

Alpine Adaptive Mechanism on Rhizosphere Microbes Recruitment of Crepis napifera (Franch.) Babc. by Multi-Omics Analysis

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials, Morphological Investigation, and Soil Characteristics
2.2. Transcriptome Sequencing and Differential Gene Expression Analysis
2.3. Metabolite Extraction and Analysis via UPLC-MS/MS
2.4. Rhizosphere and Soil Microbial DNA Extraction and 16S rRNA Sequencing
2.5. Species Abundance and Diversity Analysis
2.6. Principal Component and Pearson Correlation Analysis
2.7. KEGG Annotation and Enrichment Analysis
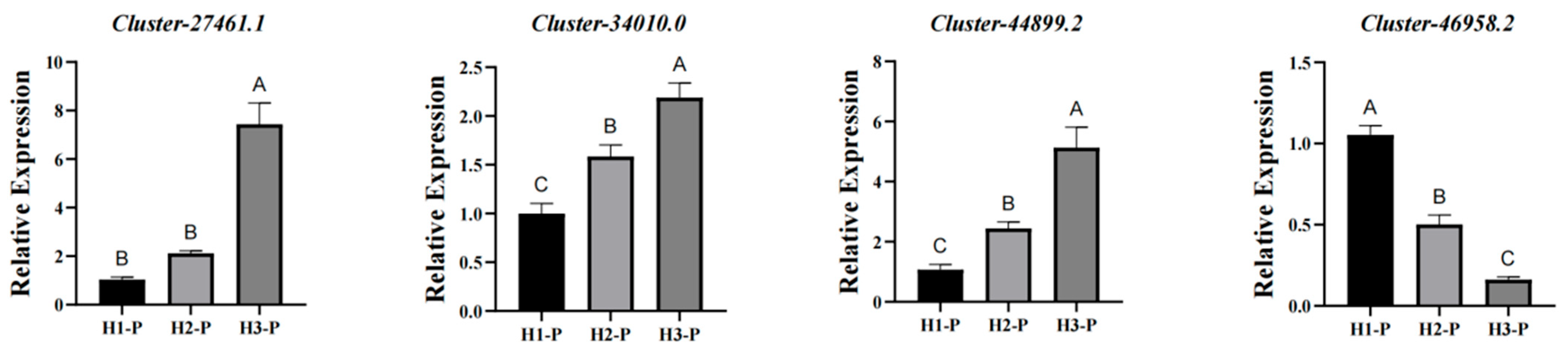
2.8. RNA Isolation and Quantitative Real-Time PCR
3. Results
3.1. Phenotypic Analysis of C. napifera (Franch.) Babc. Under Varying Altitude Conditions
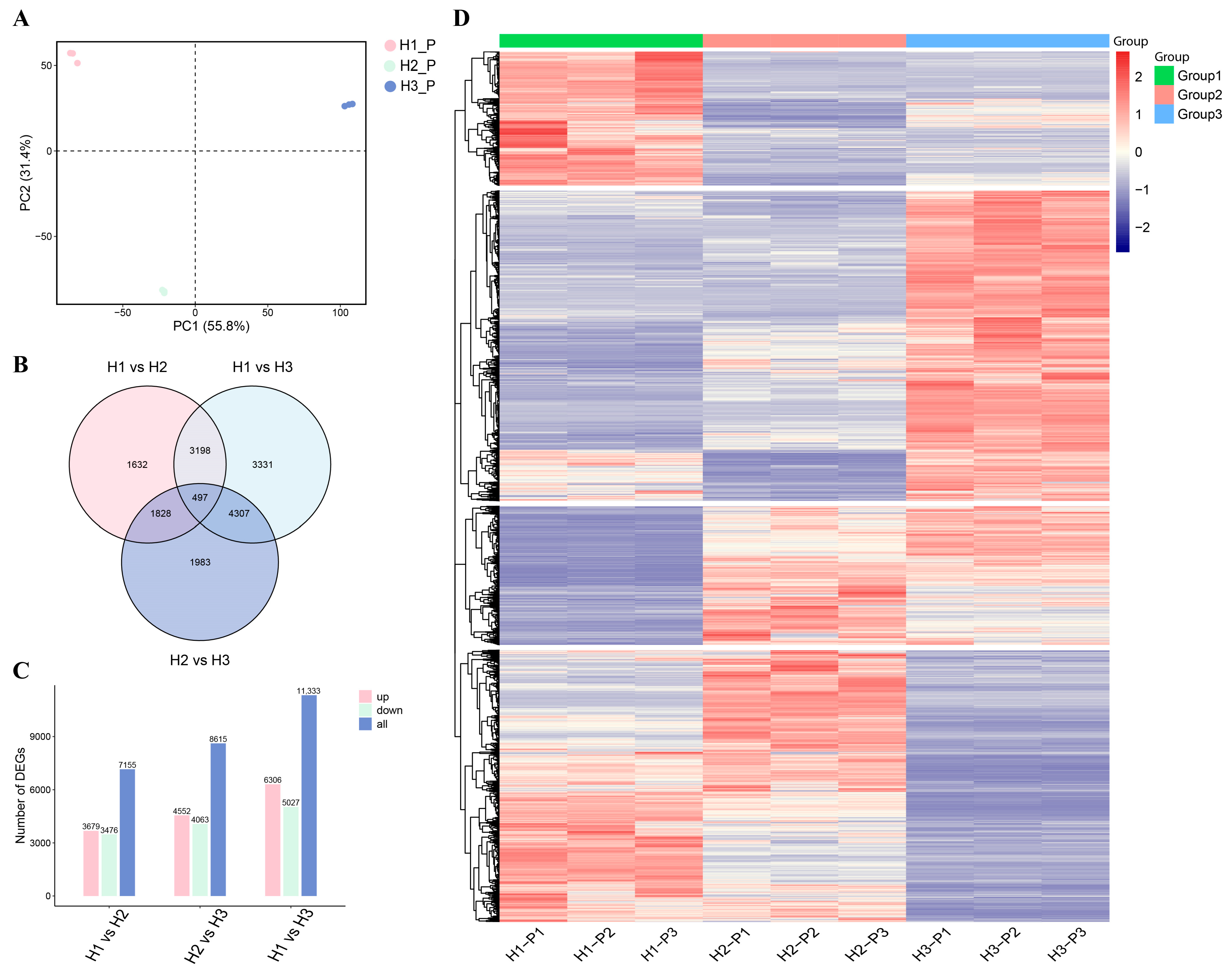
3.2. Transcriptome Analysis of C. napifera (Franch.) Babc. Under Varying Altitude Conditions
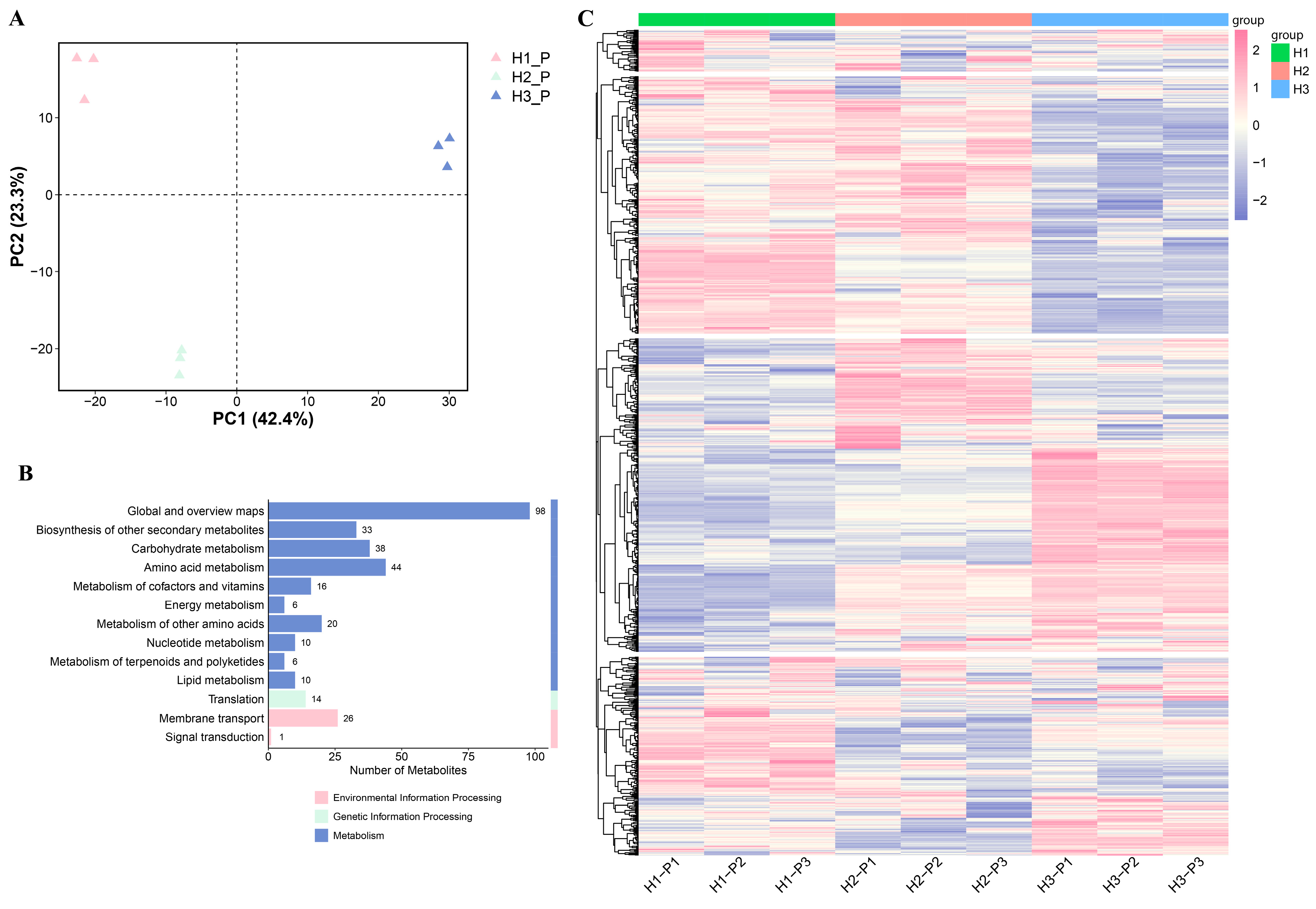
3.3. Metabolome Analysis of C. napifera (Franch.) Babc. Under Varying Altitude Conditions
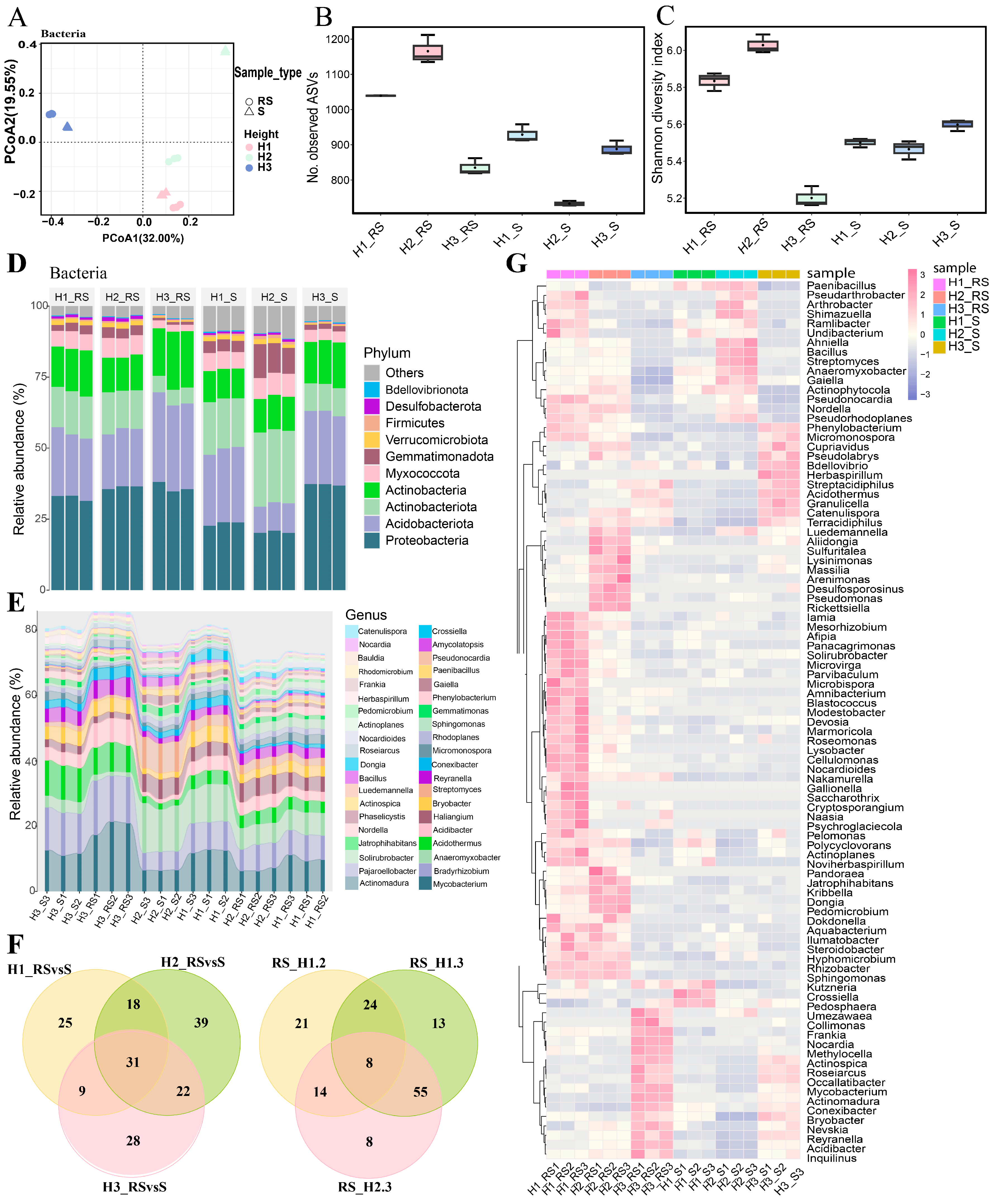
3.4. Bacterial Diversity Analysis of Roots of C. napifera (Franch.) Babc. Under Varying Altitude Conditions
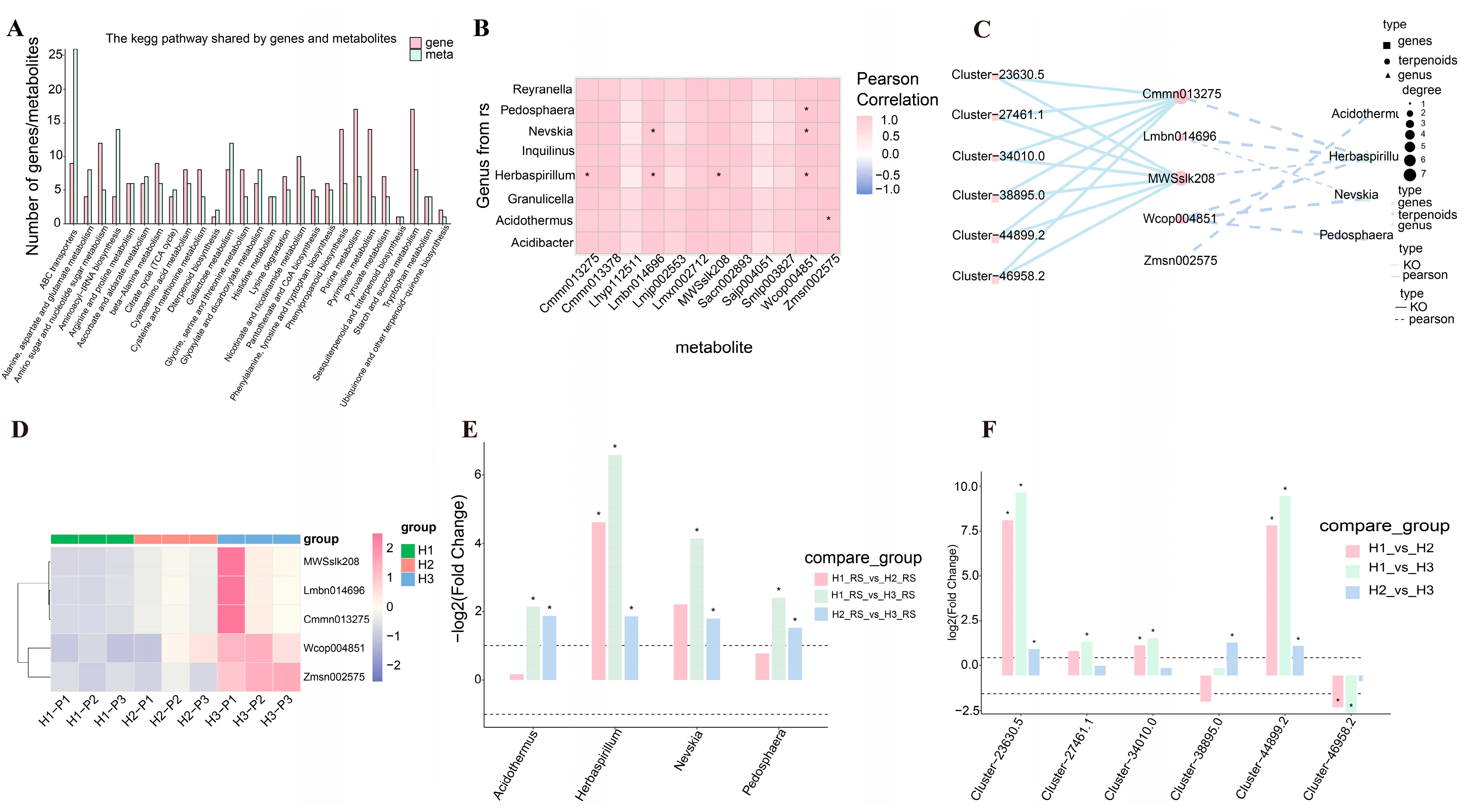
3.5. Multi-Omics Data Reveal Genes and Microbial Communities Related to the Metabolism of Terpene Compounds
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 16S rRNA | 16S ribosomal RNA |
| DEGs | Differentially expressed genes |
| DEMs | Differentially expressed metabolites |
| UV | Ultraviolet rays |
| UPLC-MS/MS | Ultra performance liquid chromatography–tandem mass spectrometry |
| MRM | Multiple reaction monitoring |
| OPLS-DA | Orthogonal partial least squares discriminant analysis |
| RS | Rhizosphere soil |
| PCA | Principal component analysis |
