
Research Progress on the Mechanism, Monitoring, and Prevention of Cardiac Injury Caused by Antineoplastic Drugs—Anthracyclines

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Pathological Mechanisms of Cardiac Injury Due to Anthracyclines
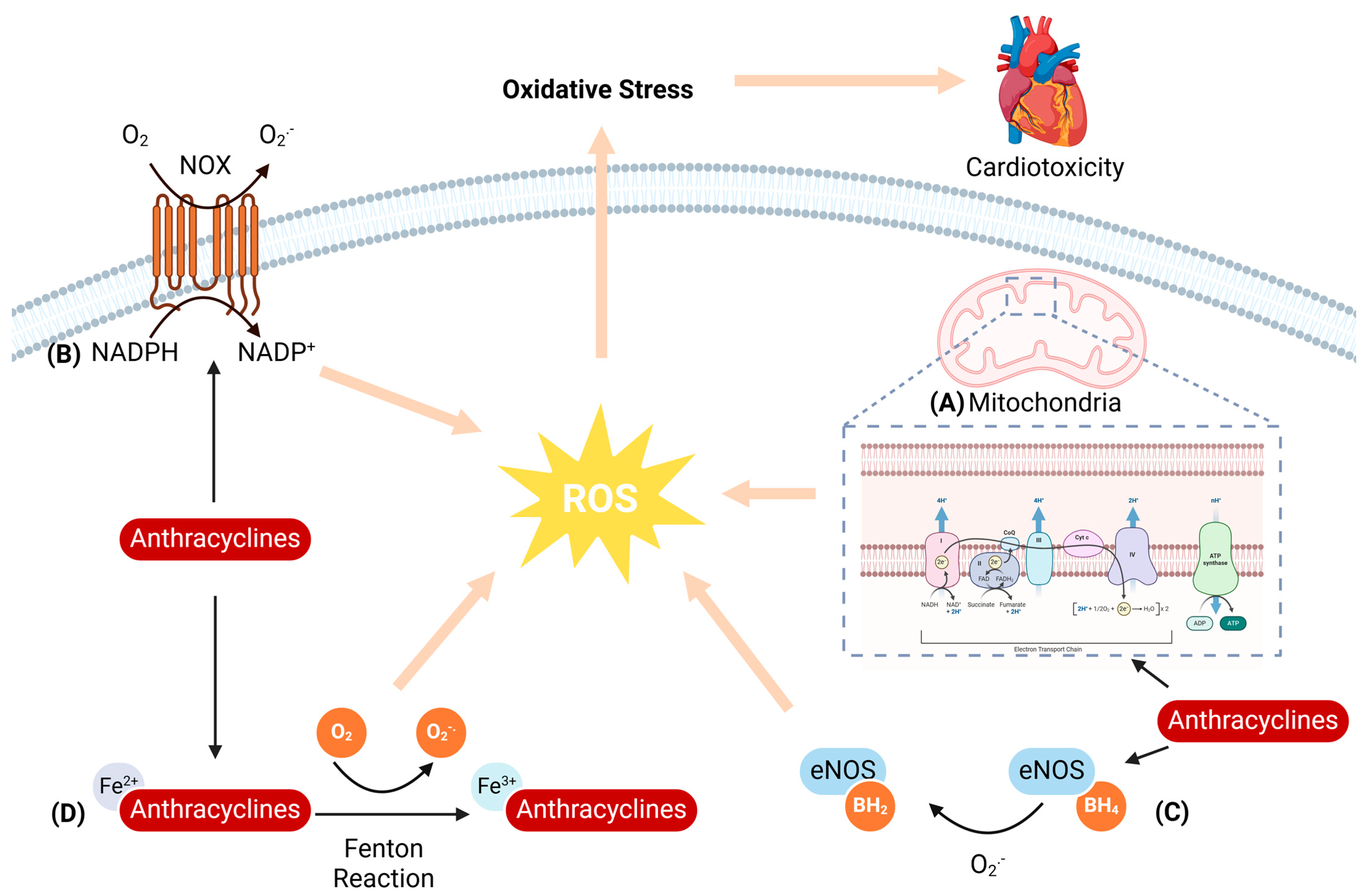
2.1. Oxidative Stress
2.1.1. Mitochondria-Mediated ROS Generation
2.1.2. NADPH Oxidase-Mediated ROS Generation
2.1.3. NOS-Mediated ROS Generation
2.1.4. Iron-Mediated ROS Generation
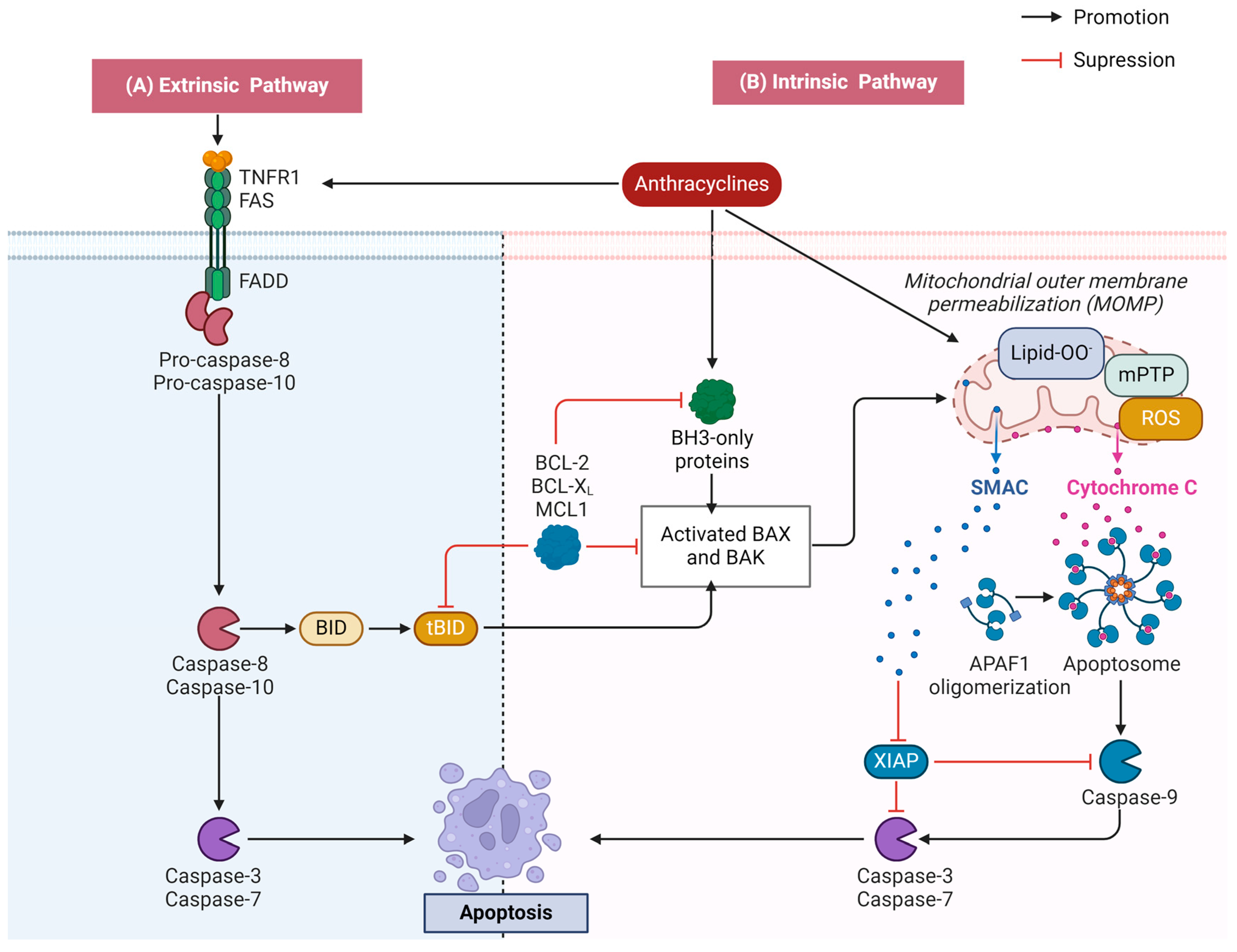
2.2. Apoptosis
2.2.1. Activation of the Mitochondrial Apoptotic Pathway
2.2.2. Activation of the Death Receptor Apoptotic Pathway
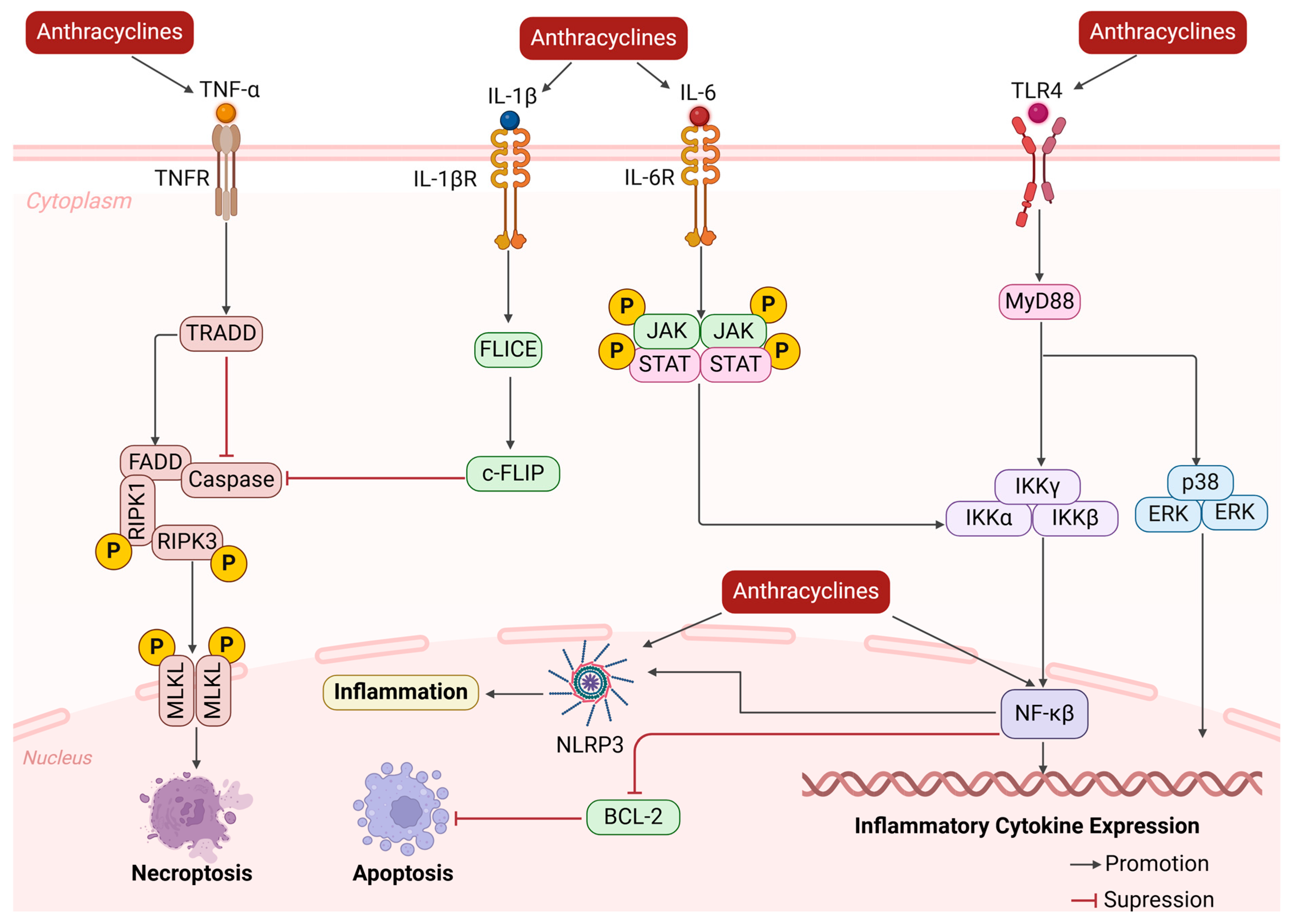
2.3. Change in Cardiac Microenvironment
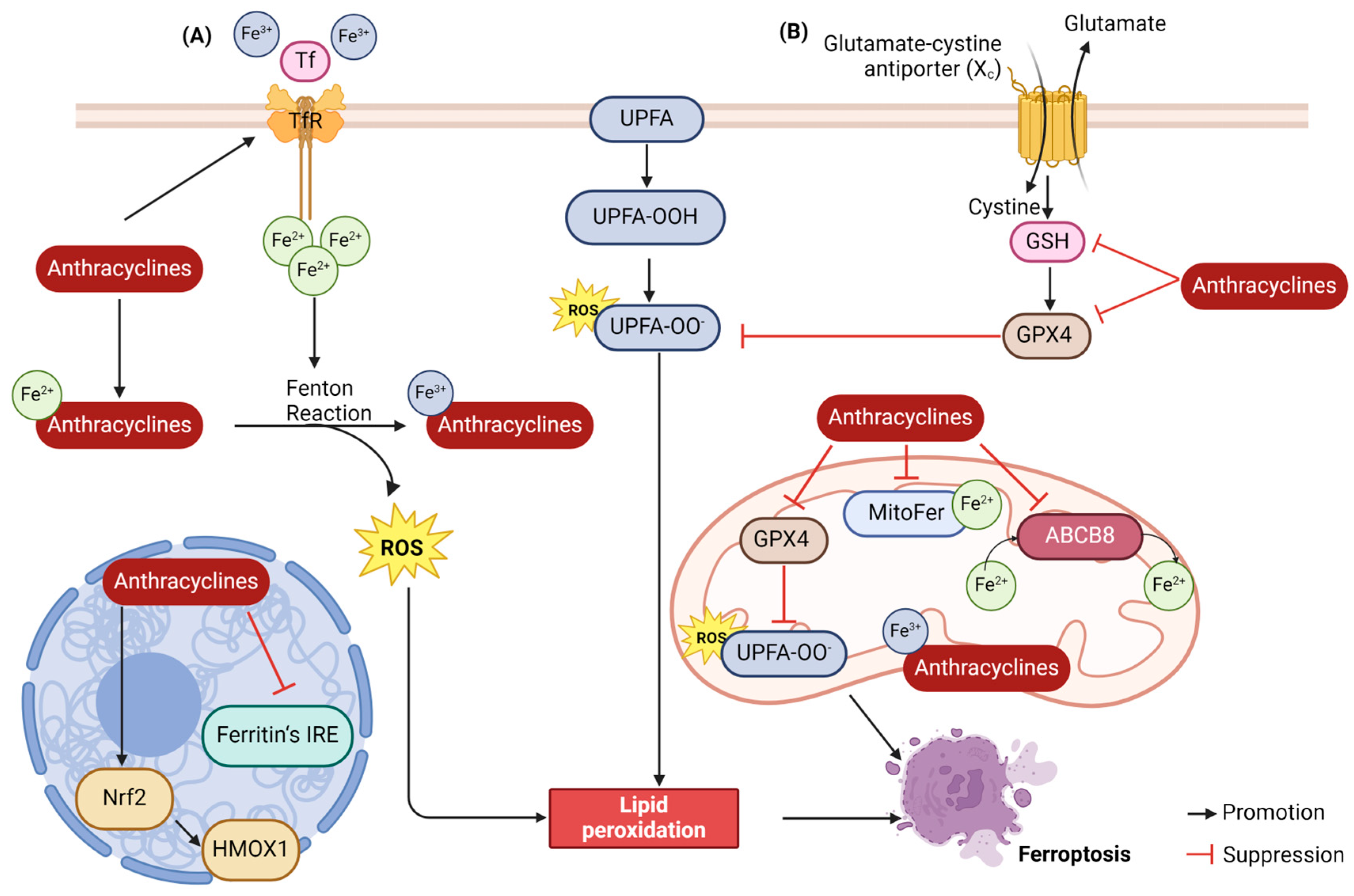
2.4. Ferroptosis
2.4.1. Lipid Peroxidation
2.4.2. Iron Accumulation
2.5. Autophagy
2.6. Other
3. Methods of Monitoring AIC
3.1. Electrocardiogram (ECG)
3.2. Echocardiography
3.3. Biomarkers
3.4. Cardiac Magnetic Resonance Imaging (CMRI)
3.5. Radionuclide Ventriculography
3.6. Other Monitoring Tools
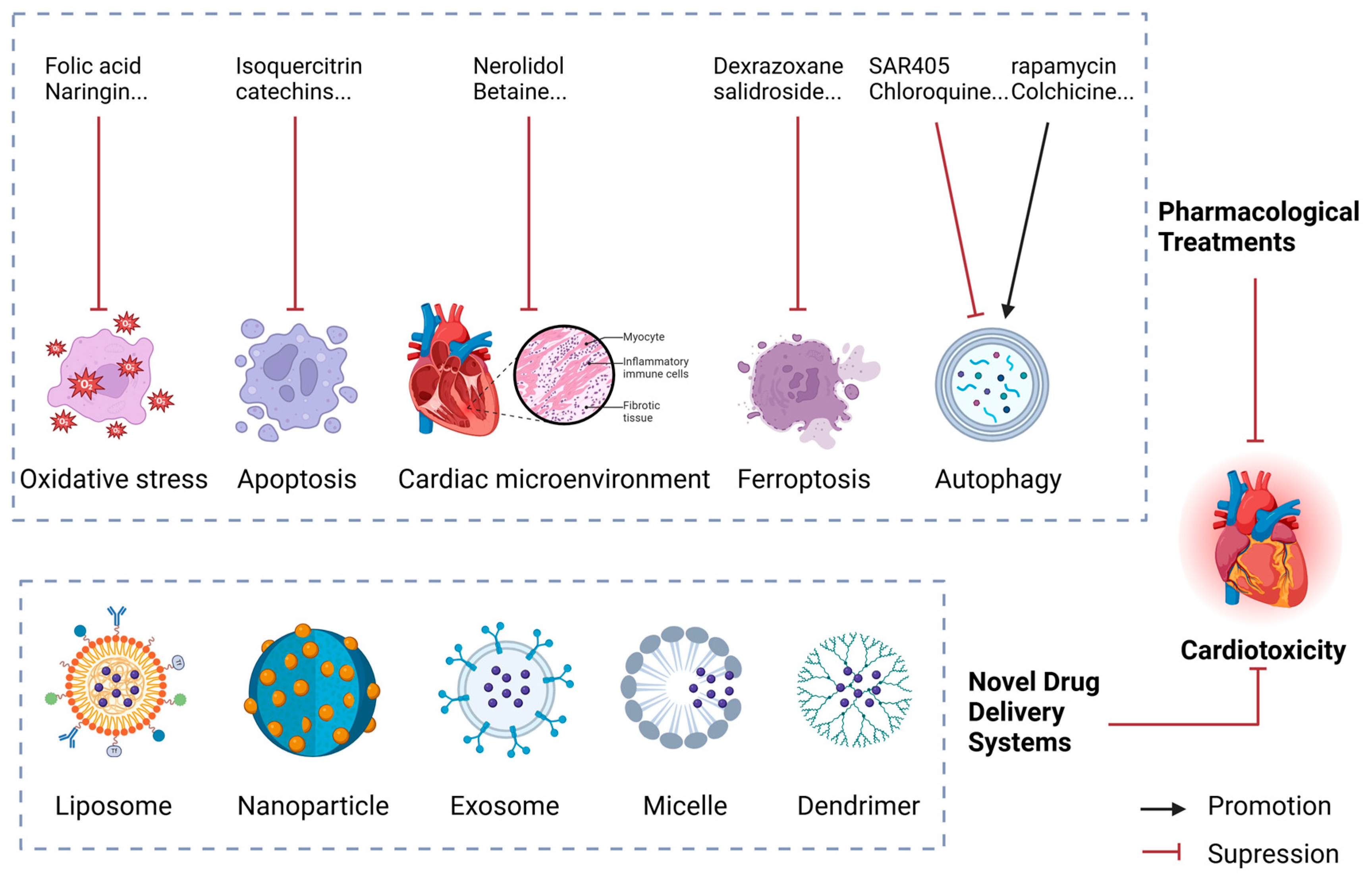
4. Advances in the Treatment of Anthracycline-Induced Cardiotoxicity
4.1. Pharmacological Treatments
4.1.1. Inhibiting Oxidative Stress
4.1.2. Inhibiting Apoptosis
4.1.3. Regulating the Cardiac Microenvironment
4.1.4. Inhibiting Ferroptosis
4.1.5. Modulating Autophagy
4.2. Novel Drug Delivery Systems
4.3. Non-Pharmacological Interventions
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANT | Anthracyclines |
| AIC | Anthracycline-induced cardiotoxicity |
| ROS | Reactive oxygen species |
| ETC | Electron transport chain |
| O2.− | Superoxide anion |
| OH− | Hydroxyl radical |
| Cyt c | Cytochrome C |
| mtDNA | Mitochondrial DNA |
| NOX | NADPH oxidase |
| H2O2 | Hydrogen peroxide |
| Dox | Doxorubicin |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| eNOS | Endothelial NOS |
| iNOS | Inducible NOS |
| nNOS | Neuronal NOS |
| Apaf-1 | Apoptotic protease activator 1 |
| TNFR | Tumor necrosis factor receptor |
| DISC | Death-inducing signaling complex |
| NFAT4 | Nuclear factor-activated T cell-4 |
| ECM | Extracellular matrix |
| System Xc− | Cystine/glutamate reverse transporter |
| GSH | Glutathione |
| GPX4 | Glutathione peroxidase 4 |
| Top II | Topoisomerase II |
| ECG | Electrocardiogram |
| LVEF | Left ventricular ejection fraction |
| STI | Speckle tracking imaging |
| GAS | Global area strain |
| GLS | Global longitudinal strain |
| GRS | Global radial strain |
| GCS | Global circumferential strain |
| CTn | Cardiac troponin |
| CK-MB | Creatine kinase isoenzymes |
| BNP | Brain natriuretic peptide |
| CMRI | Cardiac magnetic resonance imaging |
| RV | Radionuclide ventriculography |
| GWASs | Genome-wide association studies |
References
- Soerjomataram, I.; Bray, F. Planning for tomorrow: Global cancer incidence and the role of prevention 2020-2070. Nat. Rev. Clin. Oncol. 2021, 18, 663–672. [Google Scholar] [CrossRef]
- Kocarnik, J.M.; Compton, K.; Dean, F.E.; Fu, W.; Gaw, B.L.; Harvey, J.D.; Henrikson, H.J.; Lu, D.; Pennini, A.; Xu, R.; et al. Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life Years for 29 Cancer Groups From 2010 to 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. JAMA Oncol. 2022, 8, 420–444. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Nogueira, L.; Devasia, T.; Mariotto, A.B.; Yabroff, K.R.; Jemal, A.; Kramer, J.; Siegel, R.L. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin. 2022, 72, 409–436. [Google Scholar] [CrossRef]
- Sieswerda, E.; van Dalen, E.C.; Postma, A.; Cheuk, D.K.; Caron, H.N.; Kremer, L.C. Medical interventions for treating anthracycline-induced symptomatic and asymptomatic cardiotoxicity during and after treatment for childhood cancer. Cochrane Database Syst. Rev. 2011, Cd008011. [Google Scholar] [CrossRef]
