
The Therapeutic Potential of Dalbergia pinnata (Lour.) Prain Essential Oil in Alzheimer’s Disease: EEG Signal Analysis In Vivo, SH-SY5Y Cell Model In Vitro, and Network Pharmacology

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. In Vitro and In Vivo Materials
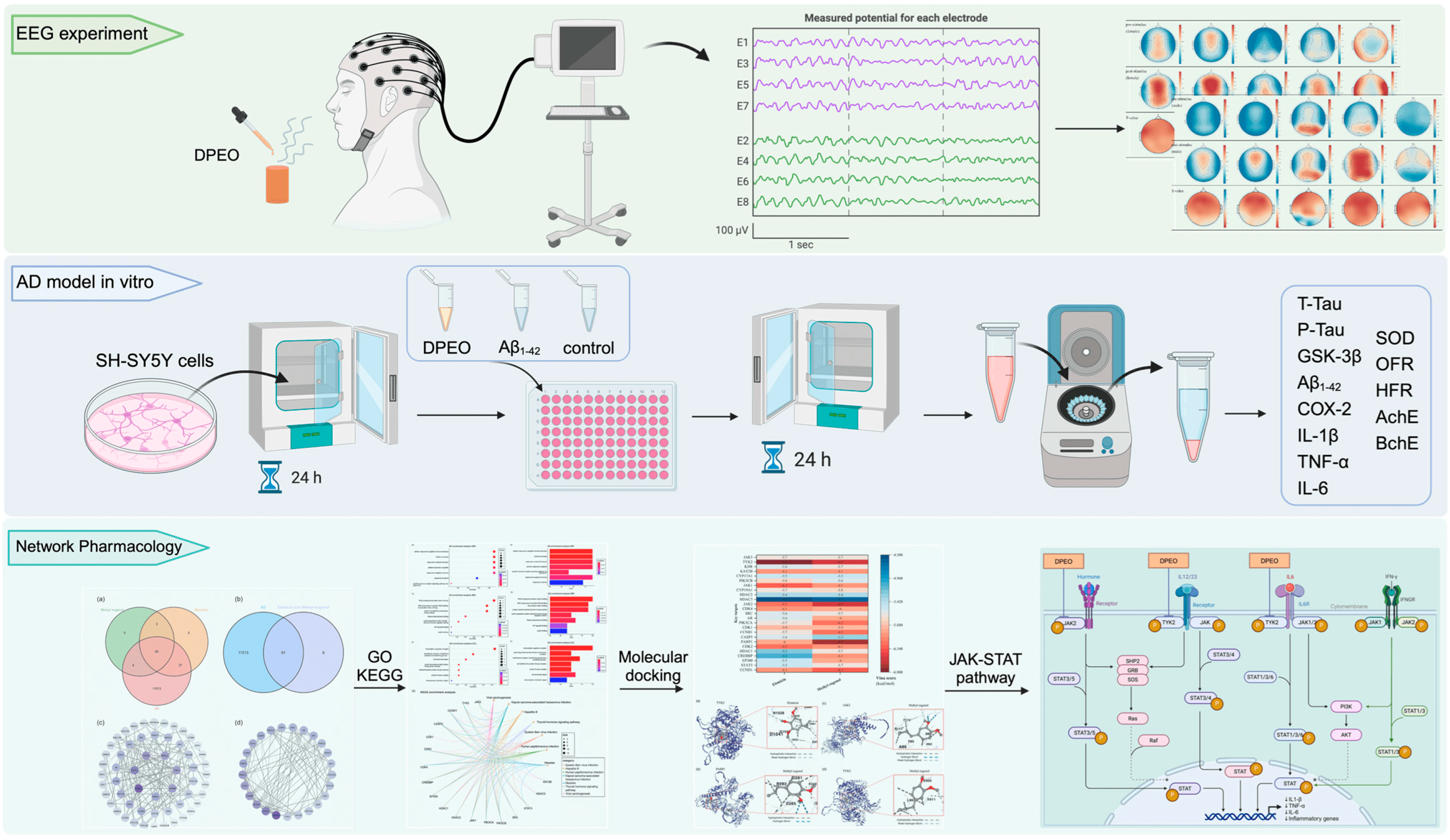
2.2. EEG Experiment Prepare
2.3. EEG Experiment Process
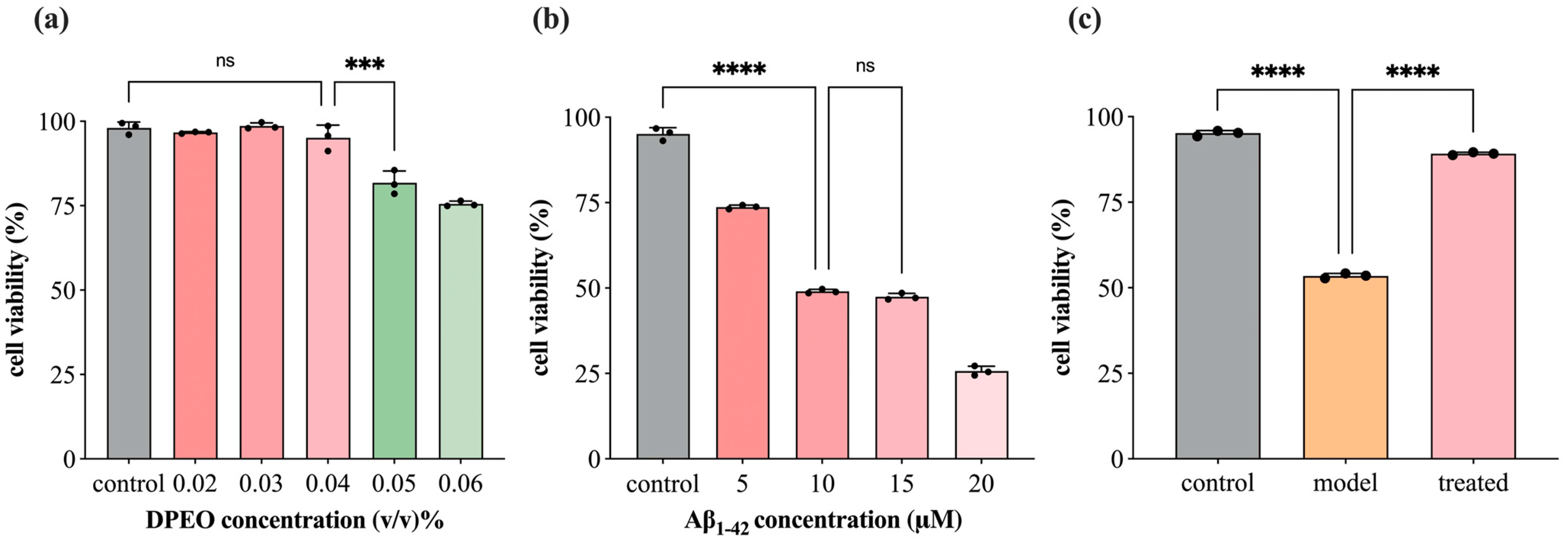