| MSEA | Metabolite sets enrichment analysis |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| WGCNA | Weighted gene co-expression network analysis |
| KEGG | Kyoto encyclopedia of genes and genomes |
References
- Badalamenti, N.; Sottile, F.; Bruno, M. Ethnobotany, phytochemistry, biological, and nutritional properties of genus Crepis-A review. Plants 2022, 11, 519. [Google Scholar] [CrossRef] [PubMed]
- Barda, C.; Anastasiou, K.; Tzara, A.; Grafakou, M.E.; Kalpoutzakis, E.; Heilmann, J.; Rallis, M.; Kourounakis, A.P.; Skaltsa, H. A Bio-guided screening for antioxidant, antiinflammatory and hypolipidemic potential supported by non-targeted metabolomic analysis of Crepis spp. Molecules 2022, 27, 6173. [Google Scholar] [CrossRef]
- Zhao, A.H.; Peng, X.Y.; Tang, C.J.; Zhang, R.P. Isolation and identification of sesquiterpenes from Crepis napifera. Acta Pharm. Sin. 2000, 35, 442–444. [Google Scholar] [CrossRef]
- Tsoukalas, M.; Gousiadou, C.; Skaltsa, H. Guaianolides and phenolic constituents from Crepis dioscoridis L., growing wild in Greece. Phytochem. Lett. 2014, 7, 202–206. [Google Scholar] [CrossRef]
- Pandey, G.; Khatoon, S.; Pandey, M.M.; Rawat, A.K.S. Altitudinal variation of berberine, total phenolics and flavonoid content in Thalictrum foliolosum and their correlation with antimicrobial and antioxidant activities. J. Ayurveda Integr. Med. 2018, 9, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.V.; Kumar, P.; Bhargava, B.; Sharma, R.K.; Irfan, M.; Chandora, R. Transcriptomic and metabolomic reprogramming to explore the high-altitude adaptation of medicinal plants: A review. J. Plant Growth Regul. 2023, 42, 7315–7329. [Google Scholar] [CrossRef]
- Gündeşli, M.A.; Uğur, R.; Yaman, M. The effects of altitude on fruit characteristics, nutrient chemicals, and biochemical properties of walnut fruits (Juglans regia L.). Horticulturae 2023, 9, 1086. [Google Scholar] [CrossRef]