- Curigliano, G.; Cardinale, D.; Dent, S.; Criscitiello, C.; Aseyev, O.; Lenihan, D.; Cipolla, C.M. Cardiotoxicity of anticancer treatments: Epidemiology, detection, and management. CA Cancer J. Clin. 2016, 66, 309–325. [Google Scholar] [CrossRef]
- Yu, H.; Qiu, Y.; Yu, H.; Wang, Z.; Xu, J.; Peng, Y.; Wan, X.; Wu, X.; Jin, R.; Zhou, F. Anthracycline Induced Cardiac Disorders in Childhood Acute Lymphoblastic Leukemia: A Single-Centre, Retrospective, Observational Study. Front. Pharmacol. 2021, 12, 598708. [Google Scholar] [CrossRef]
- Xie, S.; Sun, Y.; Zhao, X.; Xiao, Y.; Zhou, F.; Lin, L.; Wang, W.; Lin, B.; Wang, Z.; Fang, Z.; et al. An update of the molecular mechanisms underlying anthracycline induced cardiotoxicity. Front. Pharmacol. 2024, 15, 1406247. [Google Scholar] [CrossRef] [PubMed]
- Pantazi, D.; Tselepis, A.D. Cardiovascular toxic effects of antitumor agents: Pathogenetic mechanisms. Thromb. Res. 2022, 213 (Suppl. 1), S95–S102. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, S.; Yang, L.; Song, P.; Liu, Z.; Liu, X.; Yan, X.; Dong, Q. Roles of reactive oxygen species in inflammation and cancer. MedComm 2024, 5, e519. [Google Scholar] [CrossRef] [PubMed]
- Stěrba, M.; Popelová, O.; Vávrová, A.; Jirkovský, E.; Kovaříková, P.; Geršl, V.; Simůnek, T. Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid. Redox Signal. 2013, 18, 899–929. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wu, R.; Chen, L.; Yang, Z.; Yan, D.; Li, M. Understanding Anthracycline Cardiotoxicity From Mitochondrial Aspect. Front. Pharmacol. 2022, 13, 811406. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Korge, P.; Calmettes, G.; John, S.A.; Weiss, J.N. Reactive oxygen species production induced by pore opening in cardiac mitochondria: The role of complex III. J. Biol. Chem. 2017, 292, 9882–9895. [Google Scholar] [CrossRef]
- Goormaghtigh, E.; Huart, P.; Praet, M.; Brasseur, R.; Ruysschaert, J.M. Structure of the adriamycin-cardiolipin complex. Role in mitochondrial toxicity. Biophys. Chem. 1990, 35, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhao, H.P.; Yang, L.G.; Li, L.; Wang, A.L.; Zhang, X.J.; Wang, K.; Yang, B.; Zhu, Z.F.; Zhang, P.J.; et al. NADPH oxidase 2 mediates cardiac sympathetic denervation and myocyte autophagy, resulting in cardiac atrophy and dysfunction in doxorubicin-induced cardiomyopathy. Sci. Rep. 2024, 14, 6971. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; McLaughlin, D.; Robinson, E.; Harvey, A.P.; Hookham, M.B.; Shah, A.M.; McDermott, B.J.; Grieve, D.J. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with Doxorubicin chemotherapy. Cancer Res. 2010, 70, 9287–9297. [Google Scholar] [CrossRef]
- Fonoudi, H.; Jouni, M.; Cejas, R.B.; Magdy, T.; Blancard, M.; Ge, N.; Shah, D.A.; Lyra-Leite, D.M.; Neupane, A.; Gharib, M.; et al. Functional Validation of Doxorubicin-Induced Cardiotoxicity-Related Genes. JACC CardioOncology 2024, 6, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.M.; Ai, N.; Ke, M.; Tan, Y.; Huang, Z.; Li, Y.; Lu, J.H.; Ge, W.; Su, H. Roles of Nitric Oxide Synthase Isoforms in Neurogenesis. Mol. Neurobiol. 2018, 55, 2645–2652. [Google Scholar] [CrossRef]
- Kong, C.Y.; Guo, Z.; Song, P.; Zhang, X.; Yuan, Y.P.; Teng, T.; Yan, L.; Tang, Q.Z. Underlying the Mechanisms of Doxorubicin-Induced Acute Cardiotoxicity: Oxidative Stress and Cell Death. Int. J. Biol. Sci. 2022, 18, 760–770. [Google Scholar] [CrossRef]
- Neilan, T.G.; Blake, S.L.; Ichinose, F.; Raher, M.J.; Buys, E.S.; Jassal, D.S.; Furutani, E.; Perez-Sanz, T.M.; Graveline, A.; Janssens, S.P.; et al. Disruption of nitric oxide synthase 3 protects against the cardiac injury, dysfunction, and mortality induced by doxorubicin. Circulation 2007, 116, 506–514. [Google Scholar] [CrossRef]
