
Forecasting the Pharmacological Mechanisms of Plumbago zeylanica and Solanum xanthocarpum in Diabetic Retinopathy Treatment: A Network Pharmacology, Molecular Docking, and Molecular Dynamics Simulation Study
 ,
,  ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Active Phytoconstituents of SX and PZ and Their Related Target Screening
2.2. Network Construction of Active Phytoconstituents and Related Targets
2.3. Collection of Potential DR-Associated Targets
2.4. Screening Phytoconstituent-Disease Overlapping Targets
2.5. Network Construction of Phytoconstituent–Disease Common Targets
2.6. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Enrichment Analysis
2.7. Construction of PZ and SX Phytoconstituents–Targets–Pathways Network
2.8. Molecular Docking
2.9. MD Simulation
2.10. Free Energy Calculation (MM-GBSA)
2.11. Principal Component Analysis (PCA)
3. Results
3.1. Active Phytoconstituents of SX and PZ
3.2. Phytoconstituent–Target Network Construction
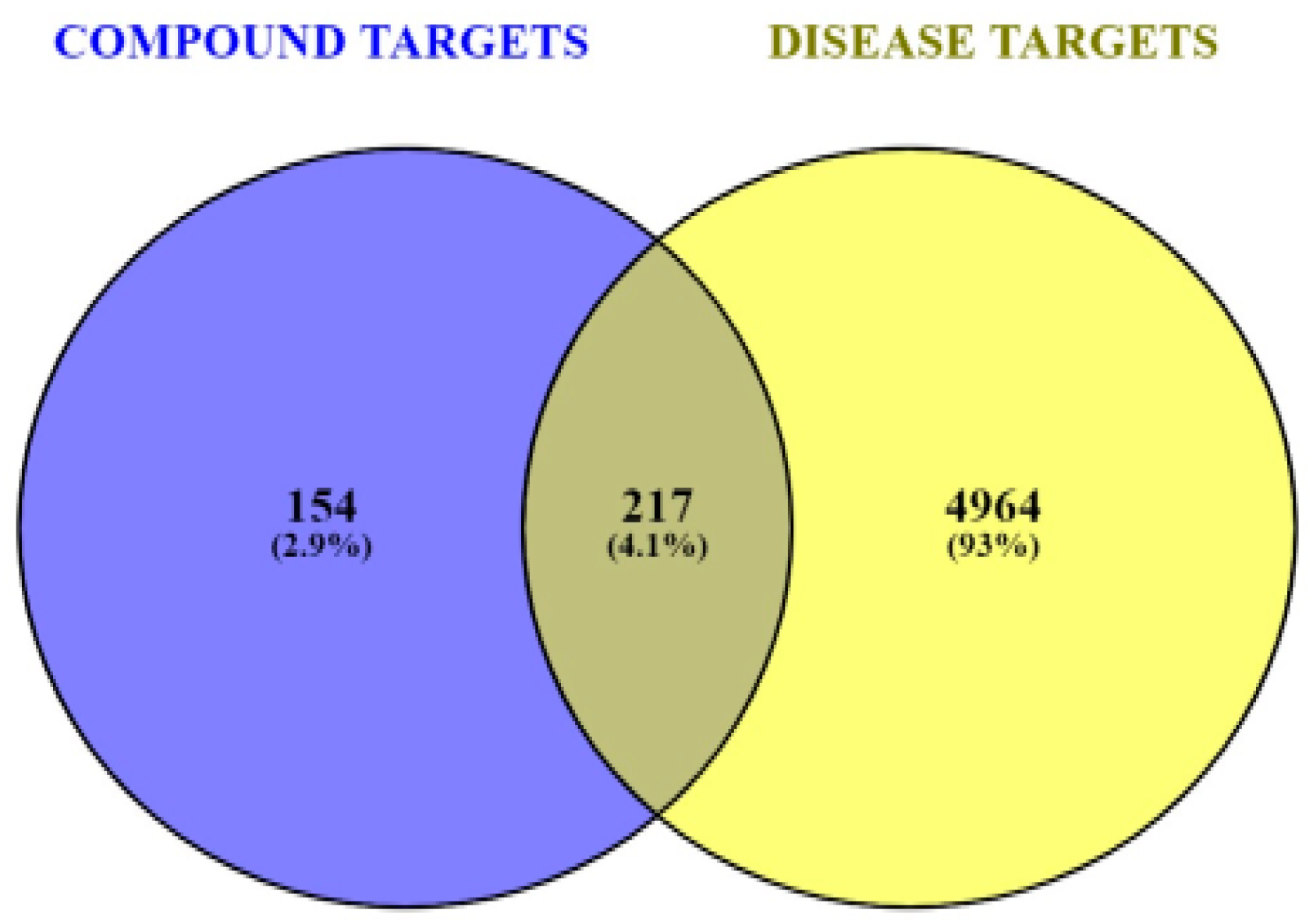
3.3. Predicting DR-Related Targets
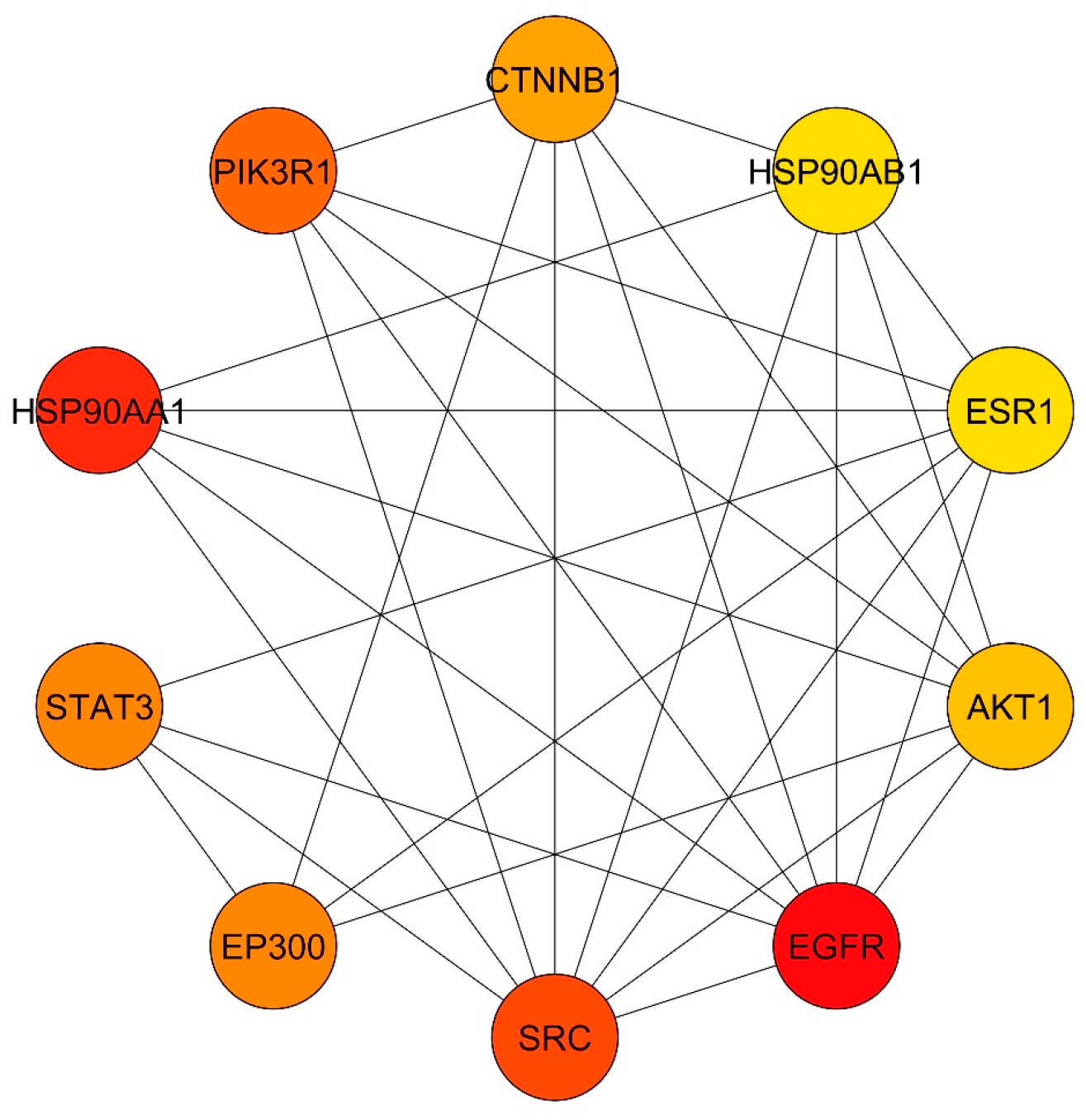
3.4. Common Targets PPI Network
3.5. GO and KEGG Enrichment Analyses
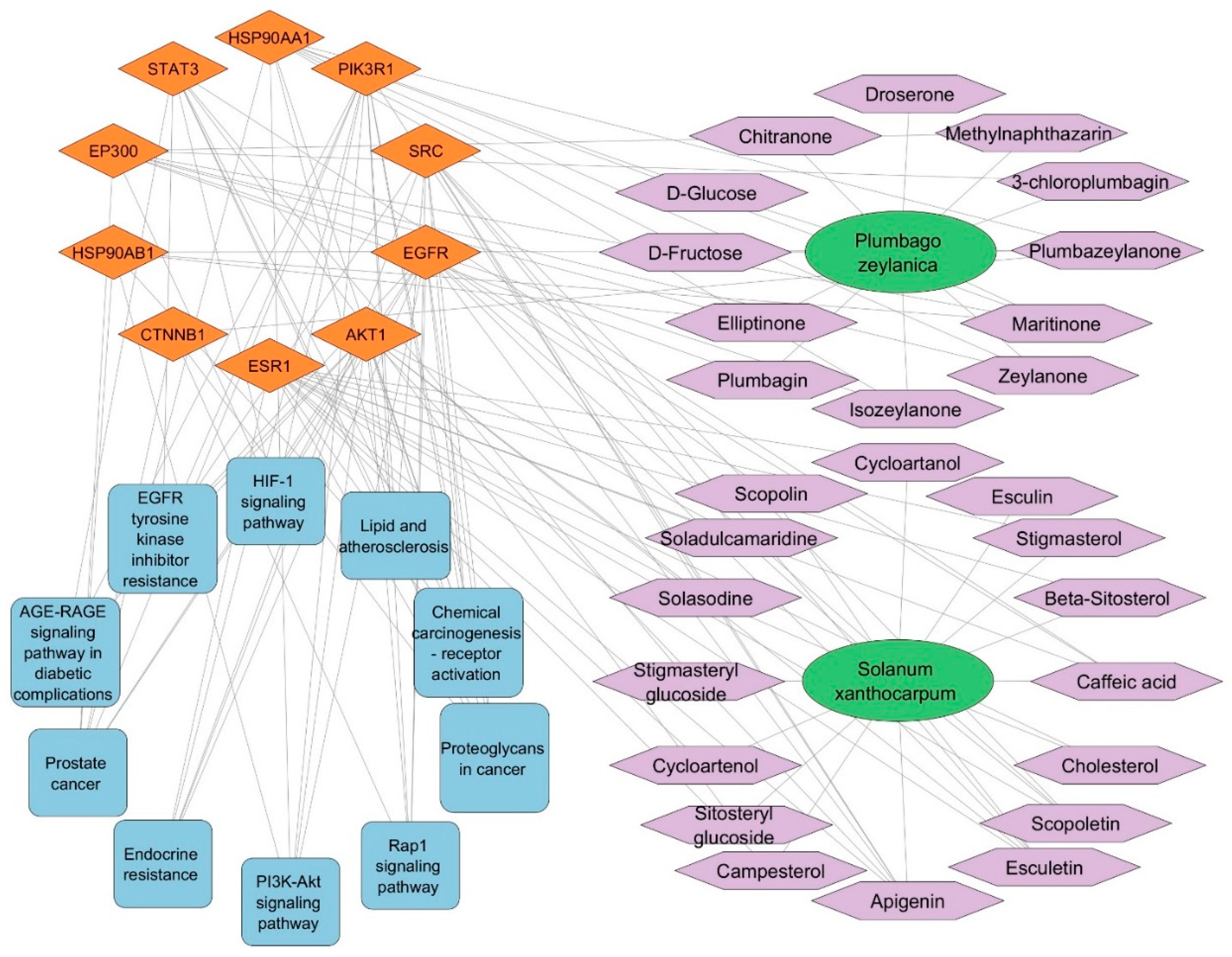
3.6. SX and PZ Phytoconstituents–Targets–Pathways Network Construction
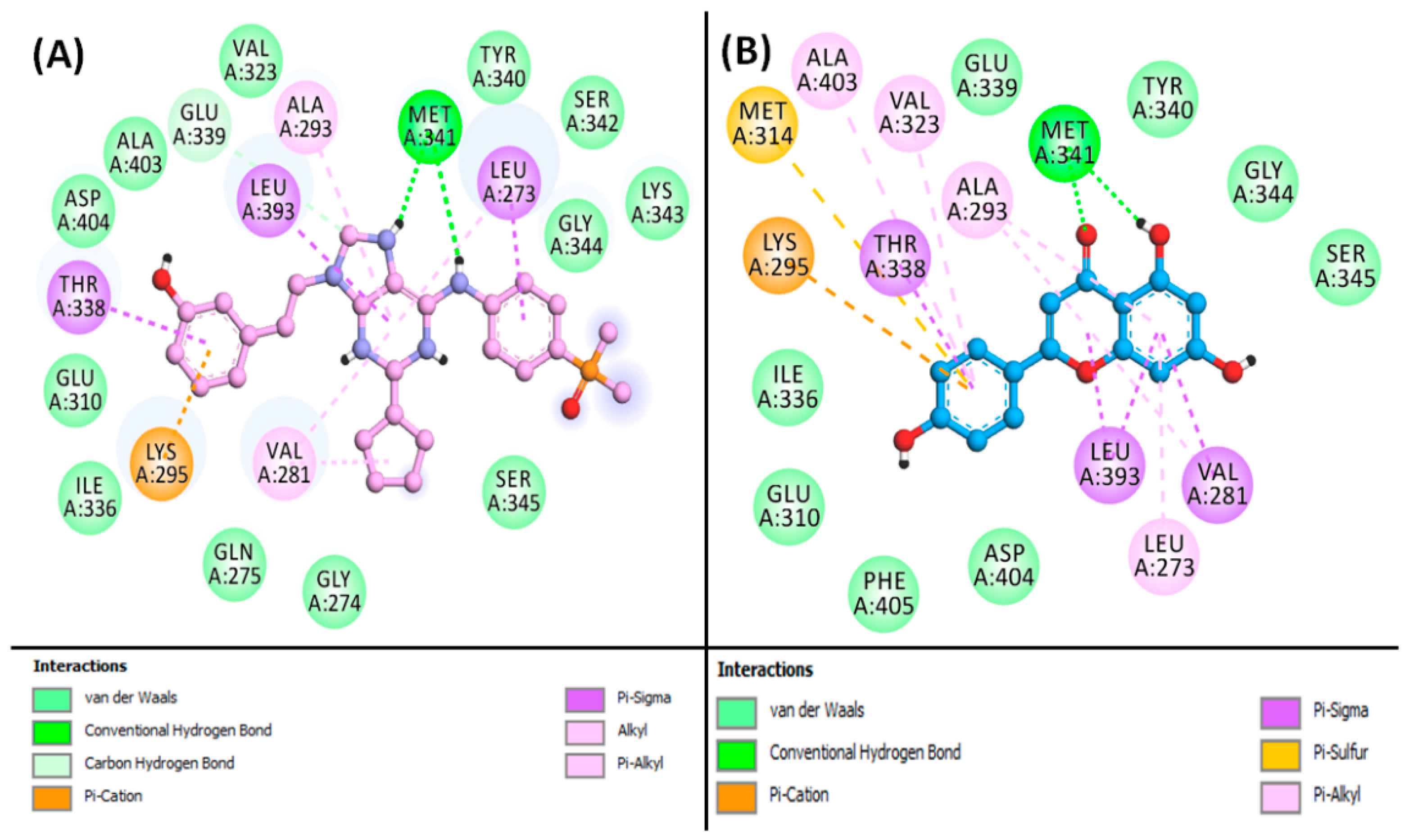
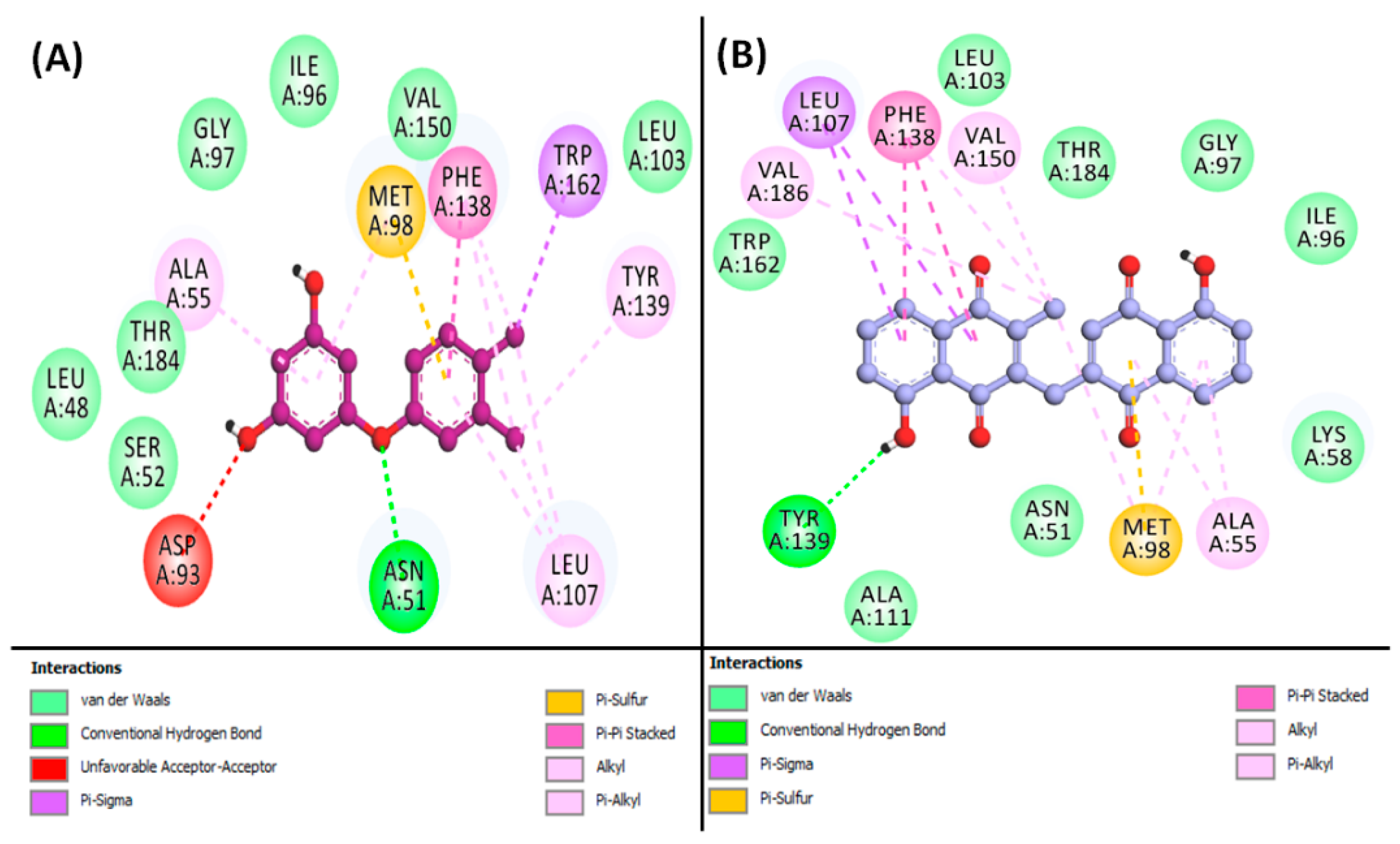
3.7. Molecular Docking Simulation of Phytoconstituents and Targets
3.8. Analysis of MD Simulation
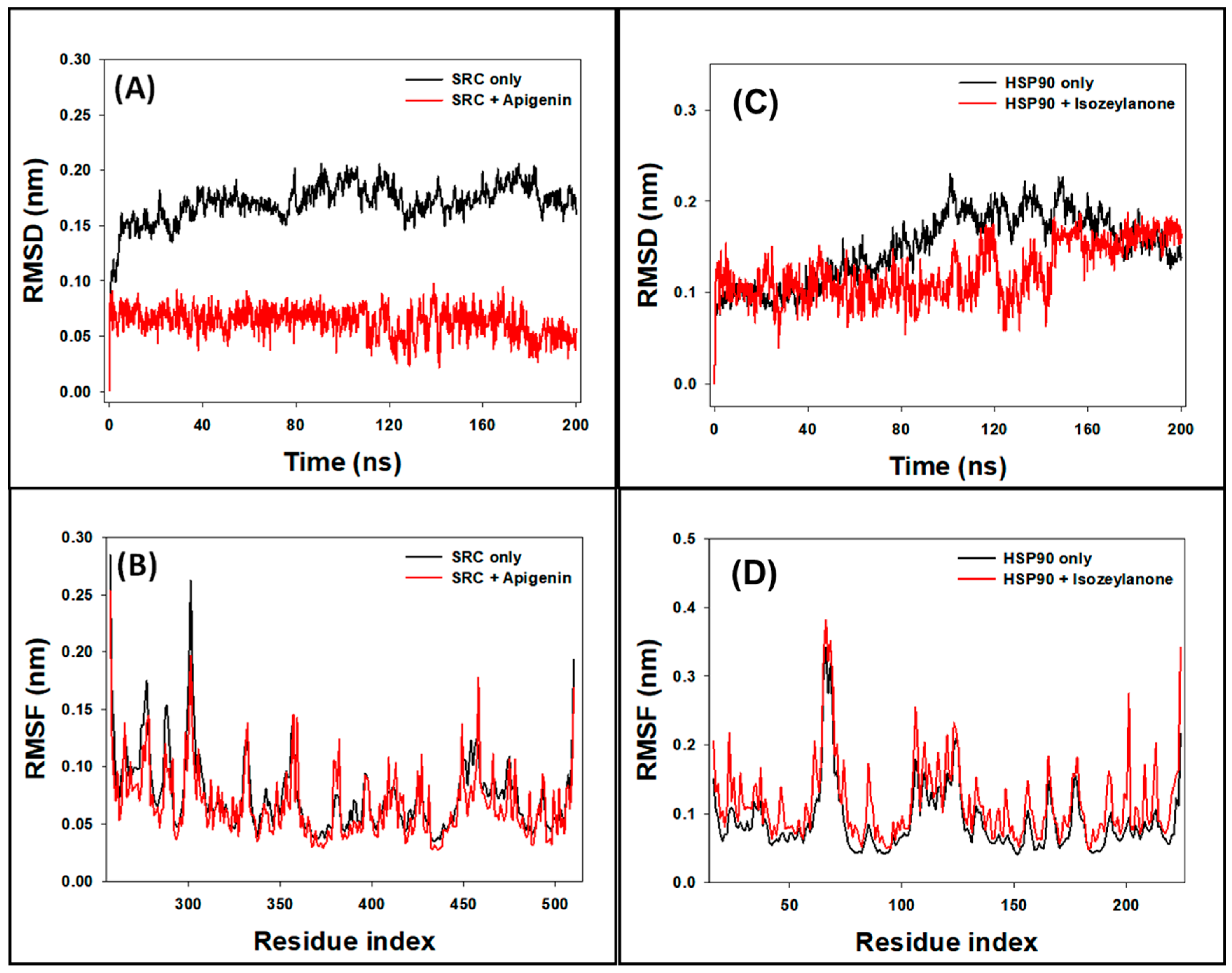
3.8.1. RMSD Analysis
3.8.2. RMSF Analysis
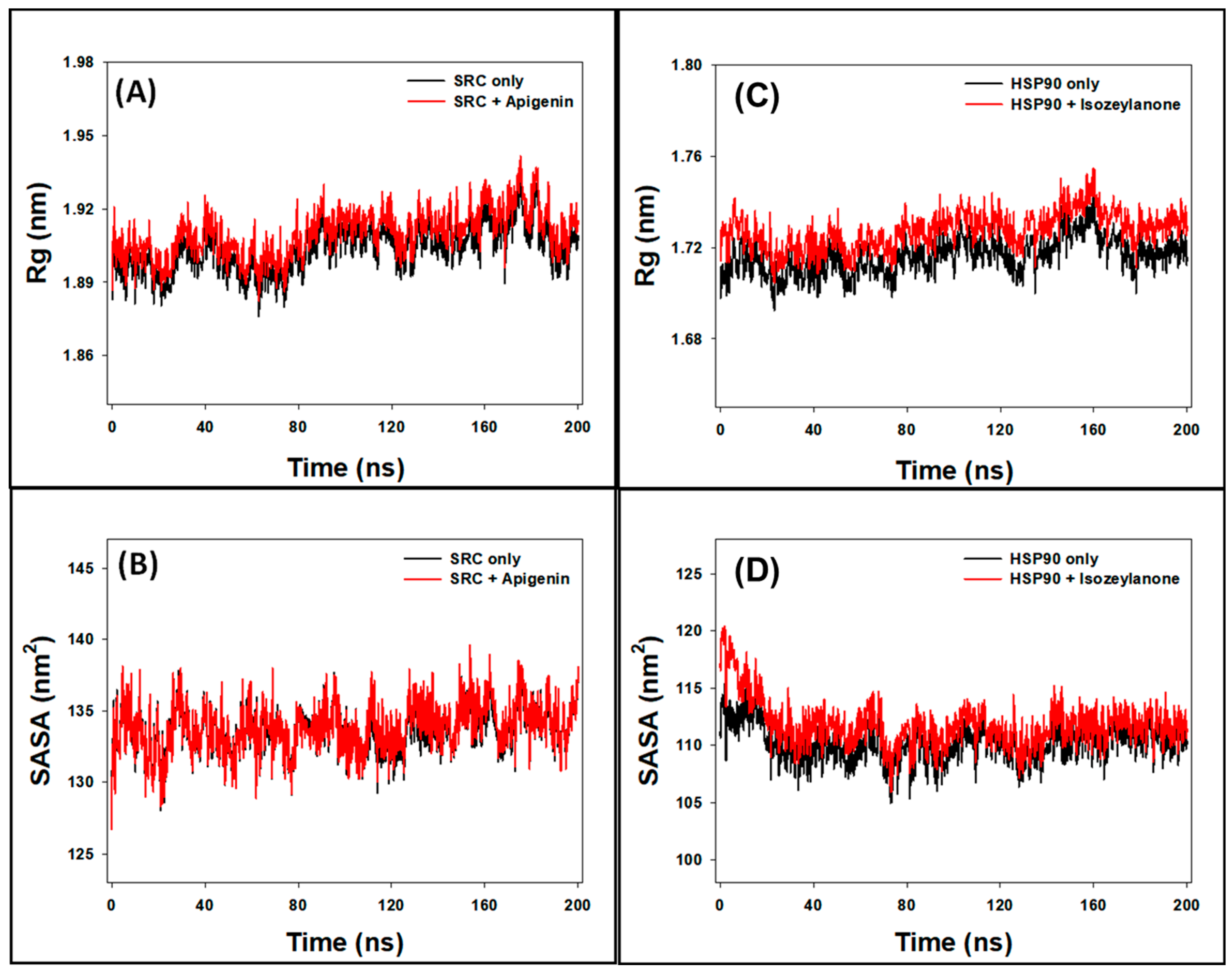
3.8.3. Rg Analysis
3.8.4. SASA
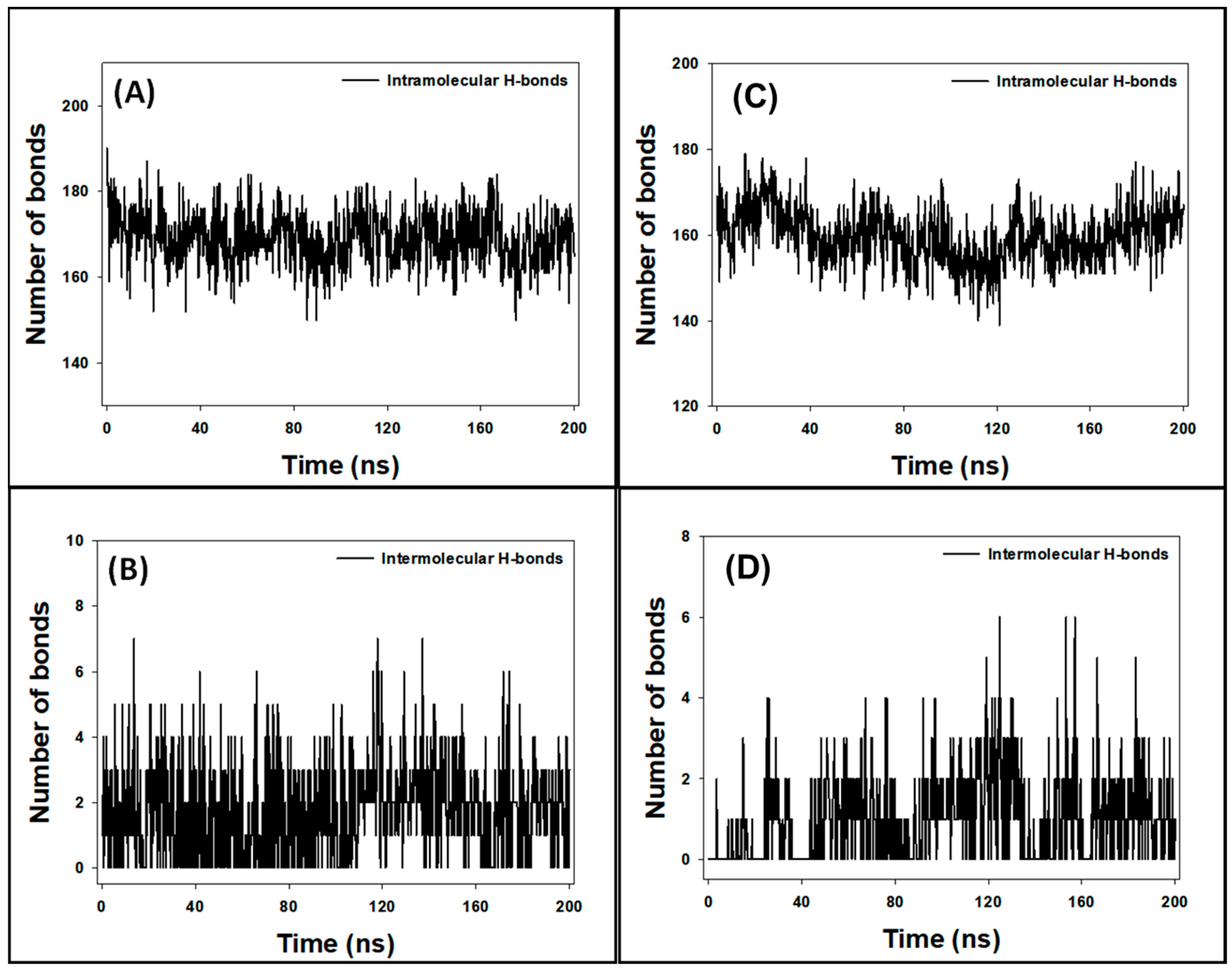
3.9. Analysis of Hydrogen Bonds
3.10. Analysis of Free Energy Calculations (MM/GBSA)
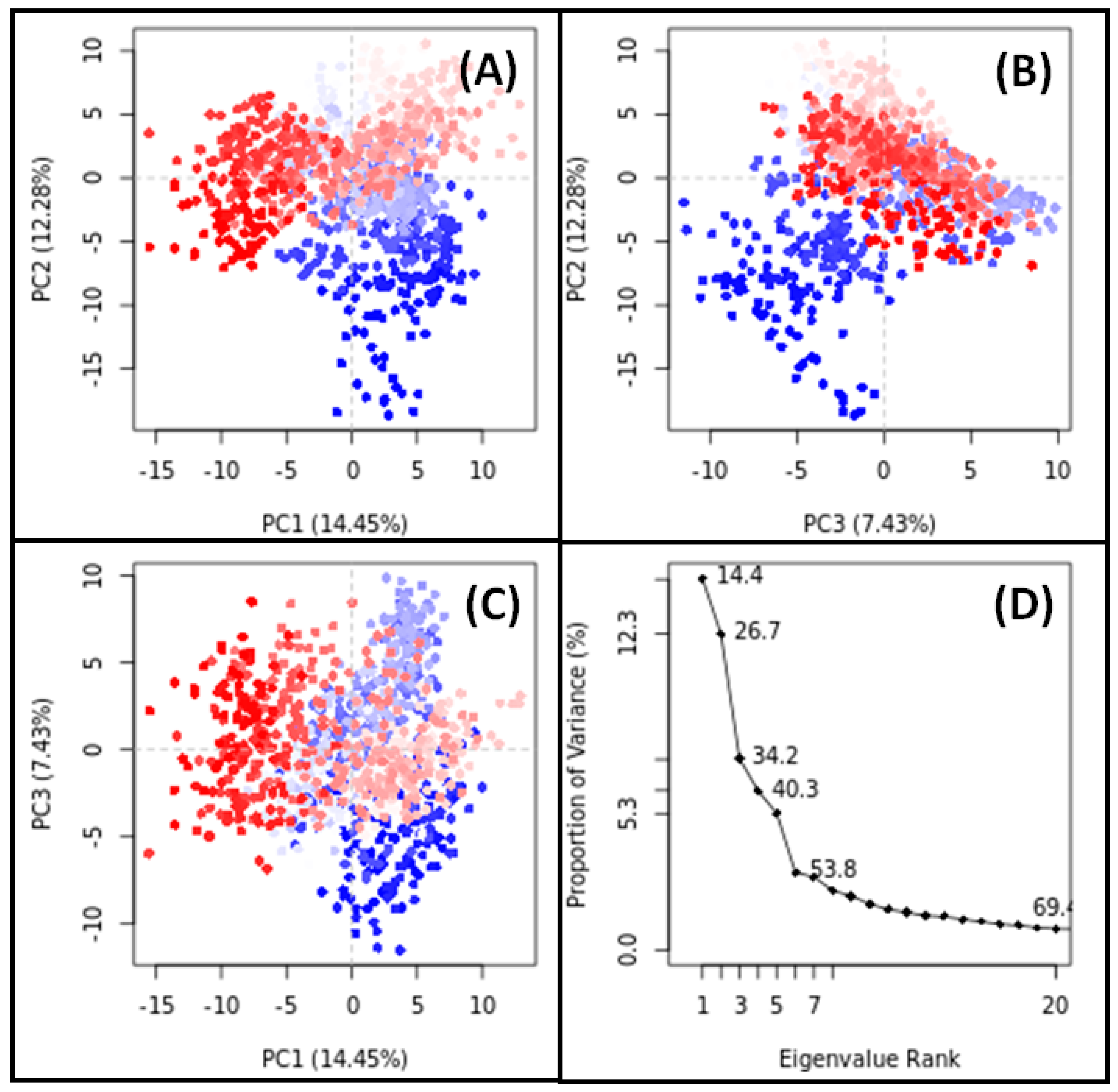
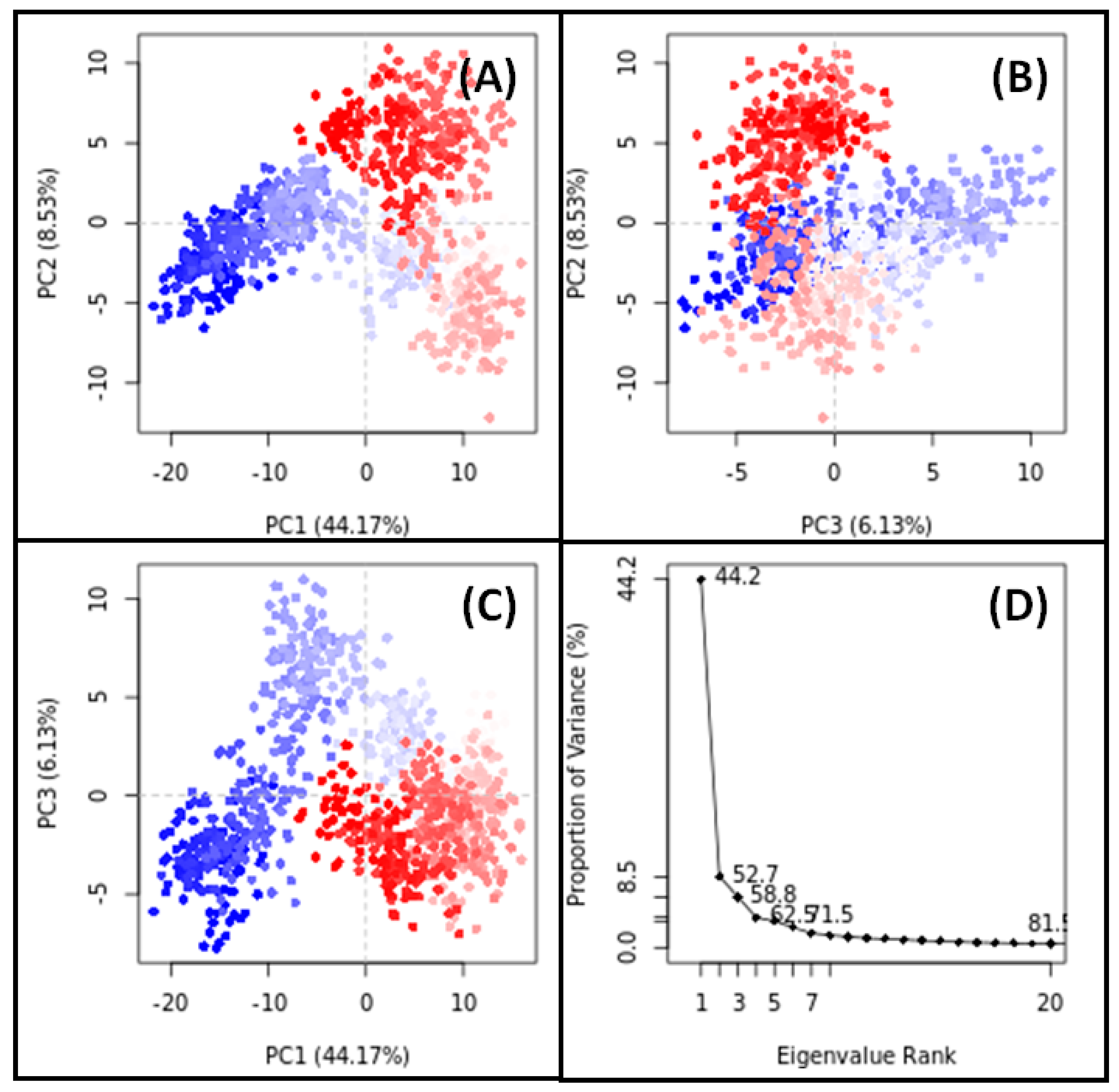
3.11. PCA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kropp, M.; Golubnitschaja, O.; Mazurakova, A.; Koklesova, L.; Sargheini, N.; Vo, T.T.K.S.; de Clerck, E.; Polivka, J.; Potuznik, P.; Polivka, J.; et al. Diabetic Retinopathy as the Leading Cause of Blindness and Early Predictor of Cascading Complications—Risks and Mitigation. EPMA J. 2023, 14, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Teo, Z.L.; Tham, Y.C.; Yu, M.; Chee, M.L.; Rim, T.H.; Cheung, N.; Bikbov, M.M.; Wang, Y.X.; Tang, Y.; Lu, Y.; et al. Global Prevalence of Diabetic Retinopathy and Projection of Burden through 2045: Systematic Review and Meta-Analysis. Ophthalmology 2021, 128, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Song, Z.; Li, G.; Zhang, C. Antivascular Endothelial Growth Factor for Macular Oedema Secondary to Retinal Vein Occlusion: A Systematic Review and Meta-Analysis. BMJ Open Ophthalmol. 2022, 7, e001086. [Google Scholar] [CrossRef]
- Tan, L.; Wang, Z.; Okoth, K.; Toulis, K.A.; Denniston, A.K.; Singh, B.M.; Crowe, F.L.; Sainsbury, C.; Wang, J.; Nirantharakumar, K. Associations of Antidiabetic Drugs with Diabetic Retinopathy in People with Type 2 Diabetes: An Umbrella Review and Meta-Analysis. Front. Endocrinol. 2023, 14, 1303238. [Google Scholar] [CrossRef] [PubMed]
