
Genetic Structure and Selection Signals for Extreme Environment Adaptation in Lop Sheep of Xinjiang
 , , , , , , , and
, , , , , , , and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
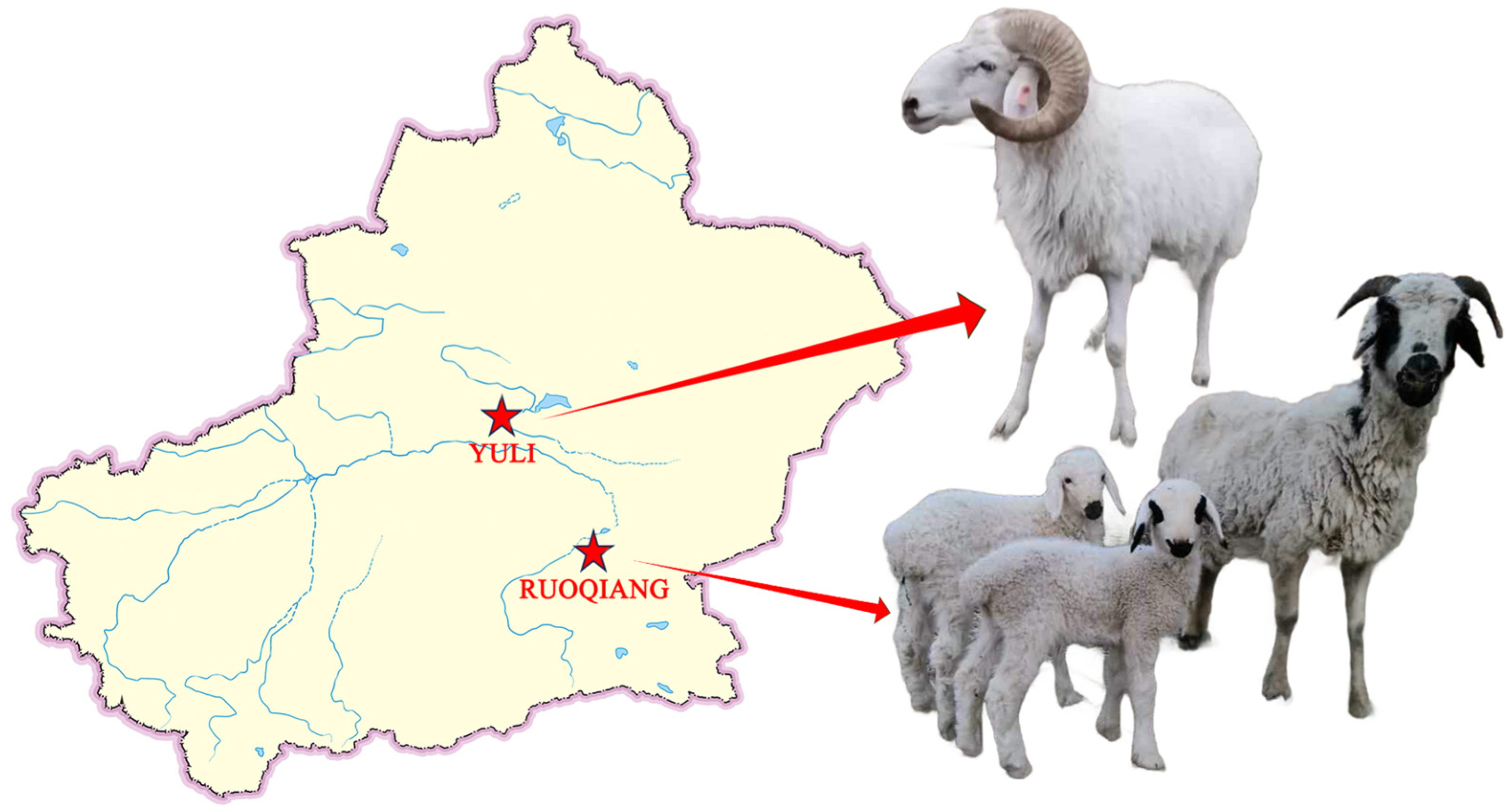
2.1. Animal Care
2.2. Sample Collection and Sequencing
2.3. Genotyping and Quality Control
2.4. Genetic Diversity and Population Structure
2.5. Selection of Signal Analyses
2.6. Gene Functional Enrichment Analysis
3. Results
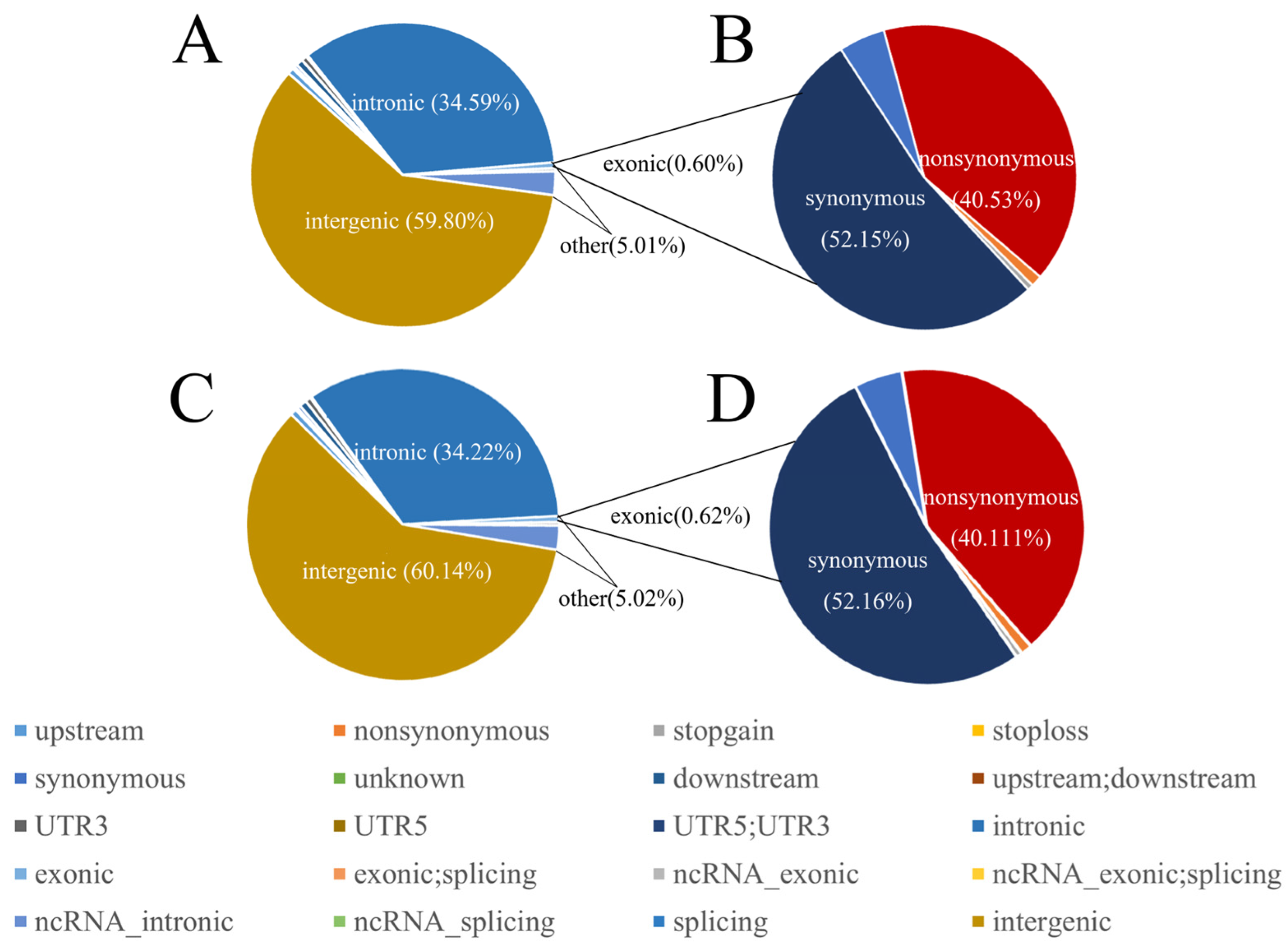
3.1. Sequencing and Identification of SNPs
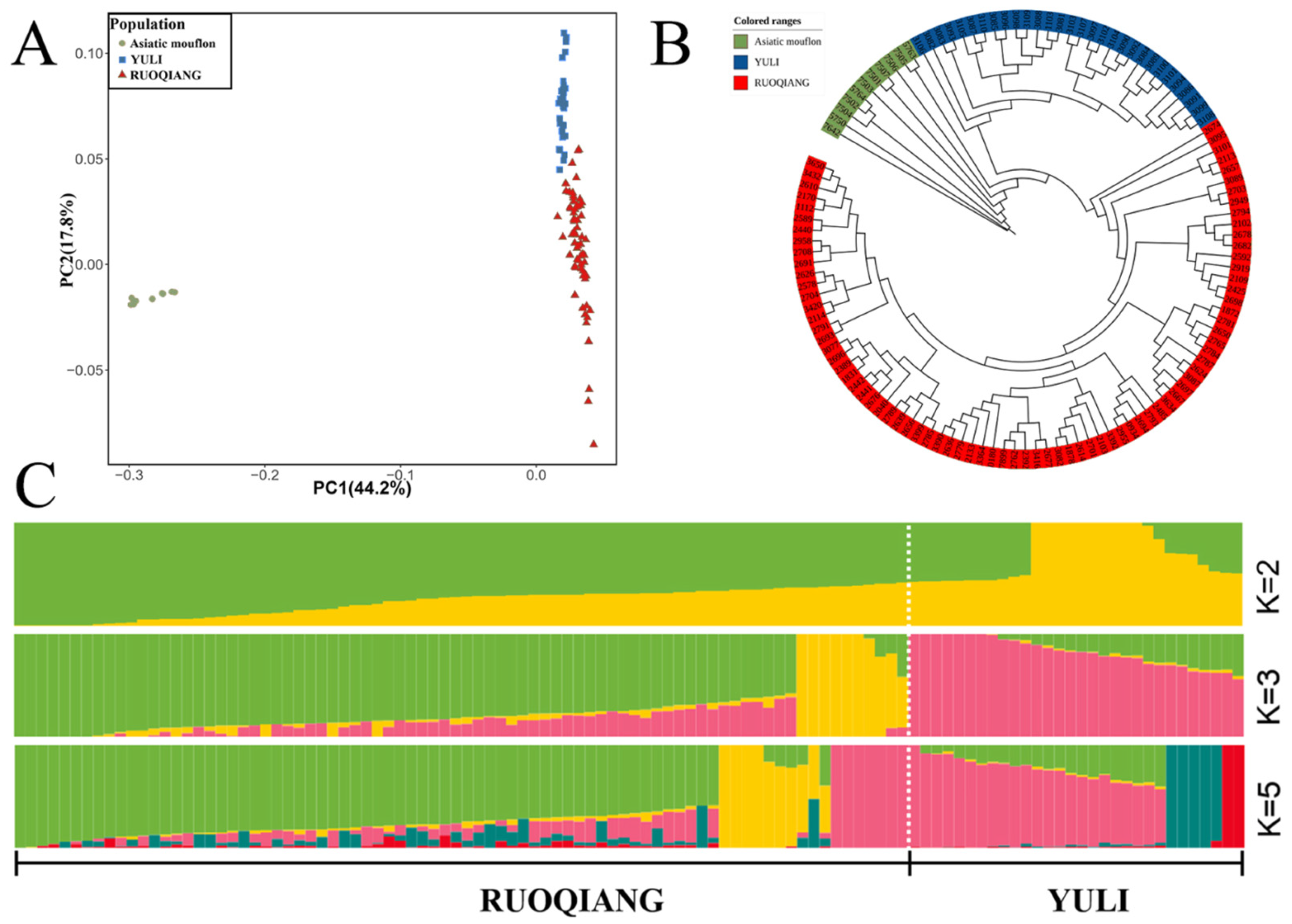
3.2. Analysis of Genetic Diversity and Linkage Disequilibrium in Lop Sheep Populations
3.3. Comparison of Population Structure Characteristics of Lop Sheep in Two Regions
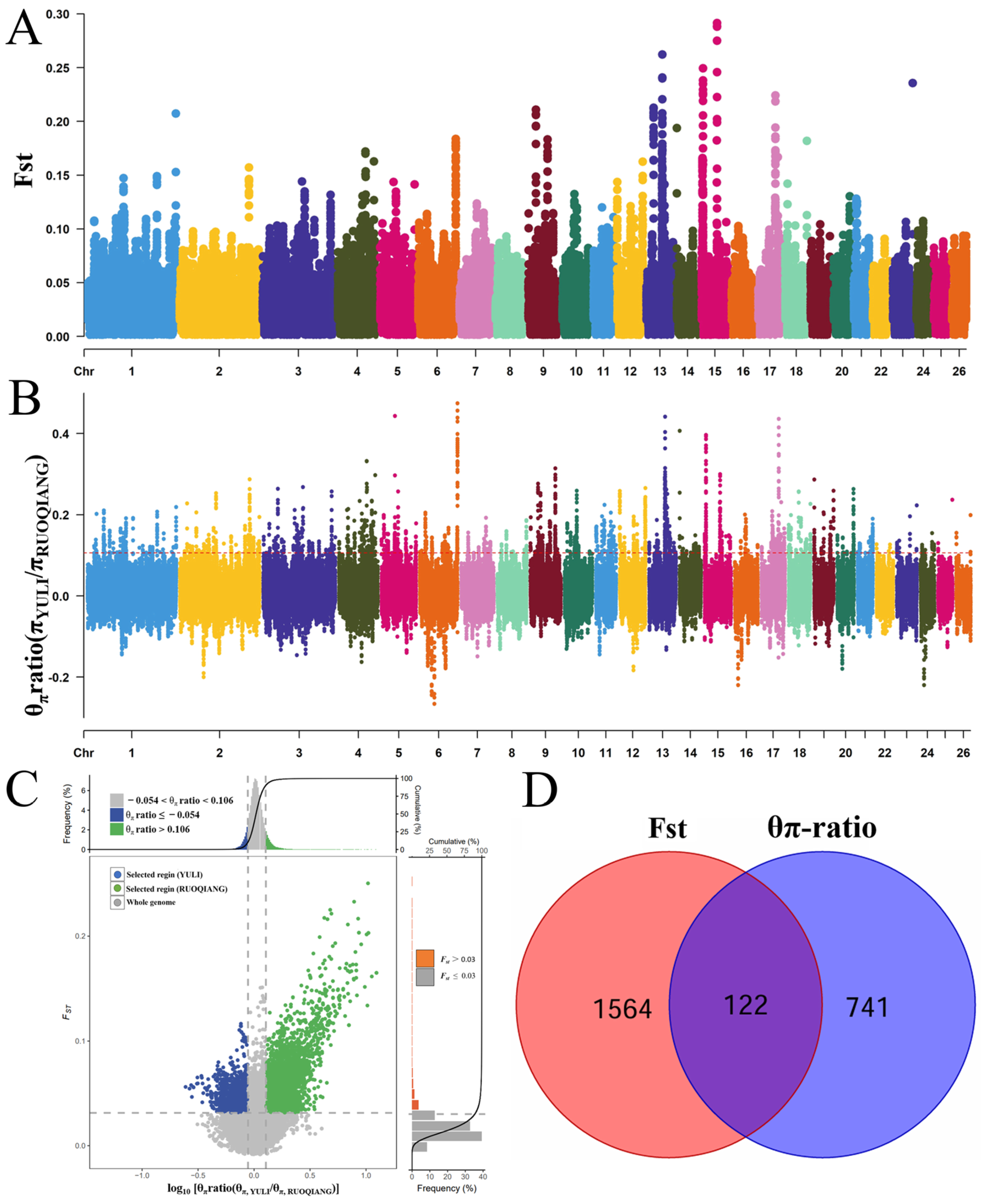
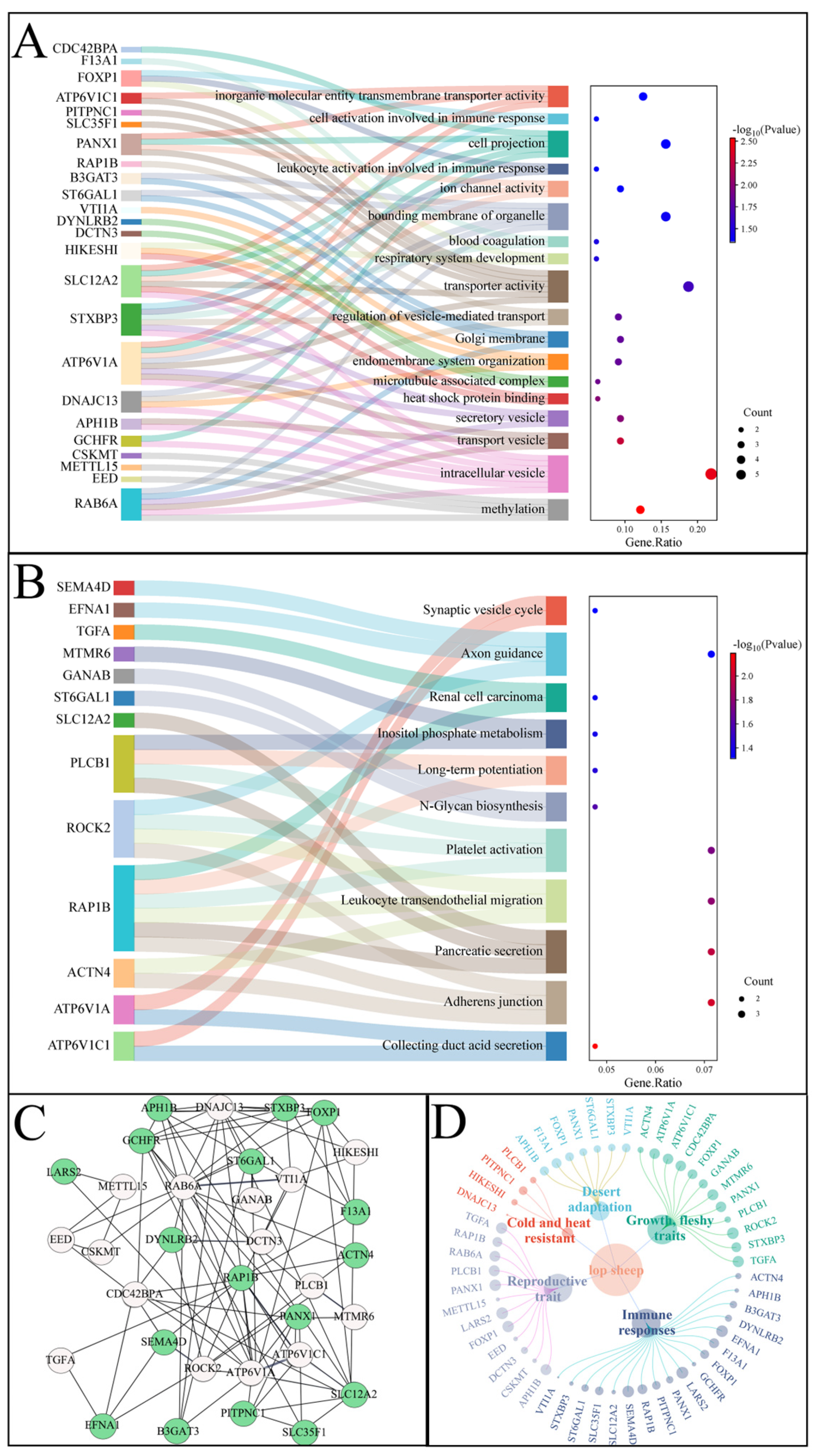
3.4. Selective Signal Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qu, Y.; Hu, Y.; Rao, H.; Abuduresule, I.; Li, W.; Hu, X.; Jiang, H.; Wang, C.; Yang, Y. Diverse lifestyles and populations in the Xiaohe culture of the Lop Nur region, Xinjiang, China. Archaeol. Anthropol. Sci. 2017, 10, 2005–2014. [Google Scholar] [CrossRef]
- Li, W.; Mu, G.; Lin, Y.; Zhang, D. Abrupt climatic shift at ~4000 cal. yr B.P. and late Holocene climatic instability in arid Central Asia: Evidence from Lop Nur saline lake in Xinjiang, China. Sci. Total Environ. 