- Karagiannis, E.; Michailidis, M.; Tanou, G.; Scossa, F.; Sarrou, E.; Stamatakis, G.; Samiotaki, M.; Martens, S.; Fernie, A.R.; Molassiotis, A. Decoding altitude-activated regulatory mechanisms occurring during apple peel ripening. Hortic Res. 2020, 7, 120. [Google Scholar] [CrossRef]
- Karagiannis, E.; Tanou, G.; Samiotaki, M.; Michailidis, M.; Diamantidis, G.; Minas, I.S.; Molassiotis, A. Comparative physiological and proteomic analysis reveal distinct regulation of peach skin quality traits by altitude. Front. Plant Sci. 2016, 7, 1689. [Google Scholar] [CrossRef]
- Zoratti, L.; Karppinen, K.; Luengo Escobar, A.; Häggman, H.; Jaakola, L. Light-controlled flavonoid biosynthesis in fruits. Front. Plant Sci. 2014, 5, 534. [Google Scholar] [CrossRef]
- Lu, M.; He, W.; Xu, Z.; Lu, Y.; Crabbe, M.J.C.; De, J. The effect of high altitude on ephedrine content and metabolic variations in two species of Ephedra. Front. Plant Sci. 2023, 14, 1236145. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, H.; Yan, J.; Li, X.; Li, J.; Hu, J.; Shang, X.; Yang, H. Deciphering the efficacy and mechanism of Astragalus membranaceus on high altitude polycythemia by integrating network pharmacology and in vivo experiments. Nutrients 2022, 14, 4968. [Google Scholar] [CrossRef]
- Kapoor, B.; Kumar, P.; Verma, V.; Irfan, M.; Sharma, R.; Bhargava, B. How plants conquered land: Evolution of terrestrial adaptation. J. Evol. Biol. 2023, 36, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Satyakam; Joshi, R.; Kumar, R. Unique metabolic shift reveals potential mechanism of cold and freezing acclimatization. J. Plant Growth Regul. 2023, 42, 5763–5779. [Google Scholar] [CrossRef]