2.4. AD Cell Model Establishment and Cell Viability
2.5. Morphological Observation
2.6. Assay of AD Cells Model
2.7. Network Pharmacology Database and Analysis Platform
2.8. Statistical Analysis
3. Results
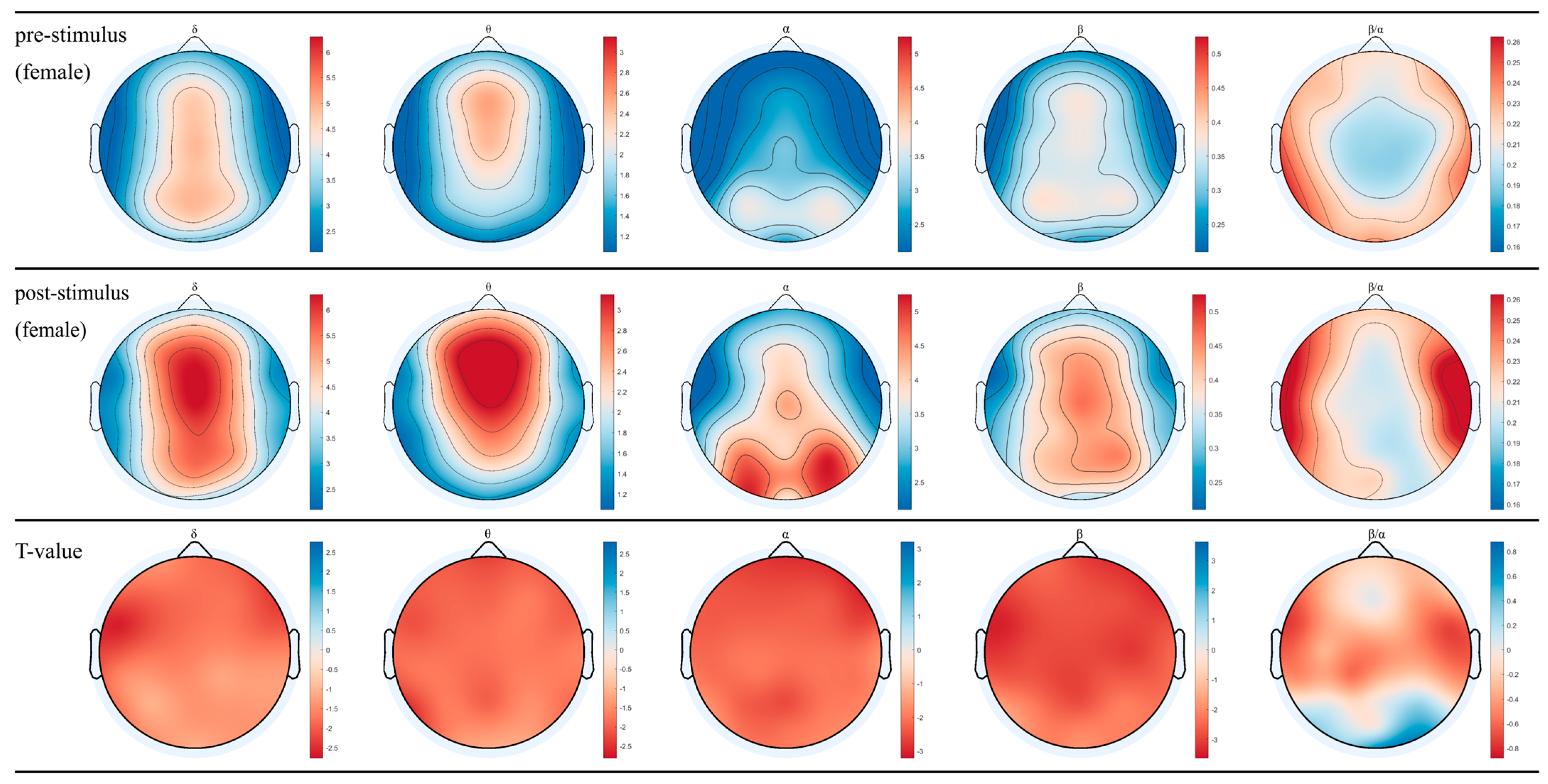