2021, 784, 147202. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Yu, L.; Zhu, J.; Fraedrich, K.; Guan, Y.; Sielmann, F.; Zhang, C.; Yu, M. The Shrinkage of Lake Lop Nur in the Twentieth Century: A Comprehensive Ecohydrological Analysis. J. Hydrometeorol. 2022, 23, 1245–1255. [Google Scholar] [CrossRef]
- Ma, C.; Wang, F.; Cao, Q.; Xia, X.; Li, S.; Li, X. Climate and environment reconstruction during the Medieval Warm Period in Lop Nur of Xinjiang, China. Sci. Bull. 2008, 53, 3016–3027. [Google Scholar] [CrossRef]
- Wang, J.; Suo, J.; Yang, R.; Zhang, L.; Li, X.; Han, Z.; Zhou, W.; Liu, S.; Gao, Q. Genetic diversity, population structure, and selective signature of sheep in the northeastern Tarim Basin. Front. Genet. 2023, 14, 1281601. [Google Scholar] [CrossRef]
- Zhang, C.L.; Zhang, J.H.; Mirenisa, T.; Zhou, W.; Han, Z.; Li, X.; Yang, R.; Zhang, L.; Zheng, L.; Liu, S. Landscape genomics reveals adaptive divergence of indigenous sheep in different ecological environments of Xinjiang, China. Sci. Total Environ. 2023, 904, 166698. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Alganmi, N.; Abusamra, H. Evaluation of an optimized germline exomes pipeline using BWA-MEM2 and Dragen-GATK tools. PLoS ONE 2023, 18, e0288371. [Google Scholar] [CrossRef]
- Charlotte, H.; Pascal, C.; Dries, D.; Reumers, J. elPrep: High-Performance Preparation of Sequence Alignment/Map Files for Variant Calling. PLoS ONE 2015, 10, e0132868. [Google Scholar] [CrossRef]