- Kakade, P.S.; Zimare, S.B. Phytochemistry and Pharmacological Studies of Plumbago Indica L.: A Medicinal Plant. Phytochem. Pharmacol. Med. Plants 2-Vol. Set 2023, 1–2, 213–225. [Google Scholar]
- Parmar, K.M.; Itankar, P.R.; Joshi, A.; Prasad, S.K. Anti-Psoriatic Potential of Solanum Xanthocarpum Stem in Imiquimod-Induced Psoriatic Mice Model. J. Ethnopharmacol. 2017, 198, 158–166. [Google Scholar] [CrossRef]
- Tilak, J.C.; Adhikari, S.; Devasagayam, T.P.A. Antioxidant Properties of Plumbago Zeylanica, an Indian Medicinal Plant and Its Active Ingredient, Plumbagin. Redox Rep. 2004, 9, 219–227. [Google Scholar] [CrossRef]
- Lai, L.; Liu, J.; Zhai, D.; Lin, Q.; He, L.; Dong, Y.; Zhang, J.; Lu, B.; Chen, Y.; Yi, Z.; et al. Plumbagin Inhibits Tumour Angiogenesis and Tumour Growth through the Ras Signalling Pathway Following Activation of the VEGF Receptor-2. Br. J. Pharmacol. 2012, 165, 1084–1096. [Google Scholar] [CrossRef]
- Allemailem, K.S.; Almatroudi, A.; Alharbi, H.O.A.; AlSuhaymi, N.; Alsugoor, M.H.; Aldakheel, F.M.; Khan, A.A.; Rahmani, A.H. Apigenin: A Bioflavonoid with a Promising Role in Disease Prevention and Treatment. Biomedicines 2024, 12, 1353. [Google Scholar] [CrossRef]
- Poongothai, K.; Ponmurugan, P.; Ahmed, K.S.Z.; Kumar, B.S.; Sheriff, S.A. Antihyperglycemic and Antioxidant Effects of Solanum Xanthocarpum Leaves (Field Grown & in Vitro Raised) Extracts on Alloxan Induced Diabetic Rats. Asian Pac. J. Trop. Med. 2011, 4, 778–785. [Google Scholar] [CrossRef]
- Martiz, R.M.; Patil, S.M.; Abdulaziz, M.; Babalghith, A.; Al-Areefi, M.; Al-Ghorbani, M.; Kumar, J.M.; Prasad, A.; Nagalingaswamy, N.P.M.; Ramu, R. Defining the Role of Isoeugenol from Ocimum Tenuiflorum against Diabetes Mellitus-Linked Alzheimer’s Disease through Network Pharmacology and Computational Methods. Molecules 2022, 27, 2398. [Google Scholar] [CrossRef] [PubMed]
- Alamri, M.A. Bioinformatics and Network Pharmacology-Based Study to Elucidate the Multi-Target Pharmacological Mechanism of the Indigenous Plants of Medina Valley in Treating HCV-Related Hepatocellular Carcinoma. Saudi Pharm. J. 2023, 31, 1125–1138. [Google Scholar] [CrossRef] [PubMed]
- Yi, P.; Zhang, Z.; Huang, S.; Huang, J.; Peng, W.; Yang, J. Integrated Meta-Analysis, Network Pharmacology, and Molecular Docking to Investigate the Efficacy and Potential Pharmacological Mechanism of Kai-Xin-San on Alzheimer’s Disease. Pharm. Biol. 2020, 58, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Tao, Y.; Xu, R.; Luo, W.; Lin, T.; Zhou, F.; Tang, L.; He, L.; He, Y. Analysis of Active Components and Molecular Mechanism of Action of Rubia cordifolia L. in the Treatment of Nasopharyngeal Carcinoma Based on Network Pharmacology and Experimental Verification. Heliyon 2023, 9, e17078. [Google Scholar] [CrossRef]
- Khairy, A.; Ghareeb, D.A.; Celik, I.; Hammoda, H.M.; Zaatout, H.H.; Ibrahim, R.S. Forecasting of Potential Anti-Inflammatory Targets of Some Immunomodulatory Plants and Their Constituents Using in Vitro, Molecular Docking and Network Pharmacology-Based Analysis. Sci. Rep. 2023, 13, 9539. [Google Scholar] [CrossRef]
