
Inflammation and Tumor Progression: The Differential Impact of SAA in Breast Cancer Models

 , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals, Housing, and Genotyping
2.1.1. Animals and Housing
2.1.2. SAADKO Genotyping
2.2. EO771 SAA1/2/3/4 mRNA Screening, Tumor Induction, Monitoring, and Assessment
2.2.1. EO771 SAA1/2/3/4 mRNA Screening
2.2.2. Cell Culture
2.2.3. Tumor Establishment
2.2.4. Assessment and Measurements
2.3. Euthanasia and Sample Collection
2.4. Tumor Growth and Species Survival
Tumor Growth Analysis
2.5. Blood Plasma Inflammatory Profiling
2.5.1. Serum Amyloid A
2.5.2. IL-1β, IL-6, IL-10, MCP-1, and TNFα
2.6. SDS-PAGE and Western Blot Analysis of Tumor Tissue
2.6.1. Protein Harvest from Tumor Tissue
2.6.2. Protein Quantification and Sample Preparation
2.6.3. SDS-PAGE and Western Blot
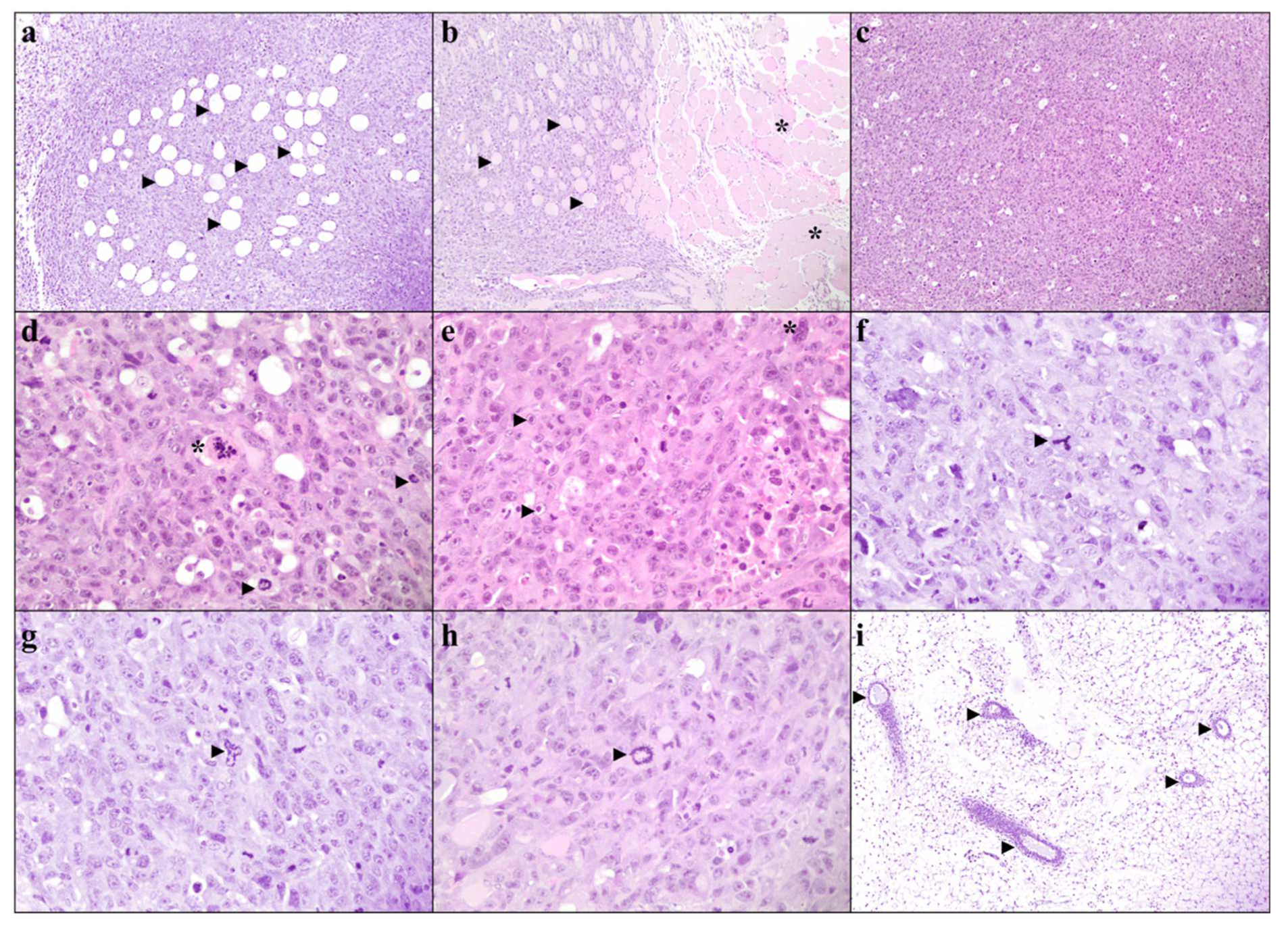
2.7. Tumor Histopathology
2.7.1. Tissue Processing and Sectioning
2.7.2. Histological Staining
2.7.3. Pathological Grading and Characterization
2.7.4. Quantitative Pathology and Bioimage Analysis (QuPath)
2.8. Data Analyses, Significance, and Presentation
3. Results
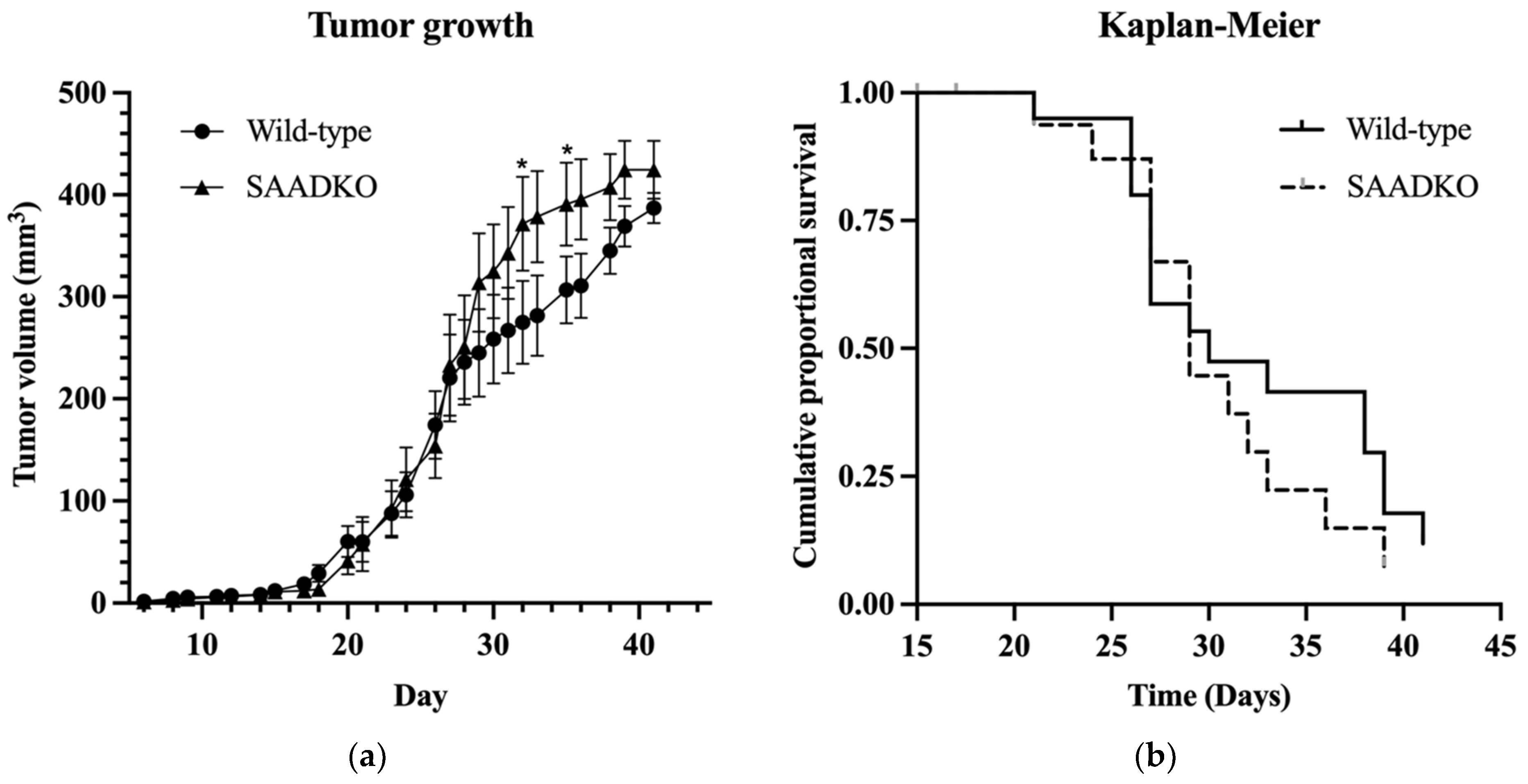
3.1. Systemic SAA1/2-Deficiency Does Not Influence Tumor Growth or Host Survival
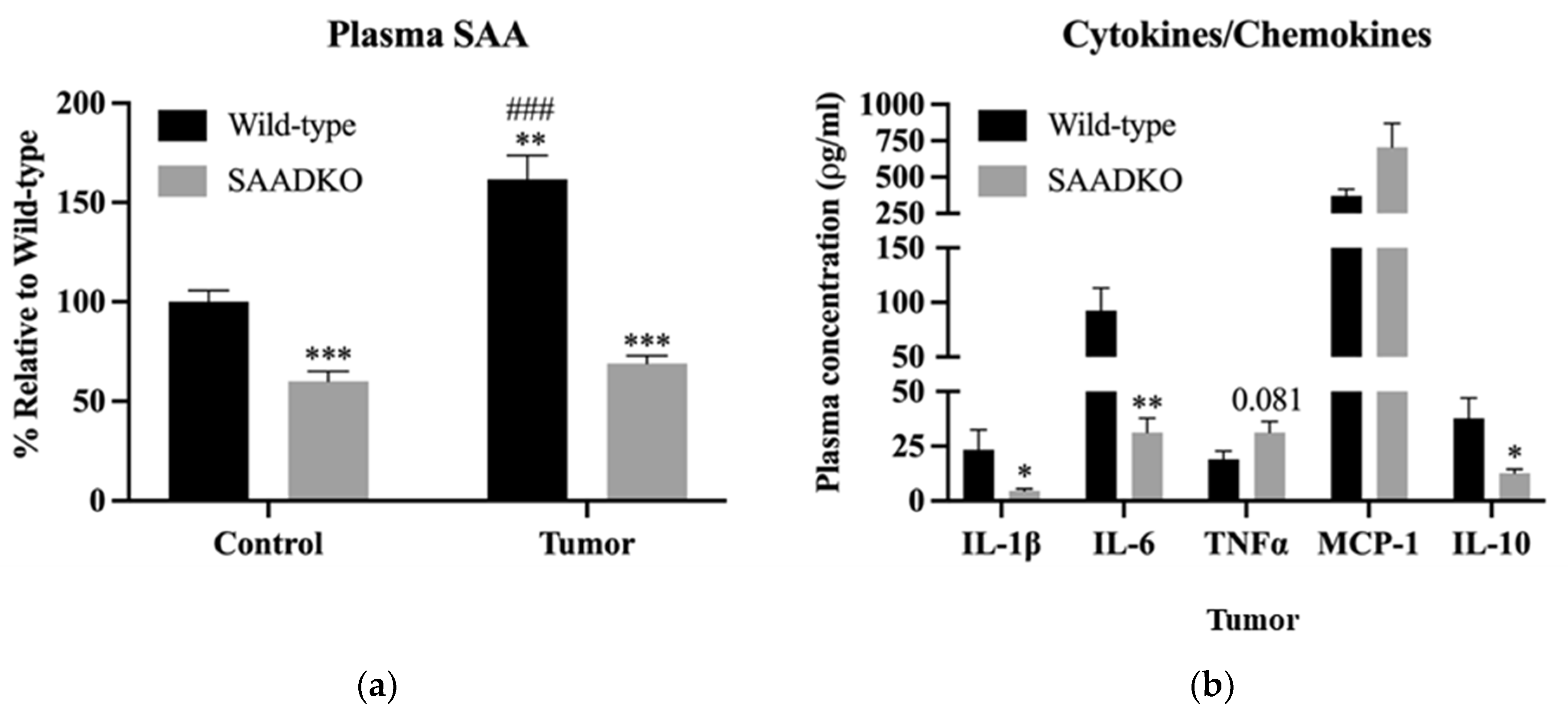
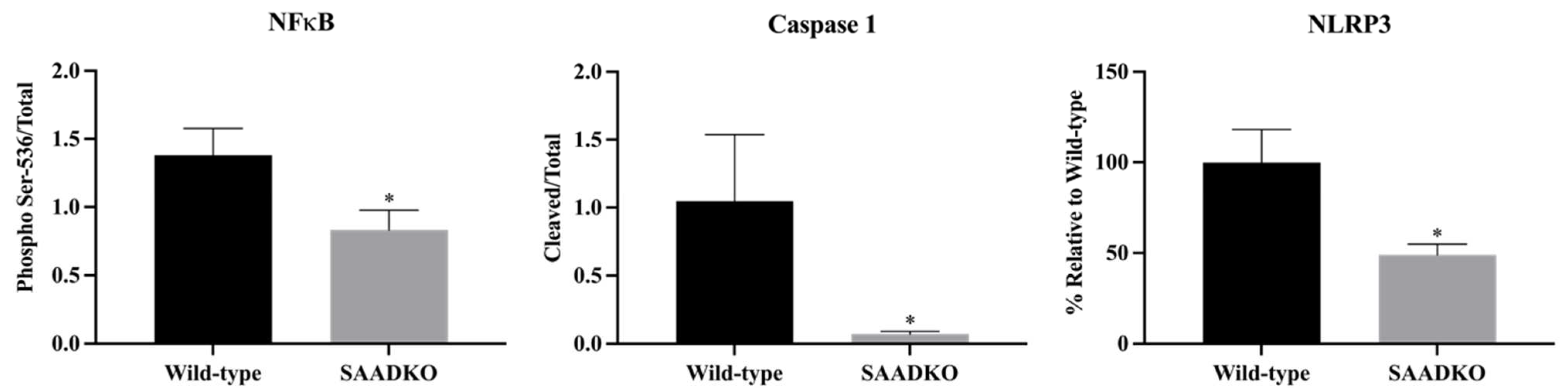
3.2. Systemic SAA1/2-Deficiency Alters Cytokine Expression and Promotes Inflammasome Signaling
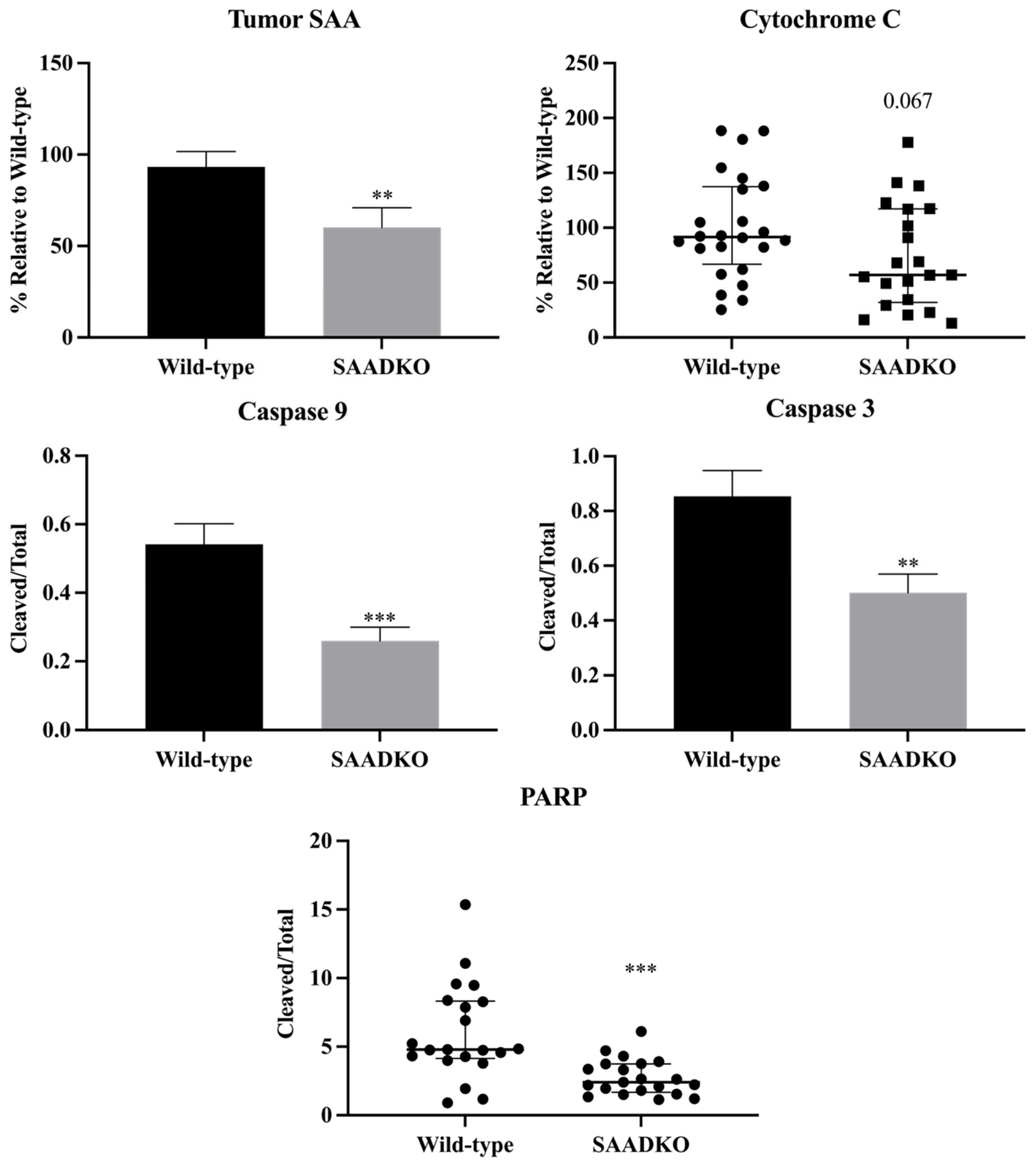
3.3. Systemic SAA1/2-Deficiency Leads to Lower SAA Levels in Tumors, Reduced Apoptosis Signaling, and Increased DNA Repair