- Niazian, M. Application of genetics and biotechnology for improving medicinal plants. Planta 2019, 249, 953–973. [Google Scholar] [CrossRef]
- Kumari, M.; Joshi, R.; Kumar, R. Metabolic signatures provide novel insights to Picrorhiza kurroa adaptation along the altitude in Himalayan region. Metabolomics 2020, 16, 77. [Google Scholar] [CrossRef]
- Tang, X.; Li, J.; Liu, L.; Jing, H.; Zuo, W.; Zeng, Y. Transcriptome analysis provides insights into Potentilla bifurca adaptation to high altitude. Life 2022, 12, 1337. [Google Scholar] [CrossRef]
- Zhao, Y.; Xu, F.; Liu, J.; Guan, F.; Quan, H.; Meng, F. The adaptation strategies of Herpetospermum pedunculosum (Ser.) Baill at altitude gradient of the Tibetan plateau by physiological and metabolomic methods. BMC Genom. 2019, 20, 451. [Google Scholar] [CrossRef]
- Dong, X.; Guo, Y.; Xiong, C.; Sun, L. Evaluation of two major Rhodiola species and the systemic changing characteristics of metabolites of Rhodiola crenulata in different altitudes by chemical methods combined with UPLC-QqQ-MS-based metabolomics. Molecules 2020, 25, 4062. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Pang, Z.; Xu, L.; Viau, C.; Lu, Y.; Salavati, R.; Basu, N.; Xia, J. MetaboAnalystR 4.0: A unified LC-MS workflow for global metabolomics. Nat. Commun. 2024, 15, 3675. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Envrion. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 1091. [Google Scholar] [CrossRef]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. microeco: An R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 2021, 97, fiaa255. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. MSEA: A web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010, 38, 71–77. [Google Scholar] [CrossRef]