- Di Silvestre, D.; Vigani, G.; Mauri, P.; Hammadi, S.; Morandini, P.; Murgia, I. Network Topological Analysis for the Identification of Novel Hubs in Plant Nutrition. Front. Plant Sci. 2021, 12, 629013. [Google Scholar] [CrossRef]
- Simó, R.; Simó-Servat, O.; Bogdanov, P.; Hernández, C. Neurovascular Unit: A New Target for Treating Early Stages of Diabetic Retinopathy. Pharmaceutics 2021, 13, 1320. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, Y.; Xu, C.; Li, G.; Song, Y.; Qiu, J.; Cui, L.; Song, X.; Yang, Y.; Sun, Y. Potential Molecular Mechanisms of Erlongjiaonang Action in Idiopathic Sudden Hearing Loss: A Network Pharmacology and Molecular Docking Analyses. Front. Neurol. 2023, 14, 1121738. [Google Scholar] [CrossRef]
- Sahu, N.; Madan, S.; Walia, R.; Tyagi, R.; Fantoukh, O.I.; Hawwal, M.F.; Akhtar, A.; Almarabi, I.; Alam, P.; Saxena, S. Multi-Target Mechanism of Solanum Xanthocarpum for Treatment of Psoriasis Based on Network Pharmacology and Molecular Docking. Saudi Pharm. J. 2023, 31, 101788. [Google Scholar] [CrossRef]
- Anqi, L.; Shijun, S. Molecular Docking, Network Pharmacology and Experimental Verification to Explore the Mechanism of Wulongzhiyangwan in the Treatment of Pruritus. Sci. Rep. 2023, 13, 361. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Kan, Z.; Yan, W.; Wang, N.; Fang, Y.; Gao, H.; Song, Y. Identification of CircRNA–MiRNA–MRNA Regulatory Network and Crucial Signaling Pathway Axis Involved in Tetralogy of Fallot. Front. Genet. 2022, 13, 917454. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Wei, W.; Wen, J.; Cao, Y.; Li, H. The Efficacy and Mechanism of Berberine in Improving Aging-Related Cognitive Dysfunction: A Study Based on Network Pharmacology. Front. Neurosci. 2023, 17, 1093180. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.X.; Zhao, Q.; Zhang, Y.; Xue, R.; Li, S.; Li, Y.; Yu, J.J.; Li, J.C.; Zhang, Y.Z. Network Pharmacology and Pharmacological Evaluation for Deciphering Novel Indication of Sishen Wan in Insomnia Treatment. Phytomedicine 2023, 108, 154500. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Duan, M.Y.; Zhong, Y.S.; Li, X.D.; Du, S.X.; Xie, P.; Zheng, G.Z.; Han, J.M. Investigating Age-induced Differentially Expressed Genes and Potential Molecular Mechanisms in Osteosarcoma Based on Integrated Bioinformatics Analysis. Mol. Med. Rep. 2019, 19, 2729–2739. [Google Scholar] [CrossRef]
- Hall, D.C.; Ji, H.F. A Search for Medications to Treat COVID-19 via in Silico Molecular Docking Models of the SARS-CoV-2 Spike Glycoprotein and 3CL Protease. Travel Med. Infect. Dis. 2020, 35, 101646. [Google Scholar] [CrossRef]
- Mercurio, I.; D’Abrosca, G.; della Valle, M.; Malgieri, G.; Fattorusso, R.; Isernia, C.; Russo, L.; Di Gaetano, S.; Pedone, E.M.; Pirone, L.; et al. Molecular Interactions between a Diphenyl Scaffold and PED/PEA15: Implications for Type II Diabetes Therapeutics Targeting PED/PEA15-Phospholipase D1 Interaction. Comput. Struct. Biotechnol. J. 2024, 23, 2001–2010. [Google Scholar] [CrossRef]