3.4. Systemic SAA1/2-Deficiency Does not Influence Histological Grading
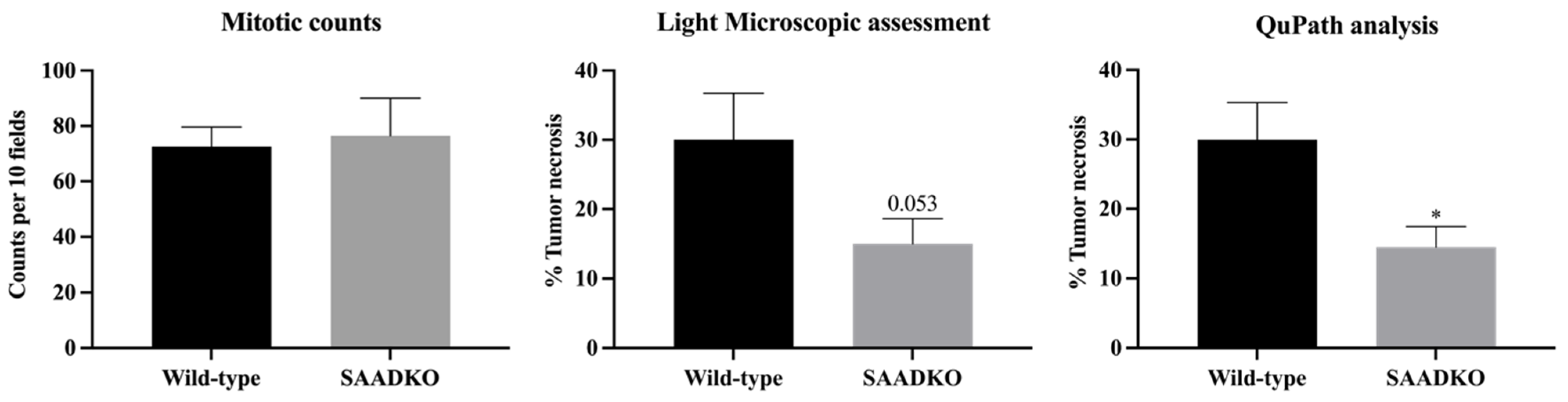
3.5. Systemic SAA1/2-Deficiency Leads to Lower Tumor Necrosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of Variance |
| ASC | Apoptosis-associated speck-like protein containing a CARD domain |
| CAF | Central analytical facilities |
| CANSA | Cancer Association of South Africa |
| DAMPs | Damage-associated molecular patterns |
| EMT | Epithelial to Mesenchymal Transition |
| GSDMD | Gasdermin D |
| H&E | Hematoxylin and Eosin |
| HBSS | Hank’s balanced salt solution |
| HIF1α | Hypoxia-inducible factor 1 alpha |
| HPF | High power field |
| IL-10 | Interleukin 10 |
| IL-18 | Interleukin 18 |
| IL-1β | Interleukin 1 beta |
| IL-6 | Interleukin 6 |
| MCP-1 | Monocyte chemotactic protein 1 |
| NaCl | Sodium Chloride |
| NBF | Neutral buffered formaldehyde |
| NFκB | Nuclear factor kappa beta |
| NLR | Nod-like receptor |
| NLRP3 | NLR family pyrin domain containing 3 |
| NPM | Nucleophosmin |
| NRF | National Research Foundation |
| PAMPs | Pathogen-associated molecular patterns |
| PARP | Poly(ADP-ribose) polymerase |
| PCR | Polymerase chain reaction |
| PRRs | Pattern recognition receptors |
| PVDF | Polyvinylidene fluoride |
| QuPath | Quantitative pathology and bioimage analysis |
| RIPA | Radioimmunoprecipitation assay |
| SAA | Serum Amyloid A |
| SAADKO | SAA1/2 Double Knockout |
| SAMRC | South African Medical Research Council |