- Brouard, J.S.; Bissonnette, N. Variant Calling from RNA-seq Data Using the GATK Joint Genotyping Workflow. Methods Mol. Biol. 2022, 2493, 205–233. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Chimusa, E.R.; Defo, J.; Thami, P.K.; Awany, D.; Mulisa, D.D.; Allali, I.; Ghazal, H.; Moussa, A.; Mazandu, G.K. Dating admixture events is unsolved problem in multi-way admixed populations. Brief. Bioinform. 2020, 21, 144–155. [Google Scholar] [CrossRef]
- Gianola, D.; Simianer, H.; Qanbari, S. A two-step method for detecting selection signatures using genetic markers. Genet. Res. 2010, 92, 141–155. [Google Scholar] [CrossRef]
- Shriver, M.D.; Kennedy, G.C.; Parra, E.J.; Lawson, H.A.; Sonpar, V.; Huang, J.; Akey, J.M.; Jones, K.W. The genomic distribution of population substructure in four populations using 8,525 autosomal SNPs. Hum. Genom. 2004, 1, 274–286. [Google Scholar] [CrossRef]
- Huang, F.; Fu, M.; Li, J.; Chen, L.; Feng, K.; Huang, T.; Cai, Y.D. Analysis and prediction of protein stability based on interaction network, gene ontology, and KEGG pathway enrichment scores. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2023, 1871, 140889. [Google Scholar] [CrossRef]