- Terefe, E.M.; Ghosh, A. Molecular Docking, Validation, Dynamics Simulations, and Pharmacokinetic Prediction of Phytochemicals Isolated from Croton Dichogamus against the HIV-1 Reverse Transcriptase. Bioinform. Biol. Insights 2022, 16, 11779322221125605. [Google Scholar] [CrossRef]
- Mooers, B.H.M. Shortcuts for Faster Image Creation in PyMOL. Protein Sci. 2020, 29, 268–276. [Google Scholar] [CrossRef]
- Rampogu, S.; Shaik, M.R.; Khan, M.; Khan, M.; Oh, T.H.; Shaik, B. CBPDdb: A Curated Database of Compounds Derived from Coumarin-Benzothiazole-Pyrazole. Database J. Biol. Databases Curation 2023, 2023, baad062. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Jairajpuri, D.S.; Hussain, A.; Nasreen, K.; Mohammad, T.; Anjum, F.; Tabish Rehman, M.; Mustafa Hasan, G.; Alajmi, M.F.; Imtaiyaz Hassan, M. Identification of Natural Compounds as Potent Inhibitors of SARS-CoV-2 Main Protease Using Combined Docking and Molecular Dynamics Simulations. Saudi J. Biol. Sci. 2021, 28, 2423–2431. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Price, D.J.; Brooks, C.L. A Modified TIP3P Water Potential for Simulation with Ewald Summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Applequist, J.; Carl, J.R.; Fung, K.K. An Atom Dipole Interaction Model for Molecular Polarizability. Application to Polyatomic Molecules and Determination of Atom Polarizabilities. J. Am. Chem. Soc. 1972, 94, 2952–2960. [Google Scholar] [CrossRef]
- Sangster, M.J.L.; Atwood, R.M. Interionic Potentials for Alkali Halides. II. Completely Crystal Independent Specification of Born-Mayer Potentials. J. Phys. C Solid State Phys. 1978, 11, 1541–1555. [Google Scholar] [CrossRef]
- Tong, J.; Peng, B.; Kontogeorgis, G.M.; Liang, X. Behavior of the Aqueous Sodium Chloride Solutions from Molecular Simulations and Theories. J. Mol. Liq. 2023, 371, 121086. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hockney, R.W.; Goel, S.P.; Eastwood, J.W. Quiet High-Resolution Computer Models of a Plasma. J. Comput. Phys. 1974, 14, 148–158. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Iqbal, D.; Rehman, M.T.; Bin Dukhyil, A.; Rizvi, S.M.D.; Al Ajmi, M.F.; Alshehri, B.M.; Banawas, S.; Khan, M.S.; Alturaiki, W.; Alsaweed, M. High-Throughput Screening and Molecular Dynamics Simulation of Natural Product-like Compounds against Alzheimer’s Disease through Multitarget Approach. Pharmaceuticals 2021, 14, 937. [Google Scholar] [CrossRef] [PubMed]
- Ichiye, T.; Karplus, M. Collective Motions in Proteins: A Covariance Analysis of Atomic Fluctuations in Molecular Dynamics and Normal Mode Simulations. Proteins Struct. Funct. Bioinform. 1991, 11, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Leng, L.; Wang, C.; Yang, Q.; Hu, Y. Analyzing the Molecular Mechanism of Scutellaria Radix in the Treatment of Sepsis Using RNA Sequencing. BMC Infect. Dis. 2024, 24, 695. [Google Scholar] [CrossRef] [PubMed]