| SANS | South African National Standard |
| SDS | Sodium dodecyl sulfate |
| SDS-PAGE | SDS–polyacrylamide gel electrophoresis |
| SEM | Standard error of the mean |
| TBS-T | Tris-buffered saline-tween-20 |
| TLR | Toll-like receptor |
| TME | Tumor microenvironment |
| TNFα | Tumor necrosis factor alpha |
| WT | Wild type |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Sack, G.H. Serum amyloid A—A review. Mol. Med. 2018, 24, 46. [Google Scholar] [CrossRef] [PubMed]
- Kaneti, J.; Winikoff, Y.; Zimlichman, S.; Shainkin-Kestenbaum, R. Importance of serum amyloid A (SAA) level in monitoring disease activity and response to therapy in patients with prostate cancer. Urol. Res. 1984, 12, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Tomita, Y.; Imai, T.; Saito, T.; Katagiri, A.; Ohara-Mikami, Y.; Matsudo, T.; Takahashi, K. Significance of serum amyloid A on the prognosis in patients with renal cell carcinoma. Cancer 2001, 92, 2072–2075. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Ahsan, M.H.; Purchio, A.F.; West, D.B. Serum amyloid A-luciferase transgenic mice: Response to sepsis, acute arthritis, and contact hypersensitivity and the effects of proteasome inhibition. J. Immunol. 2005, 174, 8125–8134. [Google Scholar] [CrossRef] [PubMed]
- Knebel, F.H.; Uno, M.; Galatro, T.F.; Bellé, L.P.; Oba-Shinjo, S.M.; Marie, S.K.N.; Campa, A. Serum amyloid A1 is upregulated in human glioblastoma. J. Neurooncol. 2017, 132, 383–391. [Google Scholar] [CrossRef]
