Analysis of Genetic Diversity in 4 Populations
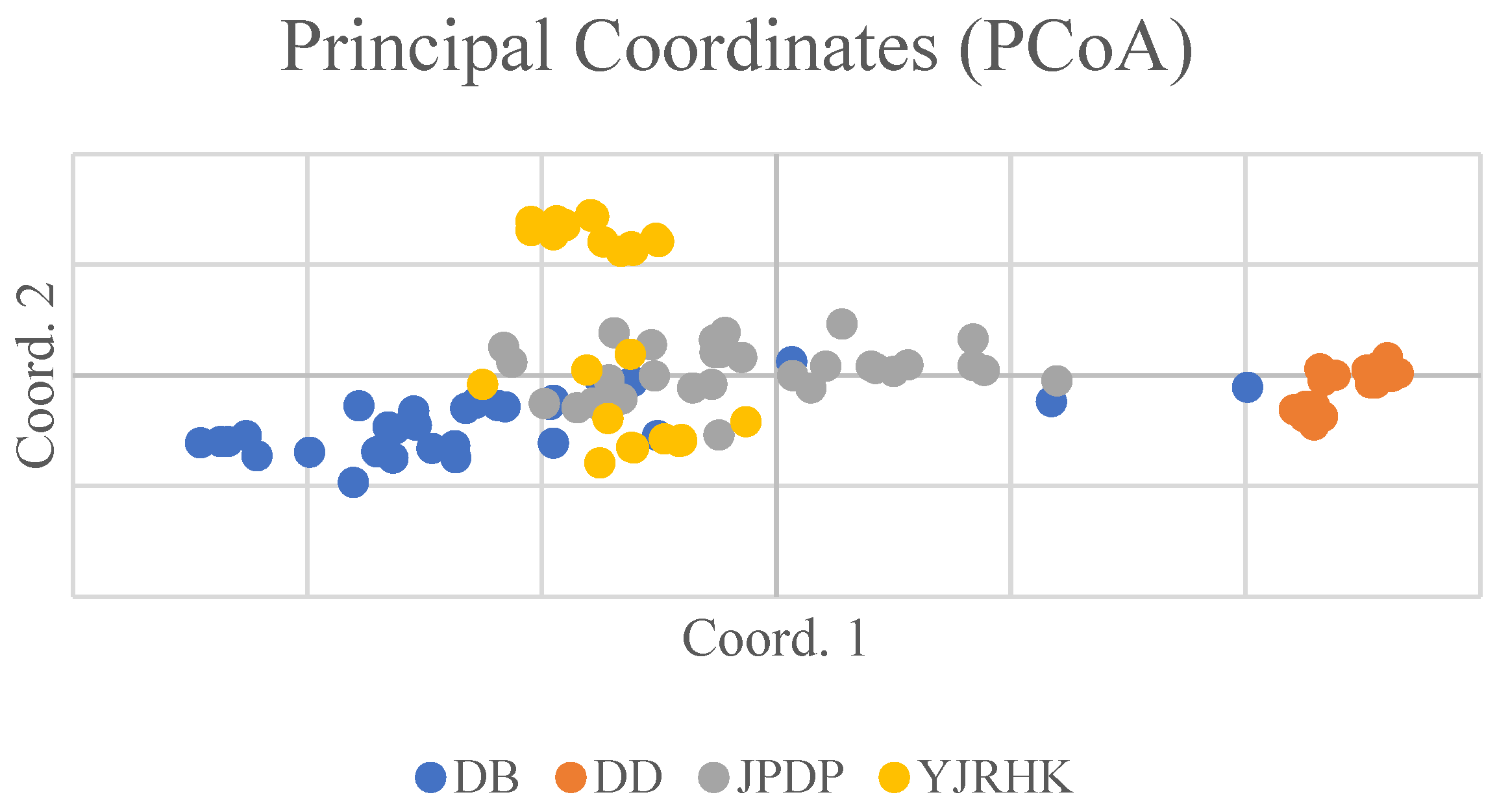
3.4. Analysis of the Genetic Structure of 4 P. australis Populations
3.5. Selection of P. australis Populations with Salinity
3.6. Distribution and Epigenetic Characteristics of Major P. australis Populations in Typical Coastal Wetlands of Nanhui Dongtan, Shanghai
4. Discussion
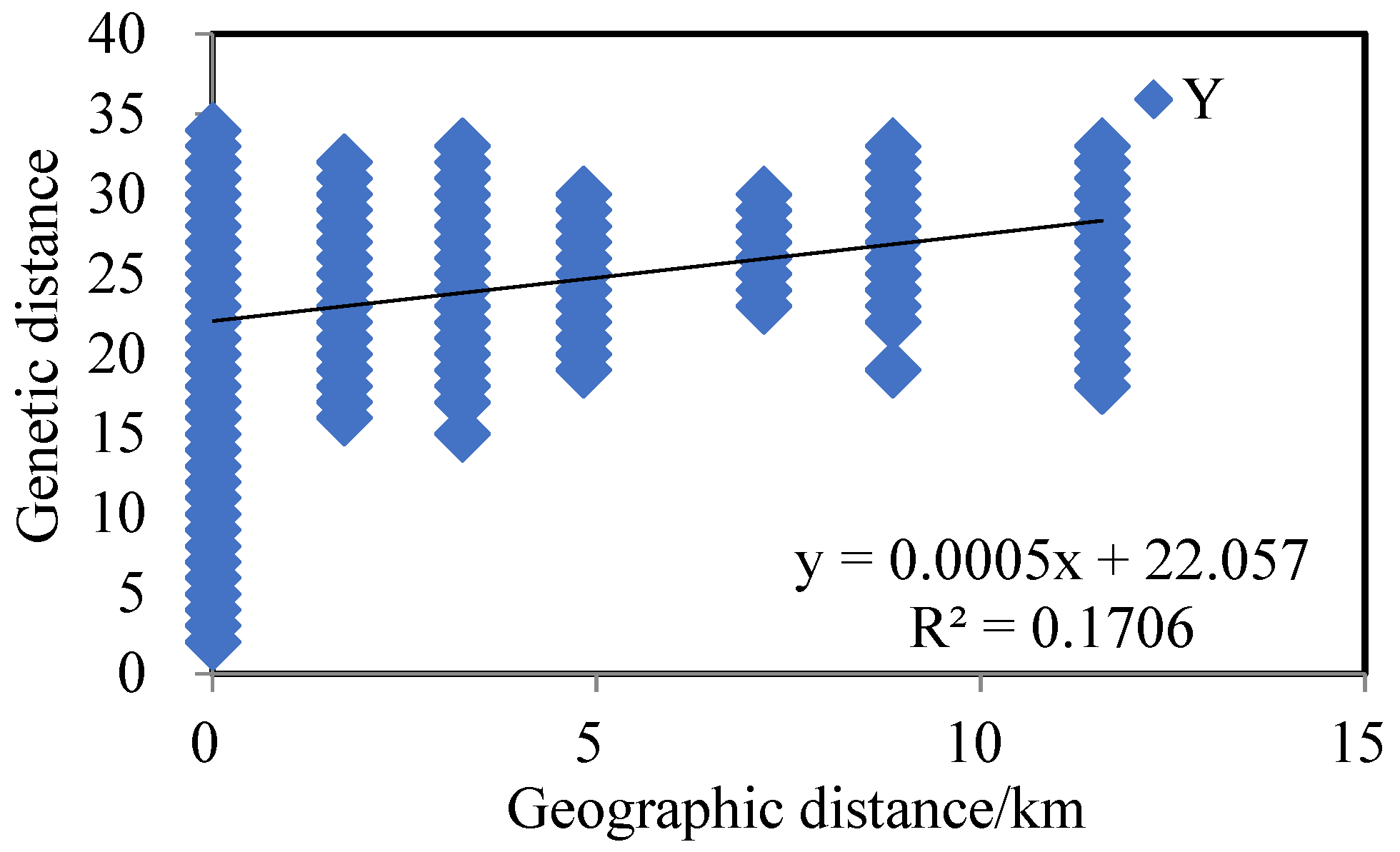
4.1. Population Genetic Diversity and Differentiation
4.2. SSR Genetic Diversity of Reed
4.3. Applicability and Prospects of SSR Markers
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kirk, H.; Paul, J.; Straka, J.; Freeland, J.R. Long-Distance dispersal and high genetic diversity are implicated in the invasive spread of the common reed, Phragmites australis (Poaceae), in Northeastern North America. Am. J. Bot. 2011, 98, 1180–1190. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zheng, G.; Li, X.; Xiao, J.F.; Xu, Z.; Tian, P.P. Habitat quality evaluation and pattern simulation of coastal salt marsh wetlands. Sci. Total Environ. 2024, 945, 174003. [Google Scholar] [CrossRef] [PubMed]
- Saltonstall, K. Microsatellite variation within and among North American lineages of Phragmites australis. Mol. Ecol. 2003, 12, 1689–1702. [Google Scholar] [CrossRef] [PubMed]
- Vymazal, J. Emergent plants used in free water surface constructed wetlands: A review. Ecol. Eng. 2013, 61, 582–592. [Google Scholar] [CrossRef]
- Canavan, K.; Paterson, I.D.; Lambertini, C.; Hill, M.P. Expansive reed populations—alien invasion or disturbed wetlands? AoB Plants 2018, 10, ply014. [Google Scholar] [CrossRef]
- Srivastava, J.; Kalra, S.J.S.; Naraian, R. Environmental perspectives of Phragmites australis (Cav.) Trin. Ex. Steudel. Appl. Water Sci. 2014, 4, 193–202. [Google Scholar] [CrossRef]
- Jiao, L.; Li, F.; Liu, X.; Wang, S.J.; Zhou, Y. Fine-Scale distribution patterns of Phragmites australis populations across an environmental gradient in the salt marsh wetland of Dunhuang, China. Sustainability 2020, 12, 1671. [Google Scholar] [CrossRef]
- Xu, M.H.; Li, X.; Ye, Q.N.; Gong, F.; He, X.L. Occurrence of dark septate endophytes in Phragmites australis in the Baiyang Lake and their resistance to Cd stress. Pedosphere 2024, 34, 484–496. [Google Scholar] [CrossRef]
- Jing, L.; Zeng, Q.; He, K.; Liu, P.Z.; Fan, R.; Lu, W.Z.; Lei, G.C.; Lu, C.; Wen, L. Vegetation dynamic in a large floodplain wetland: The effects of hydroclimatic regime. Remote Sens. 2023, 15, 2614. [Google Scholar] [CrossRef]