- Shamsi, A.; Mohammad, T.; Khan, M.S.; Shahwan, M.; Husain, F.M.; Rehman, M.T.; Hassan, M.I.; Ahmad, F.; Islam, A. Unraveling Binding Mechanism of Alzheimer’s Drug Rivastigmine Tartrate with Human Transferrin: Molecular Docking and Multi-Spectroscopic Approach towards Neurodegenerative Diseases. Biomolecules 2019, 9, 495. [Google Scholar] [CrossRef]
- Kiani, S.; Kahrizi, D.; Varmira, K.; Kassaee, S.M. Molecular and Biochemical Evaluation of Ethyl Methanesulfonate-Induced Mutant Lines in Camelina sativa L. Iran. J. Biotechnol. 2022, 20, e2948. [Google Scholar] [CrossRef]
- Choudhary, S.; Kesavan, A.K.; Juneja, V.; Thakur, S. Molecular Modeling, Simulation and Docking of Rv1250 Protein from Mycobacterium Tuberculosis. Front. Bioinform. 2023, 3, 1125479. [Google Scholar] [CrossRef]
- Belapure, J.; Sorokina, M.; Kastritis, P.L. IRAA: A Statistical Tool for Investigating a Protein–Protein Interaction Interface from Multiple Structures. Protein Sci. 2023, 32, 1–17. [Google Scholar] [CrossRef]
- Li, N.; Guo, X.L.; Xu, M.; Chen, J.L.; Wang, Y.F.; Xiao, Y.G.; Gao, A.S.; Zhang, L.C.; Liu, X.Z.; Wang, T.H. Network Pharmacology Mechanism of Scutellarin to Inhibit RGC Pyroptosis in Diabetic Retinopathy. Sci. Rep. 2023, 13, 6504. [Google Scholar] [CrossRef]
- Nakagawa, T.; Kosugi, T.; Haneda, M.; Rivard, C.J.; Long, D.A. Abnormal Angiogenesis in Diabetic Nephropathy. Diabetes 2009, 58, 1471–1478. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Sun, X.; Fan, C.; Li, R.; Zhou, S.; Yu, H. The Pathophysiological Mechanisms Underlying Diabetic Retinopathy. Front. Cell Dev. Biol. 2022, 10, 963615. [Google Scholar] [CrossRef] [PubMed]
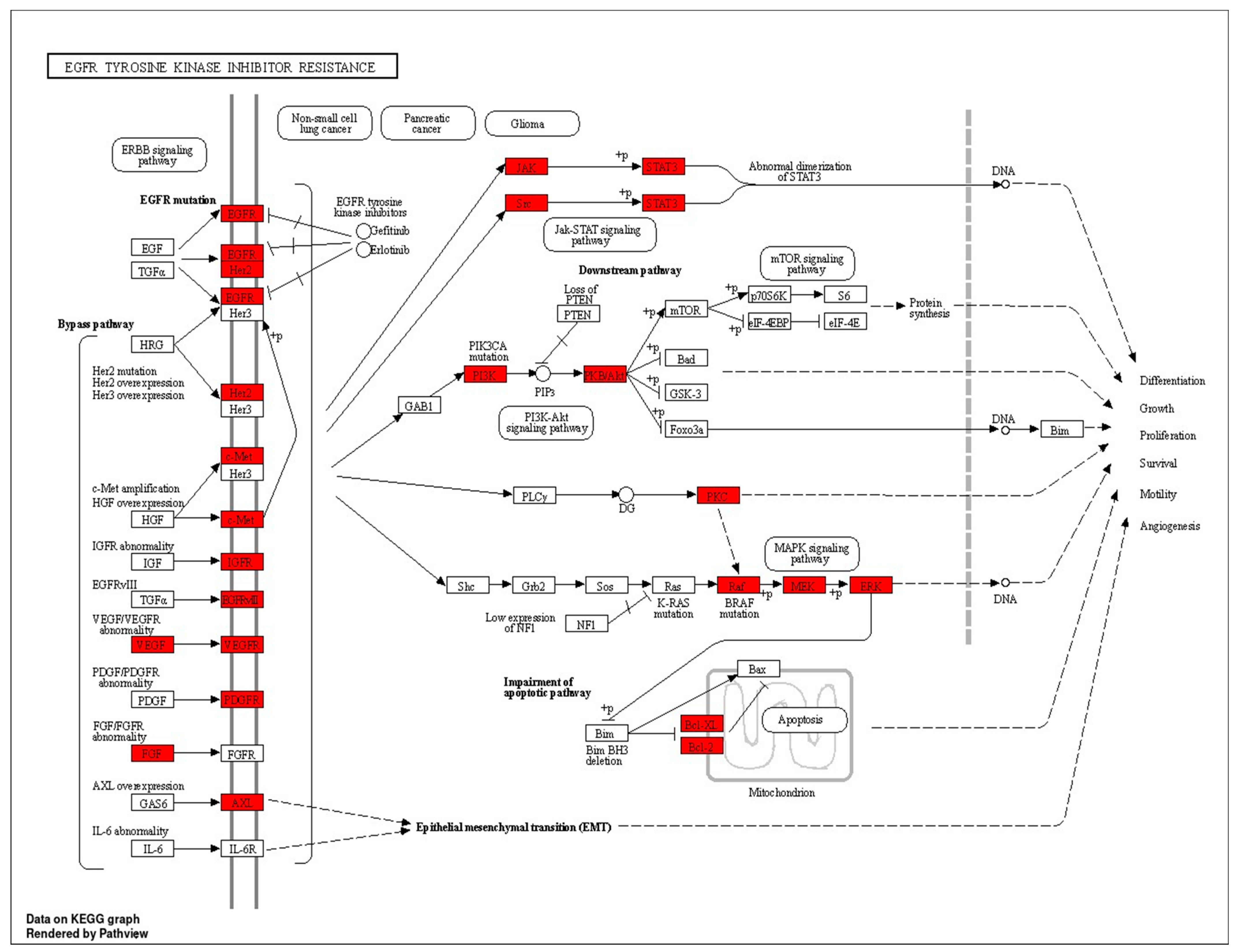
- Huang, L.; Fu, L. Mechanisms of Resistance to EGFR Tyrosine Kinase Inhibitors. Acta Pharm. Sin. B 2015, 5, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.L.; Chen, H.H.; Zheng, L.L.; Sun, L.P.; Shi, L. Angiogenic Signaling Pathways and Anti-Angiogenic Therapy for Cancer. Signal Transduct. Target. Ther. 2023, 8, 198. [Google Scholar] [CrossRef]
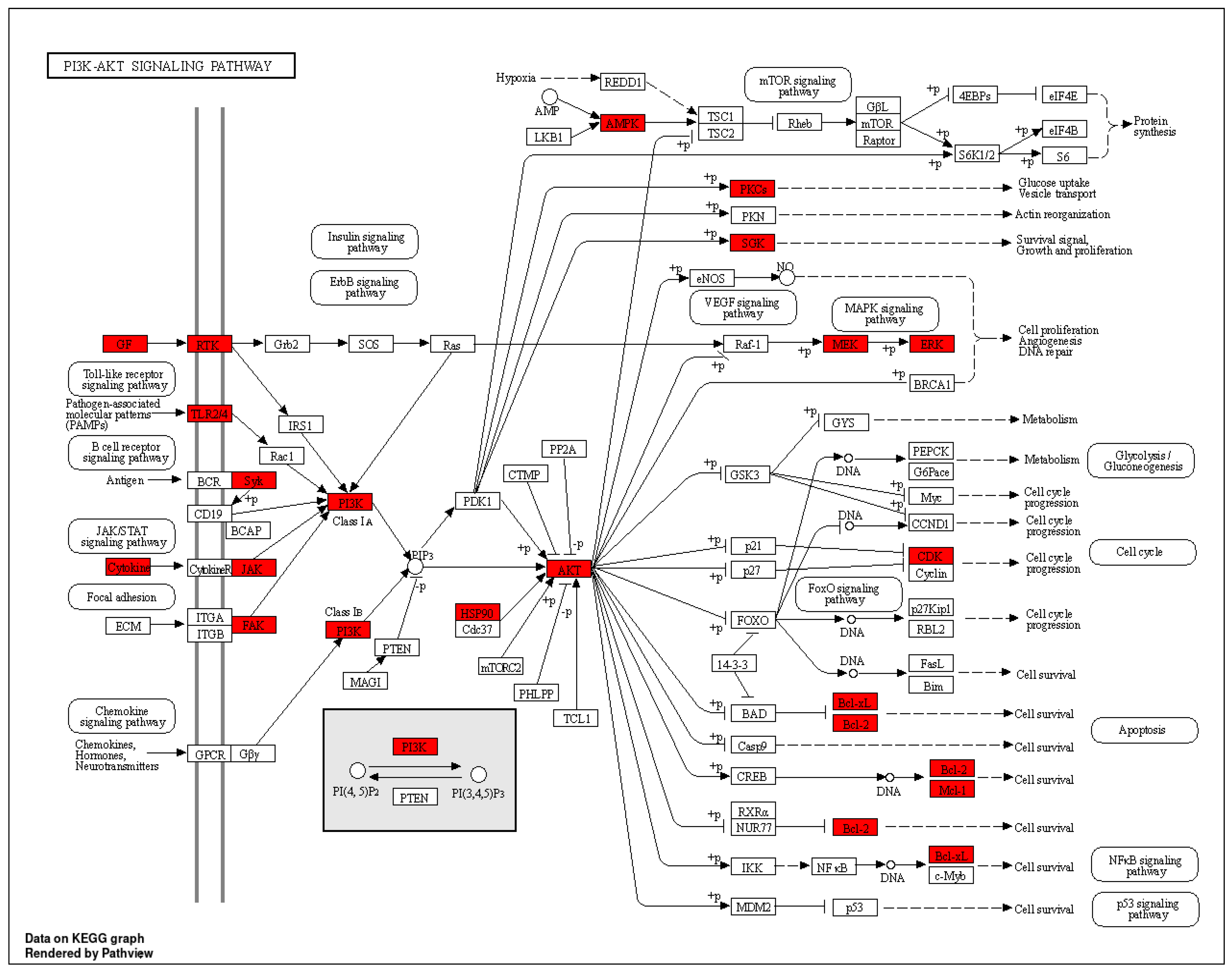
- Jacot, J.L.; Sherris, D. Potential Therapeutic Roles for Inhibition of the PI3K/Akt/MTOR Pathway in the Pathophysiology of Diabetic Retinopathy. J. Ophthalmol. 2011, 2011, 589813. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Imppat ID | Compound | DL | BS | Molecular Weight (kcal/mol) | Number Heavy Atoms | HBA | HBD | TPSA (Ų) | GIA | Log P |
| IMPHY003411 | Scopolin | 0.51 | 0.55 | 354.31 | 25 | 9 | 4 | 138.82 | Low | 1.34 |
| IMPHY003656 | Soladulcamaridine | 0.53 | 0.55 | 413.64 | 30 | 3 | 2 | 41.49 | High | 4.15 |
| IMPHY003952 | Cycloartanol | 0.47 | 0.55 | 428.73 | 31 | 1 | 1 | 20.23 | Low | 5.26 |
| IMPHY004033 | Solasodine | 0.53 | 0.55 | 413.64 | 30 | 3 | 2 | 41.49 | High | 4.26 |
| IMPHY004661 | Apigenin | 0.63 | 0.55 | 270.24 | 20 | 5 | 3 | 90.90 | High | 1.89 |
| IMPHY005620 | Esculin | 0.43 | 0.55 | 340.28 | 24 | 9 | 5 | 149.82 | Low | 1.33 |
| IMPHY006300 | Cholesterol | 0.49 | 0.55 | 386.65 | 28 | 1 | 1 | 20.23 | Low | 4.89 |