- Malle, E.; Sodin-Semrl, S.; Kovacevic, A. Serum amyloid A: An acute-phase protein involved in tumour pathogenesis. Cell Mol. Life Sci. 2009, 66, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Wang, H.; Lu, D.; Xie, X.; Chen, X.; Peng, J.; Hu, Q.; Shi, G.; Shanling, L. Expression of serum amyloid A in uterine cervical cancer. Diagn. Pathol. 2014, 9, 16. [Google Scholar] [CrossRef]
- Li, Z.; Hou, Y.; Zhao, M.; Li, T.; Liu, Y.; Chang, J.; Li, R. Serum amyloid a, a potential biomarker both in serum and tissue, correlates with ovarian cancer progression. J. Ovarian Res. 2020, 13, 67. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ye, H.; Zhu, Q.; Qu, G.; Wang, L.; Zhao, M.; Wang, P. Serum amyloid A expression is associated with breast cancer survival. Int. J. Clin. Exp. Pathol. 2016, 9, 9853–9866. [Google Scholar]
- Yang, M.; Liu, F.; Higuchi, K.; Sawashita, J.; Fu, X.; Zhang, L.; Zhang, L.J.; Fu, L.; Tong, Z.S.; Keiichi, H. Serum amyloid A expression in the breast cancer tissue is associated with poor prognosis. Oncotarget 2016, 7, 35843–35852. [Google Scholar] [CrossRef]
- Sack, G.H. Serum Amyloid A (SAA). Proteins 2020, 94, 421–436. [Google Scholar]
- Davis, T.A.; Conradie, D.; Shridas, P.; de Beer, F.C.; Engelbrecht, A.M.; de Villiers, W.J.S. Serum Amyloid A Promotes Inflammation-Associated Damage and Tumorigenesis in a Mouse Model of Colitis-Associated Cancer. Cell Mol. Gastroenterol. Hepatol. 2021, 12, 1329–1341. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.E.; Lee, J.Y.; Yang, G.; Kang, H.C.; Cho, Y.Y.; Lee, H.S. Inhibition of NLRP3 inflammasome in tumor microenvironment leads to suppression of metastatic potential of cancer cells. Sci. Rep. 2019, 9, 12277. [Google Scholar] [CrossRef]