- Guevara-Terán, M.; Gonzalez-Paramás, A.M.; Beltrán-Noboa, A.; Giampieri, F.; Battino, M.; Tejera, E.; Alvarez-Suarez, J.M. Influence of altitude on the physicochemical composition and antioxidant capacity of strawberry: A preliminary systematic review and meta-analysis. Phytochem. Rev. 2022, 22, 1567–1584. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, D. Secondary metabolites as a survival strategy in plants of high mountain habitats. Bol. Latinoam. Y Caribe Plantas Med. Y Aromat. 2019, 18, 29. [Google Scholar] [CrossRef]
- Zheng, W.; Yu, S.; Zhang, W.; Zhang, S.; Fu, J.; Ying, H.; Pingcuo, G.; Liu, S.; Zhao, F.; Wu, Q.; et al. The content and diversity of carotenoids associated with high-altitude adaptation in Tibetan peach fruit. Food Chem. 2023, 398, 133909. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Liu, Z.; Wang, L.; Bi, Z.; Sun, C.; Yao, P.; Zhang, J.; Bai, J.; Zeng, Y. Integrated transcriptomic and metabolomic analysis revealed altitude-related regulatory mechanisms on flavonoid accumulation in potato tubers. Food Res. Int. 2023, 170, 112997. [Google Scholar] [CrossRef]
- Du, Z.; Lin, W.; Yu, B.; Zhu, J.; Li, J. Integrated metabolomic and transcriptomic analysis of the flavonoid accumulation in the leaves of Cyclocarya paliurus at different altitudes. Front. Plant Sci. 2022, 12, 794137. [Google Scholar] [CrossRef]
- Dang, H.; Yu, N.; Mou, A.; Zhang, L.; Guo, B.; Liu, Y. Metagenomic insights into direct interspecies electron transfer and quorum sensing in blackwater anaerobic digestion reactors supplemented with granular activated carbon. Bioresour. Technol. 2022, 352, 127113. [Google Scholar] [CrossRef]
- Chen, X.; Li, F.L.; Kong, C.H. Rhizosphere bacteria mediate flowering time of two genotypes of Arabidopsis with and without root-secreted signaling (−)-loliolide. Rhizosphere 2023, 27, 100774. [Google Scholar] [CrossRef]