| IMPHY011518 | Esculetin | 0.47 | 0.55 | 178.14 | 13 | 4 | 2 | 70.67 | High | 1.25 |
| IMPHY011541 | Scopoletin | 0.7 | 0.55 | 192.17 | 14 | 4 | 1 | 59.67 | High | 1.86 |
| IMPHY011642 | Cycloartenol | 0.45 | 0.55 | 426.72 | 31 | 1 | 1 | 20.23 | Low | 5.16 |
| IMPHY011933 | Caffeic acid | 0.47 | 0.56 | 180.16 | 13 | 4 | 3 | 77.76 | High | 0.97 |
| IMPHY012402 | Campesterol | 0.47 | 0.55 | 400.68 | 29 | 1 | 1 | 20.23 | Low | 4.97 |
| IMPHY014836 | beta-Sitosterol | 0.44 | 0.55 | 414.71 | 30 | 1 | 1 | 20.23 | Low | 5.05 |
| IMPHY014842 | Stigmasterol | 0.46 | 0.55 | 412.69 | 30 | 1 | 1 | 20.23 | Low | 5.08 |
| IMPHY015071 | Sitosteryl glucoside | 0.26 | 0.55 | 576.85 | 41 | 6 | 4 | 99.38 | Low | 5.17 |
| IMPHY015079 | Stigmasteryl glucoside | 0.28 | 0.55 | 574.83 | 41 | 6 | 4 | 99.38 | High | 5.24 |
| IMPHY000398 | Isozeylanone | 0.84 | 0.55 | 374.34 | 28 | 6 | 2 | 108.74 | High | 2.02 |
| IMPHY000467 | Plumbazeylanone | 0.37 | 0.55 | 576.55 | 43 | 9 | 3 | 163.11 | Low | 2.74 |
| IMPHY001191 | Plumbagin | 0.67 | 0.55 | 188.18 | 14 | 3 | 1 | 54.37 | High | 1.79 |
| IMPHY002828 | Elliptinone | 0.79 | 0.55 | 374.34 | 28 | 6 | 2 | 108.74 | High | 2.58 |
| IMPHY003551 | 3-chloroplumbagin | 0.73 | 0.55 | 222.62 | 15 | 3 | 1 | 54.37 | High | 1.89 |
| IMPHY004515 | Zeylanone | 0.73 | 0.55 | 374.34 | 28 | 6 | 2 | 108.74 | High | 2.13 |
| IMPHY004866 | Droserone | 0.63 | 0.85 | 204.18 | 15 | 4 | 2 | 74.60 | High | 1.35 |
| IMPHY007957 | Chitranone | 0.79 | 0.55 | 374.34 | 28 | 6 | 2 | 108.74 | High | 2.58 |
| IMPHY008637 | Maritinone | 0.79 | 0.55 | 374.34 | 28 | 6 | 2 | 108.74 | High | 2.45 |
| IMPHY013935 | Methylnaphthazarin | 0.63 | 0.55 | 204.18 | 15 | 4 | 2 | 74.60 | High | 1.82 |
| IMPHY014893 | D-Glucose | 0.29 | 0.55 | 180.16 | 12 | 6 | 5 | 110.3 | Low | 0.35 |
| IMPHY014916 | D-Fructose | 0.29 | 0.55 | 180.16 | 12 | 6 | 5 | 110.38 | Low | 0.61 |
| ID | Pathway Name | p-Value | GeneIDs | Count |
| hsa01521 | EGFR tyrosine kinase inhibitor resistance | 5.3115 × 10−23 | PRKCG/PRKCA/PRKCB/PIK3CA/VEGFA/FGF2/BRAF/BCL2/BCL2L1/EGFR/IGF1R/KDR/MET/AXL/PIK3R1/SRC/AKT1/ERBB2/MAPK1/PIK3CB/STAT3/PDGFRB/AKT2/AKT3/MAP2K1/JAK2 | 26 |
| hsa04066 | HIF-1 signaling pathway | 2.17615 × 10−21 | EP300/EGLN1/PRKCG/PRKCA/PRKCB/PIK3CA/VEGFA/FLT1/NOS2/BCL2/PDK1/PFKFB3/EGFR/IGF1R/PIK3R1/AKT1/TLR4/ERBB2/MAPK1/PIK3CB/STAT3/INSR/TEK/GAPDH/HK1/AKT2/AKT3/MAP2K1 | 28 |
| hsa05205 | Proteoglycans in cancer | 2.29832 × 10−17 | PRKCG/PRKCA/PRKCB/PIK3CA/VEGFA/FGF2/HPSE/TNF/BRAF/CTNNB1/ESR1/MMP9/MMP2/EGFR/IGF1R/KDR/MET/PIK3R1/SRC/PTK2/AKT1/PTPN6/SHH/TLR4/ERBB2/MAPK1/PIK3CB/STAT3/MAPK14/AKT2/AKT3/MAP2K1 | 32 |
| hsa04151 | PI3K-Akt signalling pathway | 3.08738 × 10−17 | PRKCA/PIK3CA/PRKAA1/HSP90AA1/VEGFA/FGF1/FGF2/HSP90AB1/MCL1/FLT1/SGK1/CDK2/CDK4/HSP90B1/PIK3CG/BCL2/BCL2L1/CDK6/SYK/EGFR/IGF1R/KDR/MET/PIK3R1/PTK2/AKT1/TLR4/ERBB2/MAPK1/NGFR/PIK3CB/PDGFRB/FLT4/INSR/TEK/IL2/AKT2/AKT3/MAP2K1/JAK2/CSF1R | 41 |