- Karan, D. Inflammasomes: Emerging Central Players in Cancer Immunology and Immunotherapy. Front. Immunol. 2018, 9, 3028. [Google Scholar] [CrossRef] [PubMed]
- Fourie, C.; Shridas, P.; Davis, T.; de Villiers, W.J.S.; Engelbrecht, A.M. Serum amyloid A and inflammasome activation: A link to breast cancer progression? Cytokine Growth Factor. Rev. 2021, 59, 62–70. [Google Scholar] [CrossRef]
- De Beer, M.C.; Webb, N.R.; Wroblewski, J.M.; Noffsinger, V.P.; Rateri, D.L.; der Avan, J.W.; Deneys, R.; de Beer, F.C. Impact of serum amyloid A on high density lipoprotein composition and levels. J. Lipid Res. 2010, 51, 3117–3125. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Olivier, D.W. An investigation into the role of Serum Amyloid A in breast cancer. Ph.D. Thesis, Stellenbosch University, Stellenbosch, South Africa, April 2021. [Google Scholar]
- Elston, C.W.; Ellis, I.O. pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: Experience from a large study with long-term follow-up. Histopathology 1991, 19, 403–410. [Google Scholar] [CrossRef]
- Fitzgibbons, P.L.; Connolly, J.L.; Bose, S.; Chen, Y.Y.; de Baca, M.E.; Edgerton, M. Protocol for the Examination of Resection Specimens From Patients With Invasive Carcinoma of the Breast. 2020. Available online: https://cap.objects.frb.io/protocols/public-comment-drafts/cp-breast-invasive-biopsy-19-1000-draftPC.pdf (accessed on 15 July 2024).