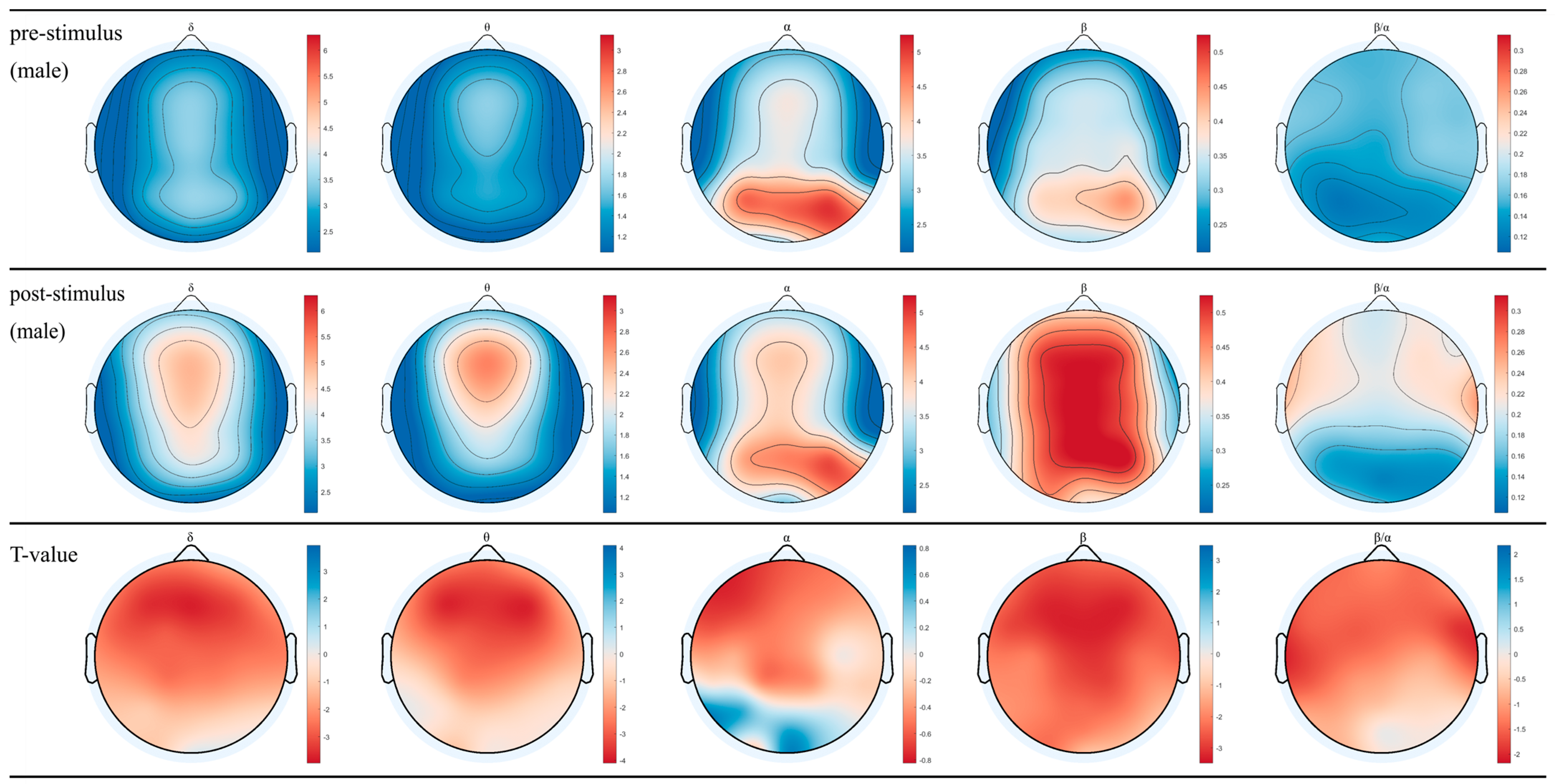
3.1. EEG for Female and Male after Inhaling DPEO
3.2. SH-SY5Y Cell Viability
3.3. Morphological Changes of SH-SY5Y Cells
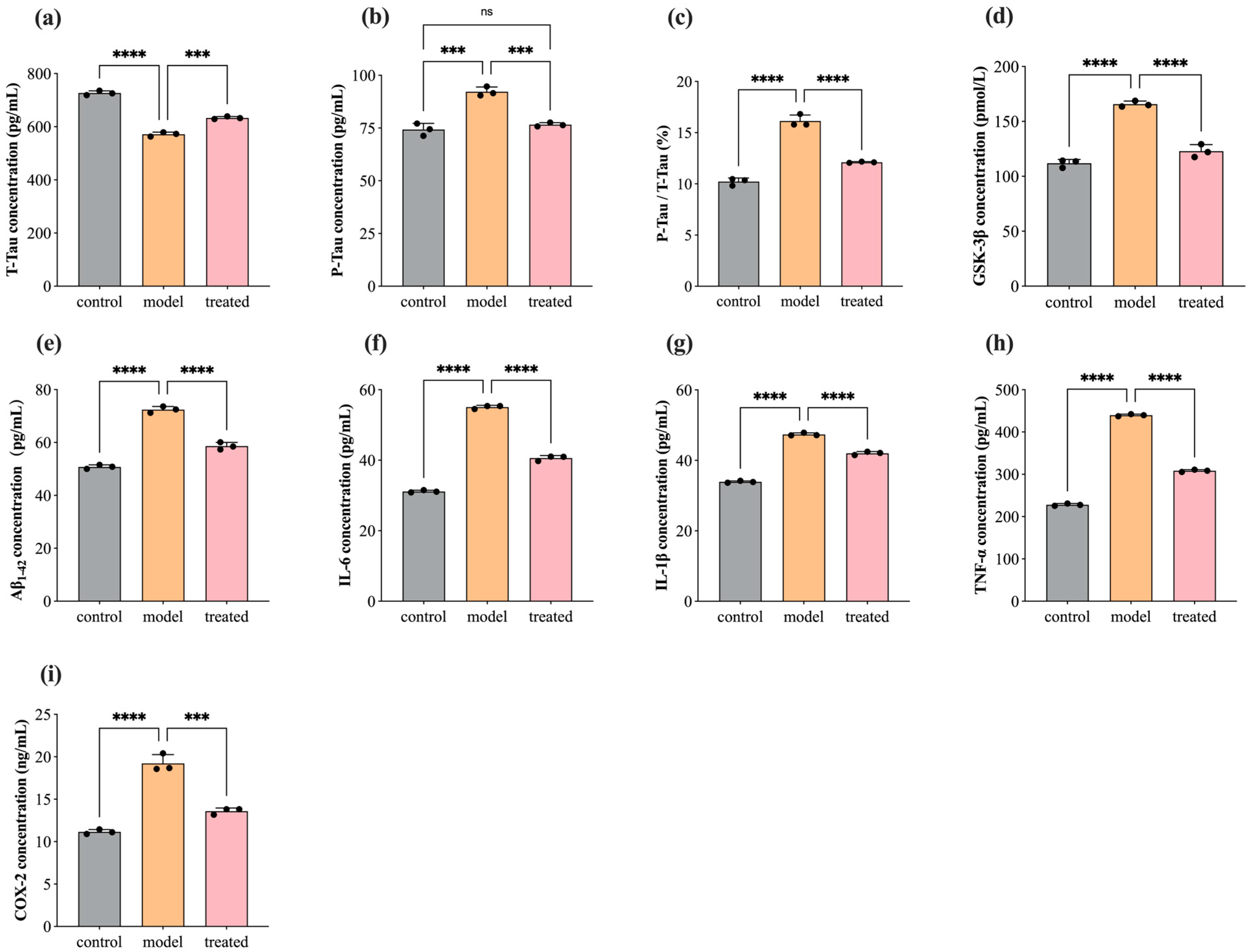