- Naval-Sanchez, M.; Nguyen, Q.; McWilliam, S.; Porto-Neto, L.R.; Tellam, R.; Vuocolo, T.; Reverter, A.; Perez-Enciso, M.; Brauning, R.; Clarke, S. Sheep genome functional annotation reveals proximal regulatory elements contributed to the evolution of modern breeds. Nat. Commun. 2018, 9, 859. [Google Scholar] [CrossRef]
- Perry, J.S.A.; Morioka, S.; Medina, C.B.; Iker Etchegaray, J.; Barron, B.; Raymond, M.H.; Lucas, C.D.; Onengut-Gumuscu, S.; Delpire, E.; Ravichandran, K.S. Interpreting an apoptotic corpse as anti-inflammatory involves a chloride sensing pathway. Nat. Cell Biol. 2019, 21, 1532–1543. [Google Scholar] [CrossRef]
- Kaminskiy, Y.; Kuznetsova, V.; Kudriaeva, A.; Zmievskaya, E.; Bulatov, E. Neglected, yet significant role of FOXP1 in T-cell quiescence, differentiation and exhaustion. Front. Immunol. 2022, 13, 971045. [Google Scholar] [CrossRef]
- Rusiecka, O.M.; Tournier, M.; Molica, F.; Kwak, B.R. Pannexin1 channels-a potential therapeutic target in inflammation. Front. Cell Dev. Biol. 2022, 10, 1020826. [Google Scholar] [CrossRef]
- Ramsey, K.A.; Bosco, A.; McKenna, K.L.; Carter, K.W.; Elliot, J.G.; Berry, L.J.; Sly, P.D.; Larcombe, A.N.; Zosky, G.R. In utero exposure to arsenic alters lung development and genes related to immune and mucociliary function in mice. Environ. Health Perspect. 2013, 121, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Awasthi, A.; White, G.C., 2nd; Chrzanowska-Wodnicka, M.; Malarkannan, S. Rap1b regulates B cell development, homing, and T cell-dependent humoral immunity. J. Immunol. 2008, 181, 3373–3383. [Google Scholar] [CrossRef]