- Zeglinski, M.; Premecz, S.; Lerner, J.; Wtorek, P.; Dasilva, M.; Hasanally, D.; Chaudhary, R.; Sharma, A.; Thliveris, J.; Ravandi, A.; et al. Congenital absence of nitric oxide synthase 3 potentiates cardiac dysfunction and reduces survival in doxorubicin- and trastuzumab-mediated cardiomyopathy. Can. J. Cardiol. 2014, 30, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Akolkar, G.; Bagchi, A.K.; Ayyappan, P.; Jassal, D.S.; Singal, P.K. Doxorubicin-induced nitrosative stress is mitigated by vitamin C via the modulation of nitric oxide synthases. Am. J. Physiol. Cell Physiol. 2017, 312, C418–C427. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Huang, S.; Liu, Y.; Liu, L.; Guo, F.; Guo, Y.; Li, D.; Cen, X.; Chen, Y.; Zhang, M.; et al. Idebenone alleviates doxorubicin-induced cardiotoxicity by stabilizing FSP1 to inhibit ferroptosis. Acta Pharm. Sin. B 2024, 14, 2581–2597. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; O’Connor, L.; Dixit, V.M. Apoptosis Signaling. Annu. Rev. Biochem. 2000, 69, 217–245. [Google Scholar] [CrossRef]
- Kitakata, H.; Endo, J.; Ikura, H.; Moriyama, H.; Shirakawa, K.; Katsumata, Y.; Sano, M. Therapeutic Targets for DOX-Induced Cardiomyopathy: Role of Apoptosis vs. Ferroptosis. Int. J. Mol. Sci. 2022, 23, 1414. [Google Scholar] [CrossRef]
- Castillo Ferrer, C.; Berthenet, K.; Ichim, G. Apoptosis—Fueling the oncogenic fire. FEBS J. 2021, 288, 4445–4463. [Google Scholar] [CrossRef]
- Zhang, P.; Lu, H.; Wu, Y.; Lu, D.; Li, C.; Yang, X.; Chen, Z.; Qian, J.; Ge, J. COX5A Alleviates Doxorubicin-Induced Cardiotoxicity by Suppressing Oxidative Stress, Mitochondrial Dysfunction and Cardiomyocyte Apoptosis. Int. J. Mol. Sci. 2023, 24, 10400. [Google Scholar] [CrossRef]
- Ergün, S.; Aslan, S.; Demir, D.; Kayaoğlu, S.; Saydam, M.; Keleş, Y.; Kolcuoğlu, D.; Taşkurt Hekim, N.; Güneş, S. Beyond Death: Unmasking the Intricacies of Apoptosis Escape. Mol. Diagn. Ther. 2024, 28, 403–423. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, C.; Kong, C.Y.; Song, P.; Wu, H.M.; Xu, S.C.; Yuan, Y.P.; Deng, W.; Ma, Z.G.; Tang, Q.Z. FNDC5 alleviates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via activating AKT. Cell Death Differ. 2020, 27, 540–555. [Google Scholar] [CrossRef]
- Li, G.M.; Chen, J.R.; Zhang, H.Q.; Sun, C.; Chen, G.R.; Xiong, Q.Y.; Cao, X.Y.; Yu, L.; Lin, Z.W.; Qin, J.Y.; et al. Rhein activated Fas-induced apoptosis pathway causing cardiotoxicity in vitro and in vivo. Toxicol. Lett. 2022, 363, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Tzahor, E.; Dimmeler, S. A coalition to heal-the impact of the cardiac microenvironment. Science 2022, 377, eabm4443. [Google Scholar] [CrossRef]
- Nagy, A.; Börzsei, D.; Hoffmann, A.; Török, S.; Veszelka, M.; Almási, N.; Varga, C.; Szabó, R. A Comprehensive Overview on Chemotherapy-Induced Cardiotoxicity: Insights into the Underlying Inflammatory and Oxidative Mechanisms. Cardiovasc. Drugs Ther. 2024. [Google Scholar] [CrossRef] [PubMed]
- Vitale, R.; Marzocco, S.; Popolo, A. Role of Oxidative Stress and Inflammation in Doxorubicin-Induced Cardiotoxicity: A Brief Account. Int. J. Mol. Sci. 2024, 25, 7477. [Google Scholar] [CrossRef]
- Sumneang, N.; Tanajak, P.; Oo, T.T. Toll-like Receptor 4 Inflammatory Perspective on Doxorubicin-Induced Cardiotoxicity. Molecules 2023, 28, 4294. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, S.; Yamada, Y.; Kawai, Y.; Osawa, T.; Furuhashi, K.; Duan, Z.; Ichihara, G. Roles of oxidative stress and Akt signaling in doxorubicin cardiotoxicity. Biochem. Biophys. Res. Commun. 2007, 359, 27–33. [Google Scholar] [CrossRef]
- Maayah, Z.; Takahara, S.; Dyck, J. The beneficial effects of reducing NLRP3 inflammasome activation in the cardiotoxicity and the anti-cancer effects of doxorubicin. Arch. Toxicol. 2021, 95, 1–9. [Google Scholar] [CrossRef]