3.4. Protein Concentration Level of T-Tau, P-Tau, GSK-3β, Aβ1–42, COX-2, IL-1β, TNF-α, and IL-6
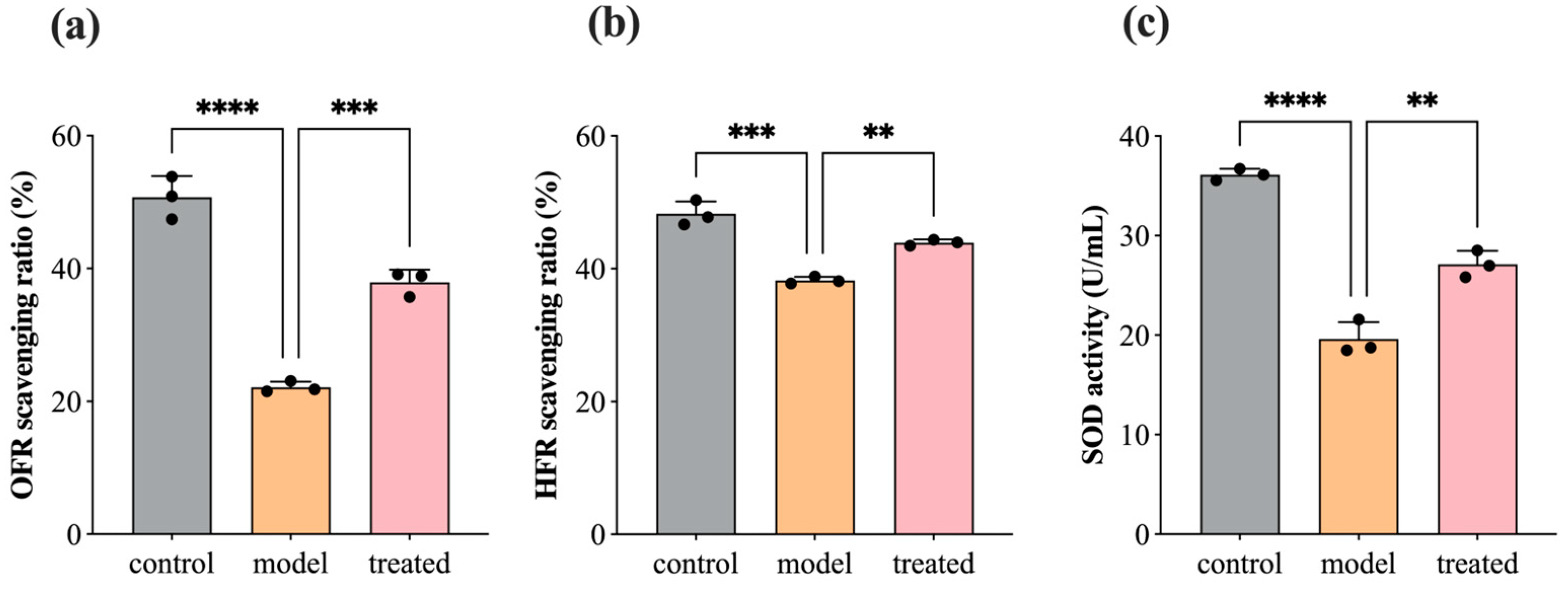
3.5. Antioxidant: SOD, OFR, and HFR
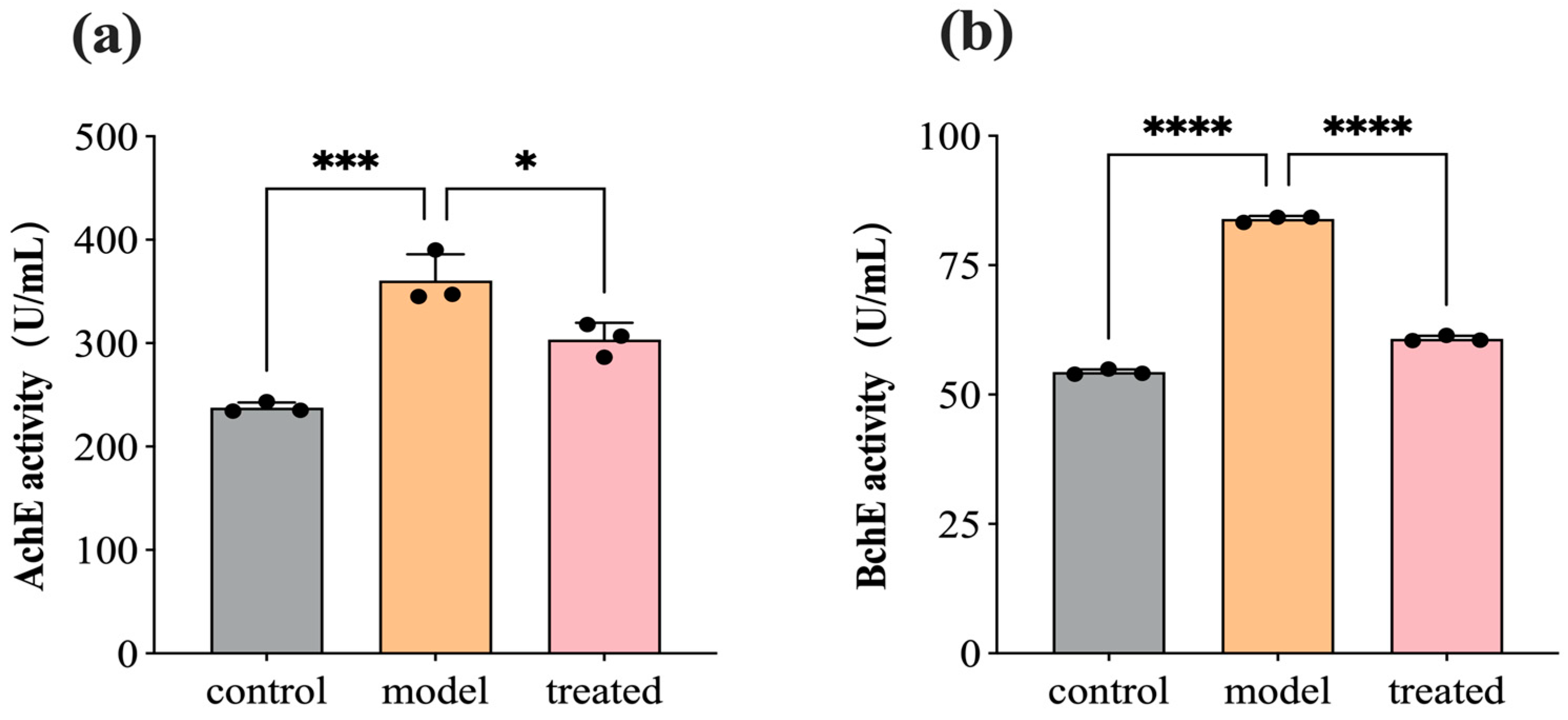