- Zhu, Z.; Luo, Y.; Yu, J.; Gao, J.; Zhang, Y.; Xiao, C.; Zhang, C.; Wang, G.; Liu, Y.; Fu, M.; et al. Sema4D is required in both the adaptive and innate immune responses of contact hypersensitivity. Mol. Immunol. 2016, 78, 98–104. [Google Scholar] [CrossRef]
- Guan, X.; Zhao, S.; Xiang, W.; Jin, H.; Chen, N.; Lei, C.; Jia, Y.; Xu, L. Genetic Diversity and Selective Signature in Dabieshan Cattle Revealed by Whole-Genome Resequencing. Biology 2022, 11, 1327. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Hu, M.; Shi, L.; Li, T.; Bai, C.; Sun, F.; Ma, H.; Zhao, Z.; Yan, S. Whole genome sequencing identified genomic diversity and candidated genes associated with economic traits in Northeasern Merino in China. Front. Genet. 2024, 15, 1302222. [Google Scholar] [CrossRef]
- Kim, E.S.; Elbeltagy, A.R.; Aboul-Naga, A.M.; Rischkowsky, B.; Sayre, B.; Mwacharo, J.M.; Rothschild, M.F. Multiple genomic signatures of selection in goats and sheep indigenous to a hot arid environment. Heredity 2016, 116, 255–264. [Google Scholar] [CrossRef]
- Aboul-Naga, A.M.; Alsamman, A.M.; El Allali, A.; Elshafie, M.H.; Abdelal, E.S.; Abdelkhalek, T.M.; Abdelsabour, T.H.; Mohamed, L.G.; Hamwieh, A. Genome-wide analysis identified candidate variants and genes associated with heat stress adaptation in Egyptian sheep breeds. Front. Genet. 2022, 13, 898522. [Google Scholar] [CrossRef]