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef]
- Ignacio, R.M.C.; Gibbs, C.R.; Kim, S.; Lee, E.S.; Adunyah, S.E.; Son, D.S. Serum amyloid A predisposes inflammatory tumor microenvironment in triple negative breast cancer. Oncotarget 2019, 10, 511–526. [Google Scholar] [CrossRef]
- Zhang, G. Serum amyloid A: A new potential serum marker correlated with the stage of breast cancer. Oncol. Lett. 2012, 3, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.C.; Yi, Y.; Fu, Y.P.; He, H.W.; Cai, X.Y.; Wang, J.X.; Zhou, J.; Fan, J.; Shuang-Jian, Q. Serum amyloid A is a novel prognostic biomarker in hepatocellular carcinoma. Asian Pac. J. Cancer Prev. 2014, 15, 10713–10718. [Google Scholar] [CrossRef]
- Biaoxue, R.; Hua, L.; Wenlong, G.; Shuanying, Y. Increased serum amyloid A as potential diagnostic marker for lung cancer: A meta-analysis based on nine studies. BMC Cancer. 2016, 16, 836. [Google Scholar] [CrossRef] [PubMed]
- Mannino, M.H.; Zhu, Z.; Xiao, H.; Bai, Q.; Wakefield, M.R.; Fang, Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett. 2015, 367, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Garza, M.T.; Cruz-Vega, E.D.; Maldonado-Bernal, C. IL10 as Cancer Biomarker. In Translational Research in Cancer; Intech Open: London, UK, 2020. [Google Scholar] [CrossRef]
- Pileczki, V.; Braicu, C.; Gherman, C.; Berindan-Neagoe, I. TNF-α Gene Knockout in Triple Negative Breast Cancer Cell Line Induces Apoptosis. Int. J. Mol. Sci. 2012, 14, 411–420. [Google Scholar] [CrossRef]