- Gu, J.L.; Luo, M.; Zhang, X.J.; Christakos, G.; Agusti, S.; Duarte, C.M.; Wu, J.P. Losses of salt marsh in China: Trends, threats and management. Estuar. Coast. Shelf Sci. 2018, 214, 98–109. [Google Scholar] [CrossRef]
- Murray, N.J.; Phinn, S.R.; DeWitt, M.; Ferrari, R.; Johnston, R.; Lyons, M.B.; Clinton, N.; Thau, D.; Fuller, R.A. The global distribution and trajectory of tidal flats. Nature 2019, 565, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.W.; Li, X.Z.; Yang, B.; Ma, Y.X.; Jiang, C.; Xue, L.M.; Wang, J.J.; Yan, Z.Z. Systematic assessments of tidal wetlands loss and degradation in Shanghai, China: From the perspectives of area, composition and quality. Glob. Ecol. Conserv. 2021, 25, e01450. [Google Scholar] [CrossRef]
- Liu, L.L.; Guo, Y.; Wu, Y.; Yin, M.Q.; Guo, X.; Eller, F.; Richards, C.L.; Brix, H.; Ju, R.T.; Guo, W.H. Revealing biogeographic patterns in genetic diversity of native and invasive plants and their association with soil community diversity in the Chinese coast. Oikos 2024, 2024, e10116. [Google Scholar] [CrossRef]
- Qiao, H.M.; Liu, W.W.; Zhang, Y.H.; Zhang, Y.Y.; Li, Q.S.Q. Genetic admixture accelerates invasion via provisioning rapid adaptive evolution. Mol. Ecol. 2019, 28, 4012–4027. [Google Scholar] [CrossRef]
- Xie, B.H.; Han, G.X. Control of invasive plant Spartina alterniflora: Concept, technology and practice. Bull. Chin. Acad. Sci. 2023, 38, 1924–1938. [Google Scholar] [CrossRef]
- Hoban, S.; Archer, F.I.; Bertola, L.D.; Bragg, J.G.; Breed, M.F.; Bruford, M.W.; Coleman, M.A.; Ekblom, R.; Funk, W.C.; Grueber, C.E.; et al. Global genetic diversity status and trends: Towards a suite of Essential Biodiversity Variables (EBVs) for genetic composition. Biol. Rev. Camb. Philos. Soc. 2022, 97, 1511–1538. [Google Scholar] [CrossRef] [PubMed]
- Milesi, P.; Kastally, C.; Dauphin, B.; Cervantes, S.; Bagnoli, F.; Budde, K.B.; Cavers, S.; Fady, B.; Faivre-Rampant, P.; Gonzalez-Martinez, S.C.; et al. Resilience of genetic diversity in forest trees over the Quaternary. Nat. Commun. 2024, 15, 8538. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.D.; Ramakant, S.S.; Kumari, D.; Kumar, P.; Jambhulkar, S.J.; Alamri, S.; Adil, M.F. Gene expression analyses of the calmodulin binding protein 60 family under water stress conditions in rice. Sci. Rep. 2025, 15, 6203. [Google Scholar] [CrossRef]
- Sheng, W.Y.; Liu, L.L.; Wu, Y.M.; Yin, M.Q.; Yu, Q.; Guo, X.; Song, H.J.; Guo, W.H. Exploring salt tolerance and indicator traits across four temperate lineages of the common wetland plant, Phragmites australis. Sci. Total Environ. 2024, 912, 169100. [Google Scholar] [CrossRef]