- Kose, S.; Imai, K.; Watanabe, A.; Nakai, A.; Suzuki, Y.; Imamoto, N. Lack of Hikeshi activates HSF1 activity under normal conditions and disturbs the heat-shock response. Life Sci. Alliance 2022, 5, e202101241. [Google Scholar] [CrossRef]
- Tang, G.; Ma, C.; Li, L.; Zhang, S.; Li, F.; Wu, J.; Yin, Y.; Zhu, Q.; Liang, Y.; Wang, R.; et al. PITPNC1 promotes the thermogenesis of brown adipose tissue under acute cold exposure. Sci. China Life Sci. 2022, 65, 2287–2300. [Google Scholar] [CrossRef]
- Alshehri, F.S.M.; Whyte, C.S.; Mutch, N.J. Factor XIII-A: An Indispensable “Factor” in Haemostasis and Wound Healing. Int. J. Mol. Sci. 2021, 22, 3055. [Google Scholar] [CrossRef] [PubMed]
- Scemes, E.; Velíšková, J. Exciting and not so exciting roles of pannexins. Neurosci. Lett. 2019, 695, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yue, L.; Zhang, F.; Surati, U.; Pukhrambam, M.; Sivalingam, J.; Kumar, A.; Sarkar, M. Roles of bovine sialoglycoproteins for anti-skin aging and accelerating skin wound healing. J. Cosmet. Dermatol. 2023, 22, 3470–3479. [Google Scholar] [CrossRef]
- Kour, A.; Niranjan, S.K.; Malayaperumal, M.; Surati, U.; Pukhrambam, M.; Sivalingam, J.; Kumar, A.; Sarkar, M. Genomic Diversity Profiling and Breed-Specific Evolutionary Signatures of Selection in Arunachali Yak. Genes 2022, 13, 254. [Google Scholar] [CrossRef]