| hsa04015 | Rap1 signaling pathway | 4.77529 × 10−17 | PRKCG/PRKCA/PRKCB/PIK3CA/VEGFA/FGF1/FGF2/FLT1/BRAF/CTNNB1/ADORA2A/EGFR/IGF1R/KDR/MET/PIK3R1/SRC/AKT1/DRD2/MAPK1/NGFR/PIK3CB/CNR1/PDGFRB/FLT4/INSR/TEK/MAPK14/AKT2/AKT3/MAP2K1/CSF1R | 32 |
| hsa05215 | Prostate cancer | 6.97404 × 10−17 | EP300/PIK3CA/HSP90AA1/HSP90AB1/MMP3/BRAF/CDK2/HSP90B1/BCL2/AR/CTNNB1/MMP9/EGFR/IGF1R/PIK3R1/AKT1/ERBB2/MAPK1/PIK3CB/PDGFRB/AKT2/AKT3/MAP2K1 | 23 |
| hsa04933 | AGE-RAGE signalling pathway in diabetic complications | 1.44583 × 10−16 | PRKCA/PRKCB/PIK3CA/NOX4/VEGFA/MAPK8/TNF/CDK4/BCL2/PRKCD/MMP2/PIK3R1/AKT1/MAPK1/PIK3CB/F3/STAT3/MAPK14/AKT2/AKT3/JAK2/TGFBR1/MAPK9 | 23 |
| hsa01522 | Endocrine resistance | 1.19061 × 10−15 | PIK3CA/MAPK8/BRAF/CDK4/BCL2/ESR1/MMP9/MMP2/EGFR/IGF1R/PIK3R1/SRC/PTK2/AKT1/ERBB2/MAPK1/PIK3CB/MAPK14/AKT2/AKT3/MAP2K1/MAPK9 | 22 |
| hsa05207 | Chemical carcinogenesis receptor activation | 4.00274 × 10−15 | PRKCG/PRKCA/PRKCB/PIK3CA/HSP90AA1/VEGFA/FGF2/HSP90AB1/CYP1B1/HSP90B1/BCL2/AR/CHRNA7/ESR1/AHR/EGFR/PIK3R1/SRC/AKT1/VDR/MAPK1/PIK3CB/CYP1A2/CYP3A4/STAT3/PPARA/AKT2/AKT3/MAP2K1/JAK2 | 30 |
| hsa05417 | Lipid and atherosclerosis | 5.91259 × 10−15 | PRKCA/PIK3CA/HSP90AA1/HSP90AB1/MMP1/MMP3/MAPK8/TNF/HSP90B1/BCL2/BCL2L1/MMP9/PIK3R1/SRC/PTK2/AKT1/TLR4/MAPK1/PIK3CB/CYP2C9/NFE2L2/STAT3/PPARG/HSPA8/HSPA5/MAPK14/AKT2/AKT3/JAK2/MAPK9 | 30 |
| Phytoconstituent Name (PubChem ID) | EGFR | STAT3 | SRC | AKT1 | HSP90AA1 |
| Scopolin (439514) | −6.588 (−0.264) | −4.505 (−0.180) | −8.420 (−0.337) | _ | −10.264 (−0.411) |
| Soladulcamaridine (91871142) | −3.365 ( −0.112) | −2.659 (−0.089) | −1.827 (−0.061) | _ | −2.865 (−0.095) |
| Cycloartanol (12760132) | −2.837 (−0.092) | _ | −1.455 (−0.047) | _ | −1.906 (−0.061) |
| Solasodine (442985) | −3.704 (−0.123) | −2.061 (−0.069) | −2.306 (−0.077) | _ | −2.568 (−0.086) |
| Apigenin (5280443) | −7.648 (−0.382) | −5.383 (−0.269) | −9.127 (−0.456) | −3.504 (−0.175) | −8.808 (−0.440) |
| Esculin (5281417) | −9.283 (−0.387) | −4.641 (−0.193) | −7.067 (−0.294) | −7.116 (−0.296) | −10.618 (−0.442) |
| Cholesterol (5997) | −3.574 (−0.128) | _ | −2.079 (−0.074) | _ | −4.643 (−0.166) |
| Esculetin (5281416) | −3.826 (−0.294) | −4.124 (−0.317) | −5.571 (−0.429) | −3.086 (−0.237) | −8.845 (−0.680) |
| Scopoletin (5280460) | −6.210 (−0.444) | −2.788 (−0.199) | −5.792 (−0.414) | −3.891 (−0.278) | −8.317 (−0.594) |
| Cycloartenol (92110) | −2.219 (−0.072) | −1.309 (−0.042) | −1.586 (−0.051) | _ | −4.014 (−0.129) |
| Caffeic acid (689043) | −4.200 (−0.323) | −3.903 (−0.300) | −7.016 (−0.540) | −4.569 (−0.351) | −7.172 (−0.552) |
| Campesterol (173183) | −4.021 (−0.139) | −2.198 (−0.076) | −2.656 (−0.092) | _ | −6.608 (−0.228) |
| Beta-Sitosterol (222284) | −3.850 (−0.128) | _ | −2.872 (−0.096) | _ | −6.621 (−0.221) |
| Stigmasterol (5280794) | −3.873 (−0.129) | _ | −2.950 (−0.098) | _ | −6.595 (−0.220) |
| Sitosteryl glucoside (70699351) | −5.998 (−0.146) | −4.970 (−0.121) | −0.067 (−0.002) | _ | −3.174 (−0.077) |
| Stigmasteryl glucoside (70699355) | −5.316 ( −0.130) | _ | _ | _ | _ |
| Isozeylanone (100947536) | −5.085 ( −0.182) | −3.472 (−0.124) | −5.396 (−0.193) | _ | −10.126 (−0.362) |