- Wei, C.; Gu, W.; Tian, R.; Xu, F.; Han, Y.; Ji, Y.; Li, T.; Zhu, Y.; Lang, P.; Wu, W. Comparative analysis of the structure and function of rhizosphere microbiome of the Chinese medicinal herb Alisma in different regions. Arch. Microbiol. 2022, 204, 448. [Google Scholar] [CrossRef]
- Huang, C.; Han, X.; Yang, Z.; Chen, Y.; Rengel, Z. Sowing methods influence soil bacterial diversity and community composition in a winter wheat-summer maize rotation system on the Loess plateau. Front. Microbiol. 2020, 11, 192. [Google Scholar] [CrossRef]
- Yang, Y.; Li, L.; He, Y.; Ma, T.; Zheng, J.; Wang, M.; Tu, W.; Fan, M.; Chen, S. Response processes to water quality changes driven by the dynamic regeneration of the surface microlayer film in slow-flowing freshwater bodies. Envrion. Pollut. 2024, 363, 125125. [Google Scholar] [CrossRef]
- Wang, B.; Teng, Y.; Xu, Y.; Chen, W.; Ren, W.; Li, Y.; Christie, P.; Luo, Y. Effect of mixed soil microbiomes on pyrene removal and the response of the soil microorganisms. Sci. Total Environ. 2018, 640–641, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Andersson, P.L.; Berg, A.H.; Bjerselius, R.; Norrgren, L.; Olsén, H.; Olsson, P.E.; Orn, S.; Tysklind, M. Bioaccumulation of selected PCBs in zebrafish, three-spined stickleback, and arctic char after three different routes of exposure. Arch. Envrion. Contam. Toxicol. 2001, 40, 519–530. [Google Scholar] [CrossRef]
- Straub, D.; Rothballer, M.; Hartmann, A.; Ludewig, U. The genome of the endophytic bacterium H. frisingense GSF30(T) identifies diverse strategies in the Herbaspirillum genus to interact with plants. Front. Microbiol. 2013, 4, 168. [Google Scholar] [CrossRef]