- Changkija, B.H.; Konwar, R. Role of interleukin-10 in breast cancer. Breast Cancer Res. Treat. 2012, 133, 11–21. [Google Scholar]
- Guo, B.; Fu, S.; Zhang, J.; Liu, B.; Li, Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 2016, 6, 36107. [Google Scholar] [CrossRef] [PubMed]
- Shridas, P.; De Beer, M.C.; Webb, N.R. High-density lipoprotein inhibits serum amyloid A–mediated reactive oxygen species generation and NLRP3 inflammasome activation. J. Biol. Chem. 2018, 293, 13257–13269. [Google Scholar] [CrossRef] [PubMed]
- Kho, Y.; Kim, S.; Yoon, B.S.; Moon, J.-H.; Kim, B.; Kwak, S.; Woo, J.; Oh, S.; Hong, K.; Kim, S.; et al. Induction of Serum Amyloid A Genes Is Associated with Growth and Apoptosis of HC11 Mammary Epithelial Cells. Biosci. Biotechnol. Biochem. 2008, 72, 70–81. [Google Scholar] [CrossRef]
- Satyanarayana, A.; Kaldis, P. Mammalian cell-cycle regulation: Several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009, 28, 2925–2939. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2021, 17, 1–382. [Google Scholar] [PubMed]
- Olivier, D.W.; Pretorius, E.; Engelbrecht, A.-M. Serum amyloid A1: Innocent bystander or active participant in cell migration in triple-negative breast cancer? Exp. Cell Res. 2021, 406, 112759. [Google Scholar] [CrossRef] [PubMed]
- Raut, P.K.; Kim, S.H.; Choi, D.Y.; Jeong, G.S.; Park, P.H. Growth of breast cancer cells by leptin is mediated via activation of the inflammasome: Critical roles of estrogen receptor signaling and reactive oxygen species production. Biochem. Pharmacol. 2019, 161, 73–88. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.Y.; Wang, Y.; Huang, H.W.; Zhuang, Y.H.; Cai, T.; Wang, F.C.; Feng, S. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivier, D.W.; Eksteen, C.; Plessis, M.d.; de Jager, L.; Engelbrecht, L.; McGregor, N.W.; Shridas, P.; de Beer, F.C.; de Villiers, W.J.S.; Pretorius, E.; et al. Inflammation and Tumor Progression: The Differential Impact of SAA in Breast Cancer Models. Biology 2024, 13, 654. https://doi.org/10.3390/biology13090654
Olivier DW, Eksteen C, Plessis Md, de Jager L, Engelbrecht L, McGregor NW, Shridas P, de Beer FC, de Villiers WJS, Pretorius E, et al. Inflammation and Tumor Progression: The Differential Impact of SAA in Breast Cancer Models. Biology. 2024; 13(9):654. https://doi.org/10.3390/biology13090654
Chicago/Turabian StyleOlivier, Daniel Wilhelm, Carla Eksteen, Manisha du Plessis, Louis de Jager, Lize Engelbrecht, Nathaniel Wade McGregor, Preetha Shridas, Frederick C. de Beer, Willem J. S. de Villiers, Etheresia Pretorius, and et al. 2024. "Inflammation and Tumor Progression: The Differential Impact of SAA in Breast Cancer Models" Biology 13, no. 9: 654. https://doi.org/10.3390/biology13090654
APA StyleOlivier, D. W., Eksteen, C., Plessis, M. d., de Jager, L., Engelbrecht, L., McGregor, N. W., Shridas, P., de Beer, F. C., de Villiers, W. J. S., Pretorius, E., & Engelbrecht, A.-M. (2024). Inflammation and Tumor Progression: The Differential Impact of SAA in Breast Cancer Models. Biology, 13(9), 654. https://doi.org/10.3390/biology13090654