- Zhang, Z.; Peng, J.; Hu, Y.; Zeng, G.; Du, W.; Shen, C. CTRP5 Attenuates Doxorubicin-Induced Cardiotoxicity Via Inhibiting TLR4/NLRP3 Signaling. Cardiovasc. Drugs Ther. 2023. [Google Scholar] [CrossRef]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, N.; Ma, X.; Li, M.; Feng, H. The dual role of ferroptosis in anthracycline-based chemotherapy includes reducing resistance and increasing toxicity. Cell Death Discov. 2023, 9, 184. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Dogon, G.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis. Int. J. Mol. Sci. 2022, 24, 449. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liang, J.; Qu, L.; Liu, S.; Qin, A.; Liu, H.; Wang, T.; Li, W.; Zou, W. Exploring the role of ferroptosis in the doxorubicin-induced chronic cardiotoxicity using a murine model. Chem.-Biol. Interact. 2022, 363, 110008. [Google Scholar] [CrossRef] [PubMed]
- Yapici, F.I.; Bebber, C.M.; von Karstedt, S. A guide to ferroptosis in cancer. Mol. Oncol. 2024, 18, 1378–1396. [Google Scholar] [CrossRef]
- Abe, K.; Ikeda, M.; Ide, T.; Tadokoro, T.; Miyamoto, H.D.; Furusawa, S.; Tsutsui, Y.; Miyake, R.; Ishimaru, K.; Watanabe, M.; et al. Doxorubicin causes ferroptosis and cardiotoxicity by intercalating into mitochondrial DNA and disrupting Alas1-dependent heme synthesis. Sci. Signal. 2022, 15, eabn8017. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef]
- Lu, L.; Wu, W.; Yan, J.; Li, X.; Yu, H.; Yu, X. Adriamycin-induced autophagic cardiomyocyte death plays a pathogenic role in a rat model of heart failure. Int. J. Cardiol. 2009, 134, 82–90. [Google Scholar] [CrossRef]
- Kobayashi, S.; Volden, P.; Timm, D.; Mao, K.; Xu, X.; Liang, Q. Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J. Biol. Chem. 2010, 285, 793–804. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Takemura, G.; Kanamori, H.; Takeyama, T.; Watanabe, T.; Morishita, K.; Ogino, A.; Tsujimoto, A.; Goto, K.; Maruyama, R.; et al. Prior starvation mitigates acute doxorubicin cardiotoxicity through restoration of autophagy in affected cardiomyocytes. Cardiovasc. Res. 2012, 96, 456–465. [Google Scholar] [CrossRef]
- Li, D.L.; Wang, Z.V.; Ding, G.; Tan, W.; Luo, X.; Criollo, A.; Xie, M.; Jiang, N.; May, H.; Kyrychenko, V.; et al. Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation 2016, 133, 1668–1687. [Google Scholar] [CrossRef] [PubMed]
- Seara, F.A.C.; Kasai-Brunswick, T.H.; Nascimento, J.H.M.; Campos-de-Carvalho, A.C. Anthracycline-induced cardiotoxicity and cell senescence: New therapeutic option? Cell. Mol. Life Sci. CMLS 2022, 79, 568. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Dessalvi, C.; Pepe, A.; Penna, C.; Gimelli, A. Sex differences in anthracycline-induced cardiotoxicity: The benefits of estrogens. Heart Fail. Rev. 2019, 24, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, V.; Cuomo, A.; Dessalvi, C.; Deidda, M. Redox Imbalances in Ageing and Metabolic Alterations: Implications in Cancer and Cardiac Diseases. An Overview from the Working Group of Cardiotoxicity and Cardioprotection of the Italian Society of Cardiology (SIC). Antioxidants 2020, 9, 641. [Google Scholar] [CrossRef]
- Franz, M.R.; Zabel, M. Electrophysiological basis of QT dispersion measurements. Prog. Cardiovasc. Dis. 2000, 42, 311–324. [Google Scholar] [CrossRef]
- Porta-Sánchez, A.; Gilbert, C.; Spears, D.; Amir, E.; Chan, J.; Nanthakumar, K.; Thavendiranathan, P. Incidence, Diagnosis, and Management of QT Prolongation Induced by Cancer Therapies: A Systematic Review. J. Am. Heart Assoc. 2017, 6, e007724. [Google Scholar] [CrossRef]
- Puppe, J.; van Ooyen, D.; Neise, J.; Thangarajah, F.; Eichler, C.; Krämer, S.; Pfister, R.; Mallmann, P.; Wirtz, M.; Michels, G. Evaluation of QTc Interval Prolongation in Breast Cancer Patients after Treatment with Epirubicin, Cyclophosphamide, and Docetaxel and the Influence of Interobserver Variation. Breast Care 2017, 12, 40–44. [Google Scholar] [CrossRef]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. Off. Publ. Am. Soc. Echocardiogr. 2015, 28, 1–39.e14. [Google Scholar] [CrossRef]
- Armenian, S.H.; Lacchetti, C.; Barac, A.; Carver, J.; Constine, L.S.; Denduluri, N.; Dent, S.; Douglas, P.S.; Durand, J.B.; Ewer, M.; et al. Prevention and Monitoring of Cardiac Dysfunction in Survivors of Adult Cancers: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 893–911. [Google Scholar] [CrossRef]