- Serrato, R.V.; Sassaki, G.L.; Cruz, L.M.; Carlson, R.W.; Muszyński, A.; Monteiro, R.A.; Pedrosa, F.O.; Souza, E.M.; Iacomini, M. Chemical composition of lipopolysaccharides isolated from various endophytic nitrogen-fixing bacteria of the genus Herbaspirillum. Can J. Microbiol. 2010, 56, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Camp, D.; Davis, R.A.; Campitelli, M.; Ebdon, J.; Quinn, R.J. Drug-like properties: Guiding principles for the design of natural product libraries. J. Nat. Prod. 2012, 75, 72–81. [Google Scholar] [CrossRef]
- Li, H.; Wei, W.; Xu, H. Drug discovery is an eternal challenge for the biomedical sciences. Acta Mater. Medica 2022, 1, 1–3. [Google Scholar] [CrossRef]
- Wang, W.P.; Jiang, K.; Zhang, P.; Shen, K.K.; Qu, S.J.; Yu, X.P.; Tan, C.H. Highly oxygenated and structurally diverse diterpenoids from Euphorbia helioscopia. Phytochemistry 2018, 145, 93–102. [Google Scholar] [CrossRef]
- Vieira, C.; Duarte, N.; Reis, M.A.; Spengler, G.; Madureira, A.M.; Molnár, J.; Ferreira, M.J. Improving the MDR reversal activity of 6,17-epoxylathyrane diterpenes. Bioorg. Med. Chem. 2014, 22, 6392–6400. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, W.; Zhu, C.; Lin, S.; Li, Y.; Xiong, L.; Wang, S.; Wang, L.; Yang, Y.; Guo, Y.; et al. Lathyrane diterpenoids from the roots of Euphorbia micractina and their biological activities. J. Nat. Prod. 2011, 74, 1221–1229. [Google Scholar] [CrossRef]
- Christina, B.; Ana, C.; Marina, S.; Michael, T.; Helen, S. Phytochemical investigation of Crepis incana Sm.(asteraceae) endemic to southern greece. Biochem. Syst. Ecol. 2018, 80, 59–62. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Augustin, J.M.; Kuzina, V.; Andersen, S.B.; Bak, S. Molecular activities, biosynthesis and evolution of triterpenoid saponins. Phytochemistry 2011, 72, 435–457. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Altitude (m) | Location | Annual Average Air Temperature (°C) | Annual Precipitation (mm) | Average Temperatures of the Warmest Months (°C) | Average Temperatures of the Coldest Months (°C) |