- Song, H.; Guo, X.; Liu, L.L.; Xu, Z.W.; Wang, N.; Liu, X.; Du, N.; Guo, W.H. Role of DNA methylation in ecophysiological responses to salinity in natural populations of Phragmites australis from coastal and inland habitats. Front. Mar. Sci. 2022, 9, 982234. [Google Scholar] [CrossRef]
- Song, H.; Guo, X.; Yang, J.C.; Yang, J.C.; Liu, L.L.; Li, M.Y.; Wang, J.F.; Guo, W.H. Phenotypic plasticity variations in Phragmites australis under different plant–plant interactions influenced by salinity. J. Plant Ecol. 2024, 17, rtae035. [Google Scholar] [CrossRef]
- Symstad, A.J.; Tilman, D.; Willson, J.; Knops, J.M.H. Species loss and ecosystem functioning: Effects of species identity and community composition. Oikos 1998, 81, 389–397. [Google Scholar] [CrossRef]
- Liu, L.L.; Yin, M.; Guo, X.; Wang, J.W.; Cai, Y.F.; Wang, C.; Yu, X.N.; Du, N.; Brix, H.; Eller, F.; et al. Cryptic lineages and potential introgression in a mixed-ploidy species (Phragmites australis) across temperate China. J. Syst. Evol. 2020, 60, 398–410. [Google Scholar] [CrossRef]
- Chen, L.P.; Chang, Y.G.; Li, J.; Wang, J.M.; Liu, J.L.; Zhi, Y.C.; Li, X.J. Application of DNA barcoding in the classification of grasshoppers (Orthoptera: Acridoidea) — a case study of grasshoppers from Hebei Province, China. Zootaxa 2018, 4497, 99–110. [Google Scholar] [CrossRef]
- Hao, J.F.; Zhang, X.H.; Wang, Y.S.; Liu, J.L.; Zhi, Y.C.; Li, X.J. Diversity investigation and application of DNA barcoding of Acridoidea from Baiyangdian Wetland. Biodivers. Sci. 2017, 25, 409–417. [Google Scholar] [CrossRef]
- Dettori, M.T.; Micali, S.; Giovinazzi, J.; Scalabrin, S.; Verde, I.; Cipriani, G. Mining microsatellites in the peach genome: Development of new long-core SSR markers for genetic analyses in five Prunus species. SpringerPlus 2015, 4, 1–18. [Google Scholar] [CrossRef]
- Song, Q.J.; Fickus, E.W.; Cregan, P.B. Characterization of trinucleotide SSR motifs in wheat. Theor. Appl. Genet. 2002, 104, 286–293. [Google Scholar] [CrossRef]
- McCormick, K.M.; Whigham, F.D.; Stapp, R.J.; Hazelton, E.L.G.; McFarland, E.K.; Kettenring, K.M. Shoreline modification affects recruitment of invasive Phragmites australis. Wetl. Ecol. Manag. 2020, 28, 1–11. [Google Scholar] [CrossRef]
- Saltonstall, K. Cryptic invasion by a non-native genotype of the common reed, Phragmites australis, into North America. Proc. Natl. Acad. Sci. USA 2002, 99, 2445–2449. [Google Scholar] [CrossRef]
- Tippery, N.P.; Pesch, J.D.; Murphy, B.J.; Bautzmann, R.L. Genetic diversity of native and introduced Phragmites (common reed) in Wisconsin. Genetica 2020, 148, 165–172. [Google Scholar] [CrossRef]