- Azzam, M.; Wasef, M.; Khalaf, H.; Al-Habbaa, A. 3D-based strain analysis and cardiotoxicity detection in cancer patients received chemotherapy. BMC Cancer 2023, 23, 760. [Google Scholar] [CrossRef]
- Urbano-Moral, J.A.; Patel, A.R.; Maron, M.S.; Arias-Godinez, J.A.; Pandian, N.G. Three-dimensional speckle-tracking echocardiography: Methodological aspects and clinical potential. Echocardiography 2012, 29, 997–1010. [Google Scholar] [CrossRef] [PubMed]
- Čelutkienė, J.; Pudil, R.; López-Fernández, T.; Grapsa, J.; Nihoyannopoulos, P.; Bergler-Klein, J.; Cohen-Solal, A.; Farmakis, D.; Tocchetti, C.G.; von Haehling, S.; et al. Role of cardiovascular imaging in cancer patients receiving cardiotoxic therapies: A position statement on behalf of the Heart Failure Association (HFA), the European Association of Cardiovascular Imaging (EACVI) and the Cardio-Oncology Council of the European Society of Cardiology (ESC). Eur. J. Heart Fail. 2020, 22, 1504–1524. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, E.K.; Kokkinidis, D.G.; Kampaktsis, P.N.; Amir, E.A.; Marwick, T.H.; Gupta, D.; Thavendiranathan, P. Assessment of Prognostic Value of Left Ventricular Global Longitudinal Strain for Early Prediction of Chemotherapy-Induced Cardiotoxicity: A Systematic Review and Meta-analysis. JAMA Cardiol. 2019, 4, 1007–1018. [Google Scholar] [CrossRef]
- Pudil, R.; Mueller, C.; Čelutkienė, J.; Henriksen, P.A.; Lenihan, D.; Dent, S.; Barac, A.; Stanway, S.; Moslehi, J.; Suter, T.M.; et al. Role of serum biomarkers in cancer patients receiving cardiotoxic cancer therapies: A position statement from the Cardio-Oncology Study Group of the Heart Failure Association and the Cardio-Oncology Council of the European Society of Cardiology. Eur. J. Heart Fail. 2020, 22, 1966–1983. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Sandri, M.T.; Martinoni, A.; Borghini, E.; Civelli, M.; Lamantia, G.; Cinieri, S.; Martinelli, G.; Fiorentini, C.; Cipolla, C.M. Myocardial injury revealed by plasma troponin I in breast cancer treated with high-dose chemotherapy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2002, 13, 710–715. [Google Scholar] [CrossRef]
- Zardavas, D.; Suter, T.M.; Van Veldhuisen, D.J.; Steinseifer, J.; Noe, J.; Lauer, S.; Al-Sakaff, N.; Piccart-Gebhart, M.J.; de Azambuja, E. Role of Troponins I and T and N-Terminal Prohormone of Brain Natriuretic Peptide in Monitoring Cardiac Safety of Patients With Early-Stage Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer Receiving Trastuzumab: A Herceptin Adjuvant Study Cardiac Marker Substudy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 878–884. [Google Scholar] [CrossRef]
- Houard, L.; Militaru, S.; Tanaka, K.; Pasquet, A.; Vancraeynest, D.; Vanoverschelde, J.L.; Pouleur, A.C.; Gerber, B.L. Test-retest reliability of left and right ventricular systolic function by new and conventional echocardiographic and cardiac magnetic resonance parameters. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 1157–1167. [Google Scholar] [CrossRef]
- Houard, L.; Benaets, M.B.; de Meester de Ravenstein, C.; Rousseau, M.F.; Ahn, S.A.; Amzulescu, M.S.; Roy, C.; Slimani, A.; Vancraeynest, D.; Pasquet, A.; et al. Additional Prognostic Value of 2D Right Ventricular Speckle-Tracking Strain for Prediction of Survival in Heart Failure and Reduced Ejection Fraction: A Comparative Study With Cardiac Magnetic Resonance. JACC Cardiovasc. Imaging 2019, 12, 2373–2385. [Google Scholar] [CrossRef]
- Oliveira, L.F.; O’Connell, J.L.; Carvalho, E.E.; Pulici, É.C.; Romano, M.M.; Maciel, B.C.; Simões, M.V. Comparison between Radionuclide Ventriculography and Echocardiography for Quantification of Left Ventricular Systolic Function in Rats Exposed to Doxorubicin. Arq. Bras. Cardiol. 2017, 108, 12–20. [Google Scholar] [CrossRef]