3.6. DPEO Inhibits Activity of AchE and BchE
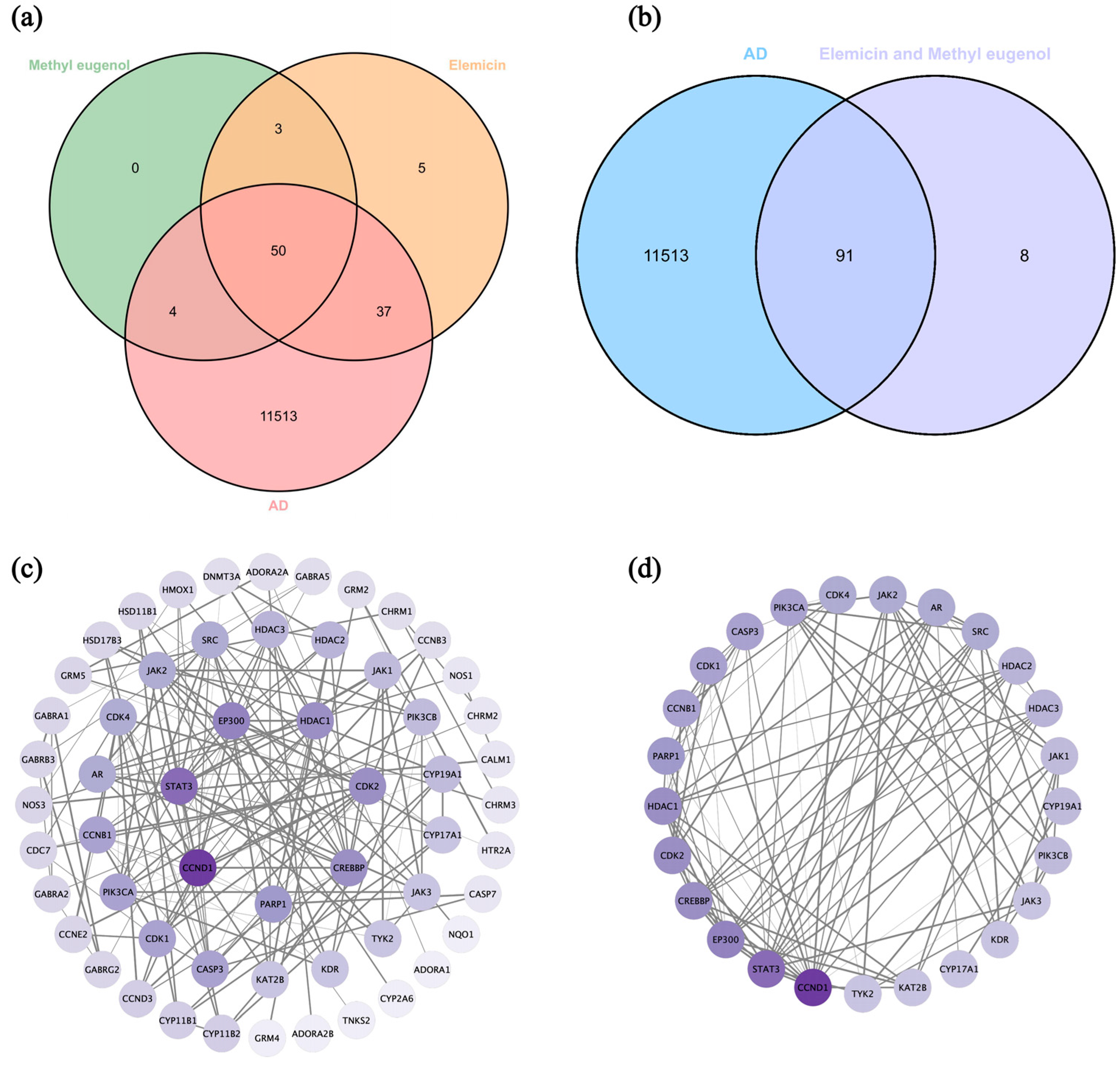
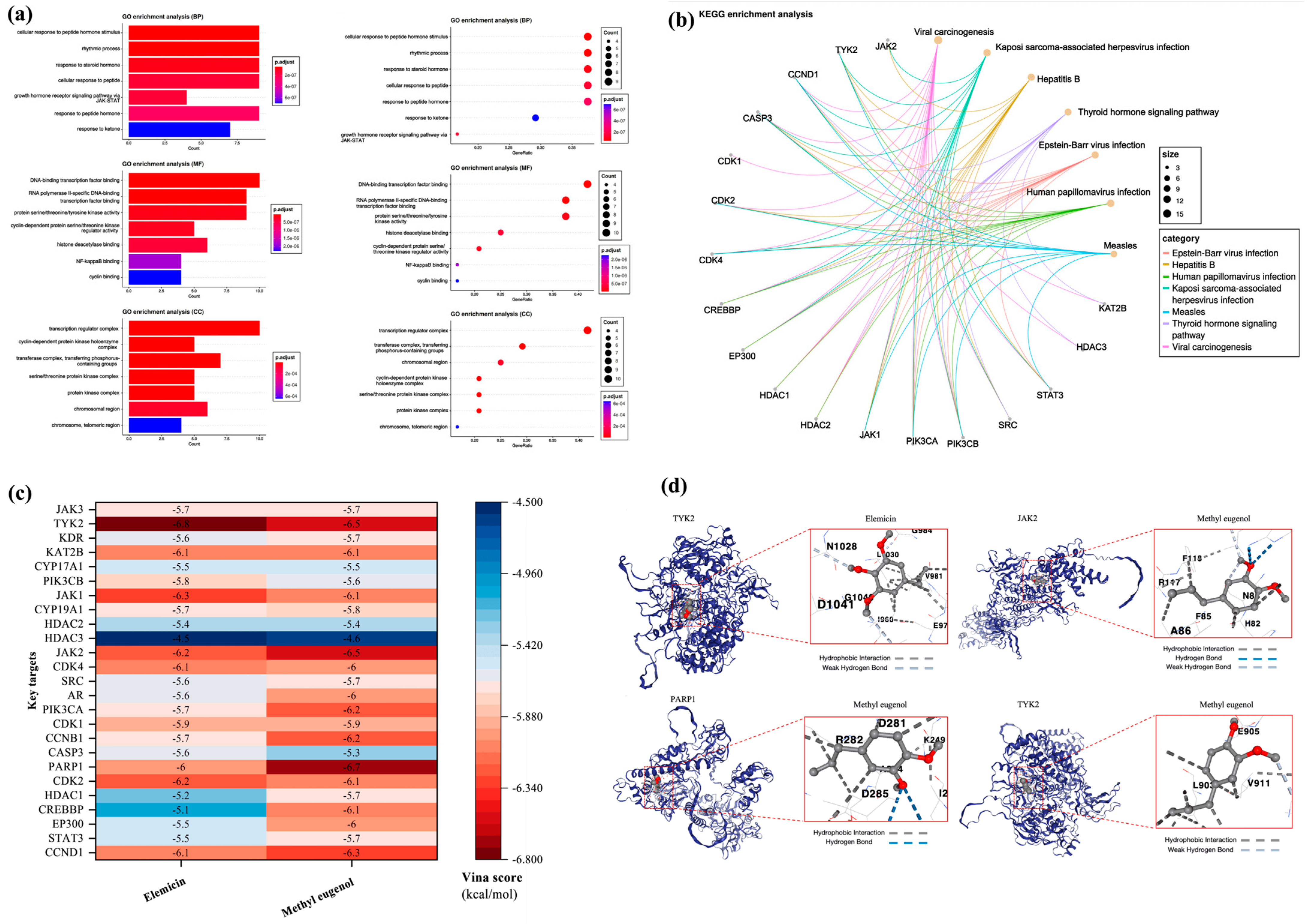
3.7. Network Pharmacology Analysis
3.7.1. Screen Targets of AD and Main Components of DPEO