- Lambertini, C.; Gustafsson, M.H.G.; Frydenberg, J.; Lissner, J.; Speranza, M.; Brix, H. A phylogeographic study of the cosmopolitan genus Phragmites (Poaceae) based on AFLPs. Plant Syst. Evol. 2006, 258, 161–182. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537. [Google Scholar] [CrossRef]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128. [Google Scholar] [CrossRef]
- Kuprina, K.; Seeber, E.; Schnittler, M.; Landeau, R.; Lambertini, C.; Bog, M. Genetic diversity of common reed in the southern Baltic Sea region—Is there an influence of disturbance? Aquat. Bot. 2022, 177, 103471. [Google Scholar] [CrossRef]
- Liu, J.K.; Yan, G.X.; Pan, Y.Y.; Zhang, Z.M.; Chen, Y.L.; Zhang, M.X. Economical energy allocation for the landward invasion of Spartina alterniflora in estuaries in the Yellow River Delta, east China. Ecol. Indic. 2023, 146, 109770. [Google Scholar] [CrossRef]
- Hu, W.M.; Kan, H.H.; Wang, W.; Xu, C.W. Statistical genetics approach for functional difference identification of allelic variations and its application. Acta Agron. Sin. 2014, 40, 72–79. [Google Scholar] [CrossRef]
- Wang, C.; Liu, L.L.; Yin, M.Q.; Liu, B.B.; Wu, Y.M.; Eller, F.; Gao, Y.Q.; Brix, H.; Wang, T.; Guo, W.H.; et al. Chromosome-level genome assemblies reveal genome evolution of an invasive plant Phragmites australis. Commun. Biol. 2024, 7, 1007. [Google Scholar] [CrossRef]
- Patamsytė, J.; Lambertini, C.; Butkuvienė, J.; Naugžemys, D.; Žvingila, D. Assessment of anthropogenic impacts on the genetic diversity of Phragmites australis in small-river habitats. Diversity 2023, 15, 1116. [Google Scholar] [CrossRef]
- Jiang, Q.; Xu, Q.; Pan, J.F.; Yao, X.H.; Cheng, Z.P. Impacts of chronic habitat fragmentation on genetic diversity of natural populations of Prunus persica in China. Plants 2022, 11, 1458. [Google Scholar] [CrossRef]
- Spens, E.A.; Douhovnikoff, V. Epigenetic variation within Phragmites australis among lineages, genotypes, and ramets. Biol. Invasions 2016, 18, 2457–2462. [Google Scholar] [CrossRef]
- Liu, B.B.; Xing, J.Q.; Gong, X.J.; Pu, T.L. Genetic differences of different reed ecotypes adapted to individual habitats. Acta Prataculturae Sin. 2009, 18, 250–255. Available online: http://cyxb.magtech.com.cn/CN/Y2009/V18/I5/250 (accessed on 24 February 2025).
- Gao, L.; Tang, S.Q.; Zhuge, L.Q.; Nie, M.; Zhu, Z.; Li, B.; Yang, J. Spatial genetic structure in natural populations of Phragmites australis in a mosaic of saline habitats in the Yellow River Delta, China. PLoS ONE 2012, 7, e43334. [Google Scholar] [CrossRef]