- Cascales, A.; Pastor-Quirante, F.; Sánchez-Vega, B.; Luengo-Gil, G.; Corral, J.; Ortuño-Pacheco, G.; Vicente, V.; de la Peña, F.A. Association of anthracycline-related cardiac histological lesions with NADPH oxidase functional polymorphisms. Oncologist 2013, 18, 446–453. [Google Scholar] [CrossRef]
- Wu, X.; Shen, F.; Jiang, G.; Xue, G.; Philips, S.; Gardner, L.; Cunningham, G.; Bales, C.; Cantor, E.; Schneider, B.P. A non-coding GWAS variant impacts anthracycline-induced cardiotoxic phenotypes in human iPSC-derived cardiomyocytes. Nat. Commun. 2022, 13, 7171. [Google Scholar] [CrossRef]
- Bozza, W.P.; Takeda, K.; Alterovitz, W.L.; Chou, C.K.; Shen, R.F.; Zhang, B. Anthracycline-Induced Cardiotoxicity: Molecular Insights Obtained from Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes (hiPSC-CMs). AAPS J. 2021, 23, 44. [Google Scholar] [CrossRef] [PubMed]
- Octavia, Y.; Kararigas, G.; de Boer, M.; Chrifi, I.; Kietadisorn, R.; Swinnen, M.; Duimel, H.; Verheyen, F.K.; Brandt, M.M.; Fliegner, D.; et al. Folic acid reduces doxorubicin-induced cardiomyopathy by modulating endothelial nitric oxide synthase. J. Cell. Mol. Med. 2017, 21, 3277–3287. [Google Scholar] [CrossRef]
- Kwatra, M.; Kumar, V.; Jangra, A.; Mishra, M.; Ahmed, S.; Ghosh, P.; Vohora, D.; Khanam, R. Ameliorative effect of naringin against doxorubicin-induced acute cardiac toxicity in rats. Pharm. Biol. 2016, 54, 637–647. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Luo, Y.; Qiao, Y.; Zhang, Z.; Yin, D.; Yao, J.; You, J.; He, M. Curcumin attenuates doxorubicin-induced cardiotoxicity via suppressing oxidative stress and preventing mitochondrial dysfunction mediated by 14-3-3γ. Food Funct. 2018, 9, 4404–4418. [Google Scholar] [CrossRef]
- Wang, L.; Ma, J.; Chen, C.; Lin, B.; Xie, S.; Yang, W.; Qian, J.; Zhang, Y. Isoquercitrin alleviates pirarubicin-induced cardiotoxicity in vivo and in vitro by inhibiting apoptosis through Phlpp1/AKT/Bcl-2 signaling pathway. Front. Pharmacol. 2024, 15, 1315001. [Google Scholar] [CrossRef]
- Syahputra, R.A.; Harahap, U.; Harahap, Y.; Gani, A.P.; Dalimunthe, A.; Ahmed, A.; Zainalabidin, S. Vernonia amygdalina Ethanol Extract Protects against Doxorubicin-Induced Cardiotoxicity via TGFβ, Cytochrome c, and Apoptosis. Molecules 2023, 28, 4305. [Google Scholar] [CrossRef] [PubMed]
- Saleh Ahmed, A.S. Potential protective effect of catechin on doxorubicin-induced cardiotoxicity in adult male albino rats. Toxicol. Mech. Methods 2022, 32, 97–105. [Google Scholar] [CrossRef]
- Ozyurt, H.; Ozyurt, B.; Koca, K.; Ozgocmen, S. Caffeic acid phenethyl ester (CAPE) protects rat skeletal muscle against ischemia–reperfusion-induced oxidative stress. Vasc. Pharmacol. 2007, 47, 108–112. [Google Scholar] [CrossRef]
- Dantas, D.; Pereira, A.G.; Fujimori, A.S.S.; Ribeiro, A.P.D.; de Almeida Silva, C.C.V.; Monte, M.G.; Corrêa, C.R.; Fernandes, A.A.; Bazan, S.G.Z.; Azevedo, P.S.; et al. Doxycycline Attenuates Doxorubicin-Induced Cardiotoxicity by Improving Myocardial Energy Metabolism in Rats. J. Cardiovasc. Dev. Dis. 2022, 9, 254. [Google Scholar] [CrossRef] [PubMed]
- Meeran, M.F.N.; Azimullah, S.; Mamoudh, H.H.; Sharma, C.; Kumar, S.; Goyal, S.N.; Ojha, S. Nerolidol, a Sesquiterpene from the Essential Oils of Aromatic Plants, Attenuates Doxorubicin-Induced Chronic Cardiotoxicity in Rats. J. Agric. Food Chem. 2021, 69, 7334–7343. [Google Scholar] [CrossRef] [PubMed]
- Sumeet Kumar, S.; Poonam, Y.; Dhaneshvaree, P.; Sampat Singh, T.; Abhishek, S.; Amit, K.; Jasvinder Singh, B.; Umashanker, N. Betaine ameliorates doxorubicin-induced cardiomyopathy by inhibiting oxidative stress, inflammation, and fibrosis through the modulation of AMPK/Nrf2/TGF-β expression. Environ. Toxicol. 2024, 39, 4134–4147. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, C.; Zhang, N.; Wei, W.Y.; Li, L.L.; Wu, H.M.; Ma, Z.G.; Tang, Q.Z. Matrine attenuates pathological cardiac fibrosis via RPS5/p38 in mice. Acta Pharmacol. Sin. 2021, 42, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Upshaw, J.N.; Parson, S.K.; Buchsbaum, R.J.; Schlam, I.; Ruddy, K.J.; Durani, U.; Epperla, N.; Leong, D.P. Dexrazoxane to Prevent Cardiotoxicity in Adults Treated with Anthracyclines: JACC: CardioOncology Controversies in Cardio-Oncology. JACC CardioOncology 2024, 6, 322–324. [Google Scholar] [CrossRef]