|---|---|---|---|---|---|---|
| H1 | 1858 | Jin Kedu, Xingming Xingyuan Village, Longpan Township | 15.7 | 790.5 | 21.1 | 8.2 |
| H2 | 2417 | Le Shibanman, Xingwen Qiping Village, Longpan Township | 12.2 | 839.1 | 18.2 | 6.4 |
| H3 | 2893 | Jizi Village, Tai’an Township | 8.7 | 643.3 | 13.5 | 3.6 |
| Sample Name | Soil Type | Available P (μg/g) | Available K (mg/kg) | pH Value | Total C (g/kg) | Total N (g/kg) | C:N |
|---|---|---|---|---|---|---|---|
| H1 | red clay | 6.04 | 512.91 | 6.52 | 13.63 | 1.62 | 8.46 |
| H2 | red clay | 5.80 | 341.43 | 6.01 | 13.81 | 1.26 | 16.45 |
| H3 | Yellow sandy soil | 5.93 | 250.46 | 6.35 | 15.22 | 1.40 | 11.73 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.; Liu, W.; Mo, X.; Mei, Z. Alpine Adaptive Mechanism on Rhizosphere Microbes Recruitment of Crepis napifera (Franch.) Babc. by Multi-Omics Analysis. Biology 2025, 14, 345. https://doi.org/10.3390/biology14040345
Yang G, Liu W, Mo X, Mei Z. Alpine Adaptive Mechanism on Rhizosphere Microbes Recruitment of Crepis napifera (Franch.) Babc. by Multi-Omics Analysis. Biology. 2025; 14(4):345. https://doi.org/10.3390/biology14040345
Chicago/Turabian StyleYang, Genlin, Weiwei Liu, Xinchun Mo, and Zhinan Mei. 2025. "Alpine Adaptive Mechanism on Rhizosphere Microbes Recruitment of Crepis napifera (Franch.) Babc. by Multi-Omics Analysis" Biology 14, no. 4: 345. https://doi.org/10.3390/biology14040345
APA StyleYang, G., Liu, W., Mo, X., & Mei, Z. (2025). Alpine Adaptive Mechanism on Rhizosphere Microbes Recruitment of Crepis napifera (Franch.) Babc. by Multi-Omics Analysis. Biology, 14(4), 345. https://doi.org/10.3390/biology14040345