- Liu, X.; Ding, D.; Sun, Y.; Li, Y.; Yang, G.; Li, Y.; Zhu, B.; Zhao, Y.; Pan, K. Effects of Spartina alterniflora invasion on the structural dynamics of bacterial and diatom communities in the native Suaeda salsa zone. Estuar. Coast. Shelf Sci. 2025, 315, 109163. [Google Scholar] [CrossRef]
- Flowers, T.J.; Colmer, T.D. Salinity tolerance in halophytes. New Phytol. 2008, 179, 945–963. [Google Scholar] [CrossRef]
- Hurry, R.C.; James, A.E.; Thompson, M.R. Connectivity, genetic structure and stress response of Phragmites australis: Issues for restoration in a salinising wetland system. Aquat. Bot. 2013, 104, 138–146. [Google Scholar] [CrossRef]
- Govindaraju, D.R. Relationship between dispersal ability and levels of gene flow in plants. Oikos 1988, 52, 31–35. [Google Scholar] [CrossRef]
- Degirmenci, F.O.; Acar, P.; Kaya, Z. Consequences of habitat fragmentation on genetic diversity and structure of Salix alba L. populations in two major river systems of Turkey. Tree Genet. Genom. 2019, 15, 59. [Google Scholar] [CrossRef]
- Li, S.Z.; Xie, T.; Pennings, S.C.; Wang, Y.C.; Craft, C.; Hu, M.M. A Comparison of coastal habitat restoration projects in China and the United States. Sci. Rep. 2019, 9, 4388. [Google Scholar] [CrossRef]
- Hu, S.W.; He, R.J.; Zhou, Q.; Wang, S.R.; Huang, R.; Cao, X.Y.; Ding, H.; Zeng, J.; Zhao, D.Y. Genetic variation characteristics of Phragmites australis populations from the middle and lower reaches of the Yangtze River and the Yunnan-Guizhou Plateau. Ecol. Environ. Sci. 2019, 28, 1379–1387. [Google Scholar] [CrossRef]
- Lindsay, D.L.; Guan, X.; Harms, N.E.; Cronin, J.T.; Meyerson, L.A.; Lance, R.F. DNA assays for genetic discrimination of three Phragmites australis subspecies in the United States. Appl. Plant Sci. 2023, 11, e11512. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.P.; Qiu, T.H.; Li, L.; Cui, S.X. De novo full-length transcriptome analysis of two ecotypes of Phragmites australis (swamp reed and dune reed) provides new insights into the transcriptomic complexity of dune reed and its long-term adaptation to desert environments. BMC Genom. 2023, 24, 180. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zhang, Y.X.; Ren, Y.L.; Sun, Q.X. Isolation and characterization of microsatellite loci for Phragmites australis. J. Genet. 2014, 93, 89–92. [Google Scholar] [CrossRef]
- McCormick, K.M.; Brooks, A.E.H.; Whigham, F.D. Microsatellite analysis to estimate realized dispersal distance in Phragmites australis. Biol. Invasions 2016, 18, 2497–2504. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Sites | Codes | Longitude | Latitude | Altitude |