- Chen, H.; Zhu, J.; Le, Y.; Pan, J.; Liu, Y.; Liu, Z.; Wang, C.; Dou, X.; Lu, D. Salidroside inhibits doxorubicin-induced cardiomyopathy by modulating a ferroptosis-dependent pathway. Phytomedicine Int. J. Phytother. Phytopharm. 2022, 99, 153964. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Du, J.; Meng, H.; Liu, F.; Yang, N.; Deng, S.; Wan, H.; Ye, D.; Song, E.; Zeng, H. Targeting autophagy with SAR405 alleviates doxorubicin-induced cardiotoxicity. Cell Biol. Toxicol. 2023, 39, 3255–3267. [Google Scholar] [CrossRef]
- Murase, R.; Shingu, Y.; Wakasa, S. Cardioprotective effects of chloroquine pretreatment on ischemic and reperfusion injury via activation of ERK1/2 in isolated rat hearts. Mol. Biol. Rep. 2022, 49, 9429–9436. [Google Scholar] [CrossRef]
- Shimizu, M.; Ohwada, W.; Yano, T.; Kouzu, H.; Sato, T.; Osanami, A.; Ogawa, T.; Toda, Y.; Kuno, A.; Tanno, M.; et al. Rapamycin ameliorates doxorubicin-induced cardiomyopathy through suppression of RIPK1-independent necroptosis. Eur. Heart J. 2023, 44 (Suppl. 2), ehad655-3126. [Google Scholar] [CrossRef]
- Peng, Y.; Li, Z.; Zhang, J.; Dong, Y.; Zhang, C.; Dong, Y.; Zhai, Y.; Zheng, H.; Liu, M.; Zhao, J.; et al. Low-Dose Colchicine Ameliorates Doxorubicin Cardiotoxicity Via Promoting Autolysosome Degradation. J. Am. Heart Assoc. 2024, 13, e033700. [Google Scholar] [CrossRef]
- Xu, Z.; Pan, Z.; Jin, Y.; Gao, Z.; Jiang, F.; Fu, H.; Chen, X.; Zhang, X.; Yan, H.; Yang, X.; et al. Inhibition of PRKAA/AMPK (Ser485/491) phosphorylation by crizotinib induces cardiotoxicity via perturbing autophagosome-lysosome fusion. Autophagy 2024, 20, 416–436. [Google Scholar] [CrossRef]
- Lin, Z.H.; Xiang, H.Q.; Yu, Y.W.; Xue, Y.J.; Wu, C.; Lin, C.; Ji, K.T. Dihydroartemisinin alleviates doxorubicin-induced cardiotoxicity and ferroptosis by activating Nrf2 and regulating autophagy. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2024, 38, e23677. [Google Scholar] [CrossRef] [PubMed]
- Gyöngyösi, M.; Lukovic, D.; Zlabinger, K.; Spannbauer, A.; Gugerell, A.; Pavo, N.; Traxler, D.; Pils, D.; Maurer, G.; Jakab, A.; et al. Liposomal doxorubicin attenuates cardiotoxicity via induction of interferon-related DNA damage resistance. Cardiovasc. Res. 2020, 116, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Szponar, J.; Niziński, P.; Dudka, J.; Kasprzak-Drozd, K.; Oniszczuk, A. Natural Products for Preventing and Managing Anthracycline-Induced Cardiotoxicity: A Comprehensive Review. Cells 2024, 13, 1151. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Sun, B.; Zuo, S.; Li, X.; Zhou, S.; Li, L.; Luo, C.; Liu, H.; Cheng, M.; Wang, Y.; et al. Trisulfide bond-mediated doxorubicin dimeric prodrug nanoassemblies with high drug loading, high self-assembly stability, and high tumor selectivity. Sci. Adv. 2020, 6, eabc1725. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Yang, Z.; Wang, J.; She, H.; Tan, L.; Ye, Q.; Wang, F.; Feng, X.; Mo, X.; Liu, K.; et al. Exosomes miRNA-499a-5p targeted CD38 to alleviate anthraquinone induced cardiotoxicity: Experimental research. Int. J. Surg. 2024, 110, 1992–2006. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Qian, C.; Ren, P.; Yu, H.; Kong, X.; Huang, C.; Luo, H.; Chen, G. Light-Responsive Micelles Loaded with Doxorubicin for Osteosarcoma Suppression. Front. Pharmacol. 2021, 12, 679610. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Gong, N.; Sun, W.; Han, J.; Liu, Y.; Zhang, S.; Zheng, A.; Butt, H.J.; Liang, X.J.; Wu, S. Red-Light-Responsive Metallopolymer Nanocarriers with Conjugated and Encapsulated Drugs for Phototherapy Against Multidrug-Resistant Tumors. Small 2022, 18, e2201672. [Google Scholar] [CrossRef]