- Bosch, D.G.; Boonstra, F.N.; de Leeuw, N.; Pfundt, R.; Nillesen, W.M.; deLigt, J.; Gilissen, C.; Jhangiani, S.; Lupski, J.R.; Cremers, F.P.; et al. Novel genetic causes for cerebral visual impairment. Eur. J. Hum. Genet. 2016, 24, 660–665. [Google Scholar] [CrossRef]
- Hosseinzadeh, S.; Masoudi, A.A. Investigating the expression of fertility-regulating LncRNAs in multiparous and uniparous Shal ewe’s ovaries. Genome 2024, 67, 78–89. [Google Scholar] [CrossRef]
- Hou, X.; Zhang, J.; Li, L.; Ma, R.; Ge, J.; Han, L.; Wang, Q. Rab6a is a novel regulator of meiotic apparatus and maturational progression in mouse oocytes. Sci. Rep. 2016, 6, 22209. [Google Scholar] [CrossRef]
- Gholizadeh, M.; Esmaeili-Fard, S.M. Meta-analysis of genome-wide association studies for litter size in sheep. Theriogenology 2022, 180, 103–112. [Google Scholar] [CrossRef]
- Zhu, M.; Zhang, H.; Yang, H.; Zhao, Z.; Blair, H.T.; Liang, H.; Wu, P.; Yu, Q. Targeting GNAQ in hypothalamic nerve cells to regulate seasonal estrus in sheep. Theriogenology 2022, 181, 79–88. [Google Scholar] [CrossRef]
- Chang, L.; Zheng, Y.; Li, S.; Niu, X.; Huang, S.; Long, Q.; Ran, X.; Wang, J. Identification of genomic characteristics and selective signals in Guizhou black goat. BMC Genom. 2024, 25, 164. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Collection Region | Number | He | Ho | π | Froh |