|---|---|---|---|---|
| Inside the embankment (wild population) | DB | 121°53′46″ E | 30°51′18″ N | 2.03 m |
| Outside the embankment and inside open breakwater (planting population) | JPDP | 121°52′46″ E | 30°50′58″ N | 3.02 m |
| Outside the embankment and inside enclosed breakwater (planting population) | YJRHK | 121°58′23″ E | 30°54′56″ N | 2.40 m |
| Shoal outside the embankment (wild population) | DD | 121°55′36″ E | 30°51′53″ N | 1.82 m |
| Repetition Type | Total | Proportion/% | Repetitions | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | ≥15 | |||
| p2 | 47,697 | 58.46 | 0 | 15,011 | 7600 | 4963 | 3284 | 2010 | 1335 | 1080 | 887 | 797 | 10,740 |
| p3 | 21,876 | 26.81 | 14,487 | 4534 | 1532 | 568 | 254 | 106 | 85 | 58 | 41 | 37 | 174 |
| p4 | 2204 | 2.70 | 1590 | 335 | 115 | 55 | 23 | 13 | 10 | 18 | 5 | 2 | 38 |
| p5 | 651 | 0.80 | 487 | 123 | 30 | 9 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| p6 | 326 | 0.40 | 245 | 55 | 7 | 2 | 5 | 2 | 2 | 0 | 1 | 0 | 7 |
| c | 8440 | 10.34 | 0 | 0 | 0 | 0 | 0 | 35 | 30 | 101 | 103 | 114 | 8057 |
| c * | 399 | 0.49 | 0 | 0 | 0 | 0 | 0 | 3 | 17 | 16 | 23 | 22 | 318 |
| Locus | Primer Sequences | Repeating Units | Number of Bases | Range/bp |
|---|---|---|---|---|
| LW401 | F:GGAATGCTACTGTATTAATGTCGTT R:GTACCACCAAGATGCCCTCA | TGTA | 4 | 206–272 |
| LW407 | F:TTAGGACGAGAGCAAGAAGCC R:CGACTGGGACAGAGGAACG | GAG | 3 | 288–315 |
| LW423 | F:GCATCAGTCTCCTTGTACCGTT R:GTTACTCCTTCGCCGACACTT | TTG | 3 | 292–311 |
| LW424 | F:GCGTCGAGTCGTTTGAAACC R:CATCACCAGCGACCTCCG | GAG | 3 | 358–391 |
| LW442 | F:ATTACAACTTCTCGCTTCGGAT R:CAGTTGACATTCCATTTCCAGC | ATAC | 4 | 217–229 |
| LW444 | F:ACAAGAGGCTGAAACGAACG R:CGCATGACGAACCAATAACA | AAG | 3 | 268–329 |
| LW450 | F:TTGGATACACCATCATTGTTCATAG R:GCTGCTGACGGTCAACCTT | TATG | 4 | 282–407 |
| LW459 | F:TTGGCTGATGGGAAGTTGTCT R:GGAGCAGAATCAAAGCAGGC | TGC | 3 | 310–325 |
| LW467 | F:GCAACTGAACTGGGAAGACAAC R:TTCAAGTGGAGCAGTATCGTCAT | CTAT | 4 | 249–297 |
| LW474 | F:AGTCTCTCAGTGTAACCTTCCCA R:GTTGTTATTCTTGCTGATGTGTATTC | GAA | 3 | 266–321 |
| LW479 | F:CCTTCTGACTTACTGTCAAGCTCTC R:AGACCCAACTCACCAGGAAAG | TTC | 3 | 268–335 |
| LW482 | F:TCAACCACGCATTCGGTG R:CGCAAGGGACAACAGAGGG | CTT | 3 | 178–221 |
| LW542 | F:GCCAATCAGCCACACAACG R:CCAGTACGTTCCTTGACCTTG | CCT | 3 | 345–370 |
| LW543 | F:TGCTCAGATGTCAGCCAGTTG R:AGCACTTAAAGCAGCGATTGAC | CTT | 3 | 320–381 |
| LW549 | F:CCTTGCCAACTTGTCCCAG R:AGATAGGCATTCACGCAAGT | ATAC | 4 | 254–292 |