- Chowdhury, S.; Toth, I.; Stephenson, R.J. Dendrimers in vaccine delivery: Recent progress and advances. Biomaterials 2022, 280, 121303. [Google Scholar] [CrossRef]
- Hu, W.; Qiu, L.; Cheng, L.; Hu, Q.; Liu, Y.; Hu, Z.; Chen, D.; Cheng, L. Redox and pH dual responsive poly(amidoamine) dendrimer-poly(ethylene glycol) conjugates for intracellular delivery of doxorubicin. Acta Biomater. 2016, 36, 241–253. [Google Scholar] [CrossRef]
- Wu, D.; Zhao, Z.; Liu, H.; Fu, K.; Ji, Y.; Ji, W.; Li, Y.; Yan, Q.; Yang, G. Escherichia coli Nissle 1917-driven microrobots for effective tumor targeted drug delivery and tumor regression. Acta Biomater. 2023, 169, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Willeke, R.N.; David, B.; Martijn, M.S.; Neil, K.A.; Arco, J.T.; Wim, H.v.H.; Wim, G.G.; Anne, M.M. Efficacy of Physical Exercise to Offset Anthracycline-Induced Cardiotoxicity: A Systematic Review and Meta-Analysis of Clinical and Preclinical Studies. J. Am. Heart Assoc. 2021, 10, e021580. [Google Scholar] [CrossRef]
- Lee, Y.; Kwon, I.; Jang, Y.; Cosio-Lima, L.; Barrington, P. Endurance Exercise Attenuates Doxorubicin-induced Cardiotoxicity. Med. Sci. Sports Exerc. 2020, 52, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Dozic, S.; Howden, E.J.; Bell, J.R.; Mellor, K.M.; Delbridge, L.M.D.; Weeks, K.L. Cellular Mechanisms Mediating Exercise-Induced Protection against Cardiotoxic Anthracycline Cancer Therapy. Cells 2023, 12, 1312. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Kobayashi, M.; Yang, Y.; Kleinerman, E. Exercise Inhibits Doxorubicin-Induced Damage to Cardiac Vessels and Activation of Hippo/YAP-Mediated Apoptosis. Cancers 2021, 13, 2740. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Diagnostic Criteria | Superiority | Inferiority |
|---|---|---|---|
| ECG [56,57,58] | 1. ST segment change 2. QT prolongation | Accessible and economical | Insufficient specificity |
| Echocardiography | 1. A decrease in LVEF of >10% and LVEF < 50% [59,60] 2. A decrease of GLS > 15% [61,62,63,64] | Provides information about structure and function | Unable to identify subclinical cardiomyocyte structural alterations |
| Biomarkers [65,66,67] | 1. Continuous cTnI elevation 2. BNP elevation | Sensitive and repeatable | Insufficient evidence |
| CMRI [68,69] | A decrease in LVEF of >10% and LVEF < 50% | 1. Presents comprehensive information encompassing both the structure and function 2. Excellent repeatability | Expensive and stringent adherence prerequisites |
| RV [70] | A decrease in LVEF of >10% and LVEF < 50% | Surpasses standard two-dimensional echocardiography in terms of precision and reproducibility when measuring LVEF | Radiation exposure |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Yang, W.; Cui, X.; Zhang, H.; Li, L.; Fu, J.; Guo, H. Research Progress on the Mechanism, Monitoring, and Prevention of Cardiac Injury Caused by Antineoplastic Drugs—Anthracyclines. Biology 2024, 13, 689. https://doi.org/10.3390/biology13090689
Chen Y, Yang W, Cui X, Zhang H, Li L, Fu J, Guo H. Research Progress on the Mechanism, Monitoring, and Prevention of Cardiac Injury Caused by Antineoplastic Drugs—Anthracyclines. Biology. 2024; 13(9):689. https://doi.org/10.3390/biology13090689
Chicago/Turabian StyleChen, Yuanyuan, Wenwen Yang, Xiaoshan Cui, Huiyu Zhang, Liang Li, Jianhua Fu, and Hao Guo. 2024. "Research Progress on the Mechanism, Monitoring, and Prevention of Cardiac Injury Caused by Antineoplastic Drugs—Anthracyclines" Biology 13, no. 9: 689. https://doi.org/10.3390/biology13090689
APA StyleChen, Y., Yang, W., Cui, X., Zhang, H., Li, L., Fu, J., & Guo, H. (2024). Research Progress on the Mechanism, Monitoring, and Prevention of Cardiac Injury Caused by Antineoplastic Drugs—Anthracyclines. Biology, 13(9), 689. https://doi.org/10.3390/biology13090689