| Plumbazeylanone (100947539) | −2.564 (−0.060) | _ | −3.568 (−0.083) | _ | −2.814 (−0.065) |
| Plumbagin (10205) | −6.645 (−0.475) | −3.552 (−0.254) | −6.604 (−0.472) | −3.336 (−0.238) | −7.421 (−0.530) |
| Elliptinone (146680) | −5.731 (−0.205) | −3.283 (−0.117) | −6.358 (−0.227) | _ | −8.926 (−0.319) |
| 3-chloroplumbagin (338719) | −6.096 (−0.406) | −3.132 (−0.209) | −6.390 (−0.426) | _ | −7.556 (−0.504) |
| Zeylanone (5276618) | −5.072 (−0.181) | −4.041 (−0.144) | −3.791 (−0.135) | _ | −8.794 (−0.314) |
| Droserone (442739) | −6.278 (−0.419) | −3.252 (−0.217) | −4.828 (−0.322) | −2.235 (−0.149) | −6.633 (−0.442) |
| Chitranone (633072) | −5.521 (−0.197) | −1.999 (−0.071) | −8.361 (−0.299) | _ | −8.287 (−0.296) |
| Maritinone (633024) | −5.921 (−0.211) | −3.702 (−0.1329) | −3.562 (−0.127) | _ | −9.810 (−0.350) |
| Methylnaphthazarin (271296) | −6.589 (−0.439) | −4.281 (−0.285) | −7.300 (−0.487) | −3.210 (−0.214) | −7.406 (−0.494) |
| D-Glucose (5793) | −6.010 (−0.501) | −5.141 (−0.451) | −7.354 (−0.613) | −4.873 (−0.406) | −5.195 (−0.433) |
| D-Fructose (2723872) | −5.005 (−0.417) | −5.641 (−0.470) | −7.712 (−0.643) | −5.094 (−0.424) | −5.589 (−0.466) |
| System | ΔG or ΔGBind | ΔGCoulomb | ΔGCovalent | ΔGH-bond | ΔGSA or ΔGSol_Lipo | ΔGSolv or ΔGSolGB | ΔGPacking | ΔGvdW |
|---|---|---|---|---|---|---|---|---|
| SRC-Apigenin | −45.58 | −18.22 | 6.91 | −2.11 | −15.70 | 19.58 | −0.49 | −35.55 |
| HSP90AA1 Isozeylanone | −49.85 | −31.73 | 2.89 | −3.03 | −8.09 | 30.21 | −1.71 | −38.39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahu, N.; Tyagi, R.; Kumar, N.; Mujeeb, M.; Akhtar, A.; Alam, P.; Madan, S. Forecasting the Pharmacological Mechanisms of Plumbago zeylanica and Solanum xanthocarpum in Diabetic Retinopathy Treatment: A Network Pharmacology, Molecular Docking, and Molecular Dynamics Simulation Study. Biology 2024, 13, 732. https://doi.org/10.3390/biology13090732
Sahu N, Tyagi R, Kumar N, Mujeeb M, Akhtar A, Alam P, Madan S. Forecasting the Pharmacological Mechanisms of Plumbago zeylanica and Solanum xanthocarpum in Diabetic Retinopathy Treatment: A Network Pharmacology, Molecular Docking, and Molecular Dynamics Simulation Study. Biology. 2024; 13(9):732. https://doi.org/10.3390/biology13090732
Chicago/Turabian StyleSahu, Nilanchala, Rama Tyagi, Neeraj Kumar, Mohd. Mujeeb, Ali Akhtar, Perwez Alam, and Swati Madan. 2024. "Forecasting the Pharmacological Mechanisms of Plumbago zeylanica and Solanum xanthocarpum in Diabetic Retinopathy Treatment: A Network Pharmacology, Molecular Docking, and Molecular Dynamics Simulation Study" Biology 13, no. 9: 732. https://doi.org/10.3390/biology13090732
APA StyleSahu, N., Tyagi, R., Kumar, N., Mujeeb, M., Akhtar, A., Alam, P., & Madan, S. (2024). Forecasting the Pharmacological Mechanisms of Plumbago zeylanica and Solanum xanthocarpum in Diabetic Retinopathy Treatment: A Network Pharmacology, Molecular Docking, and Molecular Dynamics Simulation Study. Biology, 13(9), 732. https://doi.org/10.3390/biology13090732