| Locus | Na | Ne | I | PIC | Ho | He | H | F | Fis | Fit | Fst | Nm |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LW401 | 9.500 | 5.368 | 1.737 | 0.845 | 0.975 | 0.761 | 0.859 | −0.333 | −0.282 | −0.135 | 0.115 | 1.932 |
| LW407 | 8.000 | 5.235 | 1.778 | 0.872 | 0.967 | 0.798 | 0.883 | −0.218 | −0.211 | −0.094 | 0.097 | 2.341 |
| LW423 | 5.500 | 3.041 | 1.185 | 0.657 | 0.650 | 0.621 | 0.704 | 0.001 | −0.046 | 0.077 | 0.117 | 1.879 |
| LW424 | 4.250 | 2.641 | 1.088 | 0.639 | 0.983 | 0.615 | 0.694 | −0.608 | −0.599 | −0.416 | 0.114 | 1.938 |
| LW442 | 4.250 | 2.268 | 0.904 | 0.659 | 0.333 | 0.497 | 0.697 | 0.378 | 0.330 | 0.522 | 0.287 | 0.622 |
| LW444 | 7.500 | 4.508 | 1.443 | 0.847 | 0.517 | 0.644 | 0.860 | 0.347 | 0.198 | 0.397 | 0.248 | 0.757 |
| LW450 | 13.000 | 8.265 | 2.008 | 0.872 | 1.000 | 0.787 | 0.880 | −0.353 | −0.271 | −0.137 | 0.105 | 2.121 |
| LW459 | 6.750 | 3.737 | 1.458 | 0.778 | 0.992 | 0.718 | 0.800 | −0.394 | −0.382 | −0.240 | 0.103 | 2.183 |
| LW467 | 10.250 | 6.040 | 1.781 | 0.833 | 0.983 | 0.748 | 0.845 | −0.394 | −0.315 | −0.164 | 0.115 | 1.929 |
| LW474 | 9.250 | 6.645 | 1.784 | 0.858 | 1.000 | 0.766 | 0.870 | −0.383 | −0.305 | −0.149 | 0.119 | 1.843 |
| LW479 | 9.250 | 4.690 | 1.658 | 0.852 | 0.917 | 0.740 | 0.864 | −0.315 | −0.239 | −0.061 | 0.144 | 1.486 |
| LW482 | 3.750 | 2.384 | 0.955 | 0.577 | 0.633 | 0.558 | 0.637 | −0.220 | −0.136 | 0.006 | 0.125 | 1.755 |
| LW542 | 7.250 | 3.327 | 1.430 | 0.719 | 0.867 | 0.669 | 0.737 | −0.301 | −0.295 | −0.177 | 0.091 | 2.485 |
| LW543 | 9.000 | 5.376 | 1.776 | 0.865 | 1.000 | 0.782 | 0.875 | −0.303 | −0.279 | −0.142 | 0.107 | 2.086 |
| LW549 | 6.000 | 3.026 | 1.286 | 0.681 | 0.983 | 0.648 | 0.710 | −0.556 | −0.517 | −0.384 | 0.087 | 2.615 |
| Mean | 7.567 | 4.437 | 1.485 | 0.770 | 0.853 | 0.690 | 0.794 | −0.243 | −0.223 | −0.073 | 0.132 | 1.865 |
| Population | Na | Ne | I | Ho | He | F |
|---|---|---|---|---|---|---|
| DB | 9.400 | 5.157 | 1.747 | 0.858 | 0.754 | −0.099 |
| DD | 2.600 | 2.231 | 0.817 | 0.867 | 0.529 | −0.588 |
| JPDP | 12.667 | 6.640 | 2.020 | 0.851 | 0.797 | −0.075 |
| YJRHK | 5.600 | 3.719 | 1.355 | 0.838 | 0.681 | −0.212 |
| Mean | 7.567 | 4.437 | 1.485 | 0.853 | 0.690 | −0.243 |
| Source | Degrees of Freedom | Sum of Squares | Mean Square | Variance Component | Ratio of Variance |
|---|---|---|---|---|---|
| Among populations | 3.000 | 167.975 | 55.992 | 0.852 | 12% |
| Among individuals within populations | 116.000 | 564.017 | 4.862 | 0.000 | 0% |
| Within individuals | 120.000 | 768.000 | 6.400 | 6.400 | 88% |