|---|---|---|---|---|---|
| Yuli Lop sheep | 30 | 0.3030 | 0.2941 | 3.39 × 10−3 | −0.0205 |
| Ruoqiang Lop sheep | 80 | 0.2992 | 0.2934 | 3.32 × 10−3 | 0.0547 |
| total | 120 | 0.3011 | 0.2938 | 3.36 × 10−3 | 0.0172 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.; Wang, J.; Bi, L.; Fang, D.; Xiang, X.; Khamili, A.; Kurban, W.; Han, C.; Gao, Q. Genetic Structure and Selection Signals for Extreme Environment Adaptation in Lop Sheep of Xinjiang. Biology 2025, 14, 337. https://doi.org/10.3390/biology14040337
Yang C, Wang J, Bi L, Fang D, Xiang X, Khamili A, Kurban W, Han C, Gao Q. Genetic Structure and Selection Signals for Extreme Environment Adaptation in Lop Sheep of Xinjiang. Biology. 2025; 14(4):337. https://doi.org/10.3390/biology14040337
Chicago/Turabian StyleYang, Chenchen, Jieru Wang, Lanshu Bi, Di Fang, Xin Xiang, Abliz Khamili, Waili Kurban, Chunmei Han, and Qinghua Gao. 2025. "Genetic Structure and Selection Signals for Extreme Environment Adaptation in Lop Sheep of Xinjiang" Biology 14, no. 4: 337. https://doi.org/10.3390/biology14040337
APA StyleYang, C., Wang, J., Bi, L., Fang, D., Xiang, X., Khamili, A., Kurban, W., Han, C., & Gao, Q. (2025). Genetic Structure and Selection Signals for Extreme Environment Adaptation in Lop Sheep of Xinjiang. Biology, 14(4), 337. https://doi.org/10.3390/biology14040337