| Total | 239.000 | 1499.992 | - | 7.252 | 100% |
| DB | DD | JPDP | YJRHK | |
|---|---|---|---|---|
| DB | - | 0.443 | 0.745 | 0.693 |
| DD | 0.813 | - | 0.572 | 0.401 |
| JPDP | 0.294 | 0.558 | - | 0.717 |
| YJRHK | 0.367 | 0.915 | 0.333 | - |
| Locus | DB | DD | JPDP | YJRHK |
|---|---|---|---|---|
| LW401 | 8 | 1 | 23 | 3 |
| LW407 | 15 | 1 | 21 | 3 |
| LW423 | 10 | 1 | 16 | 2 |
| LW424 | 10 | 1 | 23 | 3 |
| LW442 | 4 | 1 | 14 | 3 |
| LW444 | 11 | 1 | 18 | 3 |
| LW450 | 9 | 1 | 22 | 3 |
| LW459 | 10 | 1 | 19 | 3 |
| LW467 | 12 | 1 | 21 | 3 |
| LW474 | 10 | 1 | 21 | 4 |
| LW479 | 8 | 1 | 20 | 3 |
| LW482 | 7 | 1 | 12 | 3 |
| LW542 | 10 | 1 | 18 | 3 |
| LW543 | 8 | 1 | 24 | 3 |
| LW549 | 9 | 1 | 23 | 3 |
| Code | Seawater Salinity/ppt | Area/hm2 | Wind Waves | Tides |
|---|---|---|---|---|
| DB | 4–10 | 2.1 | none | none |
| JPDP | 7–12 | 4.5 | present | present |
| YJRHK | 7–12 | 6.2 | present | present |
| DD | 7–12 | 3.4 | present | present |
| Morphological Characteristics | DB | JPDP | YJRHK | DD |
|---|---|---|---|---|
| Height/cm | 178.1 ± 1.9 * | 188.7 ± 2.2 * | 204.2 ± 2.4 * | 234.9 ± 3.2 * |
| Leaf length/cm | 29.2 ± 0.3 * | 36.4 ± 0.3 * | 31.7 ± 0.3 * | 43.3 ± 0.3 * |
| Shape of leaves | Lanceolate, flattened, pendulous, finer | Lanceolate, flattened, pendulous, finer | Lanceolate, flattened, pendulous, finer | flattened, pendulous, finer |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, S.; Shen, Y.; Li, M.; Jiang, R.; Cai, L.; Wu, T.; Gao, L.; Wu, M.; He, P. Establishment of Novel Simple Sequence Repeat Markers in Phragmites australis and Application in Wetlands of Nanhui Dongtan, Shanghai. Biology 2025, 14, 356. https://doi.org/10.3390/biology14040356
Ma S, Shen Y, Li M, Jiang R, Cai L, Wu T, Gao L, Wu M, He P. Establishment of Novel Simple Sequence Repeat Markers in Phragmites australis and Application in Wetlands of Nanhui Dongtan, Shanghai. Biology. 2025; 14(4):356. https://doi.org/10.3390/biology14040356
Chicago/Turabian StyleMa, Shaozu, Yifei Shen, Min Li, Ruitong Jiang, Luyi Cai, Tingting Wu, Linxi Gao, Meiqin Wu, and Peimin He. 2025. "Establishment of Novel Simple Sequence Repeat Markers in Phragmites australis and Application in Wetlands of Nanhui Dongtan, Shanghai" Biology 14, no. 4: 356. https://doi.org/10.3390/biology14040356
APA StyleMa, S., Shen, Y., Li, M., Jiang, R., Cai, L., Wu, T., Gao, L., Wu, M., & He, P. (2025). Establishment of Novel Simple Sequence Repeat Markers in Phragmites australis and Application in Wetlands of Nanhui Dongtan, Shanghai. Biology, 14(4), 356. https://doi.org/10.3390/biology14040356