3.7.2. Key Targets GO/KEGG Analysis and Molecular Docking
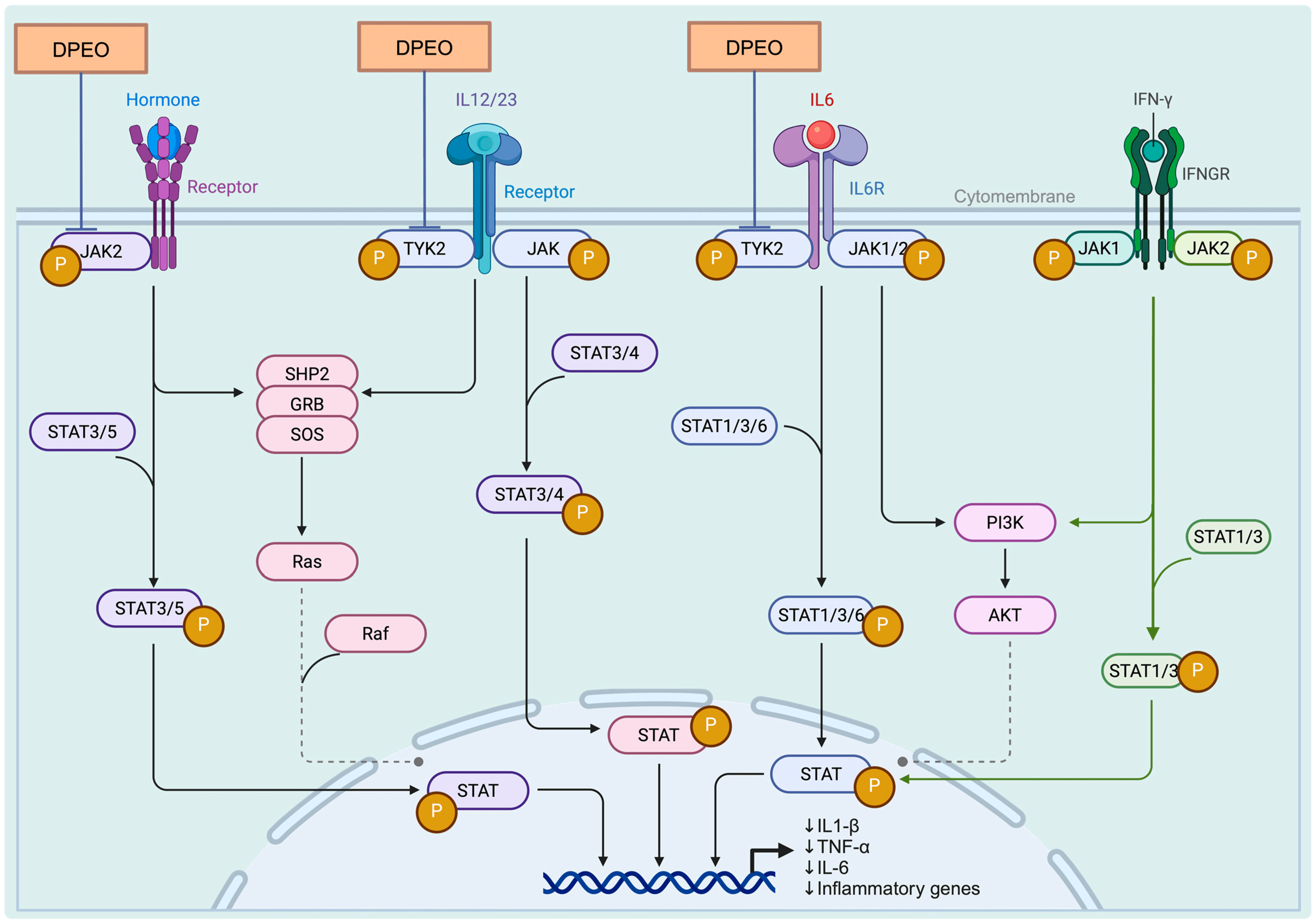
3.7.3. JAK-STAT Signaling Pathway
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Song, Z.; Yu, T.; Ge, C.; Shen, X.; Li, P.; Wu, J.; Tang, C.; Liu, T.; Zhang, D.; Li, S. Advantage effect of Dalbergia pinnata on wound healing and scar formation of burns. J. Ethnopharmacol. 2023, 317, 116872. [Google Scholar] [CrossRef]
- Zhou, W.; He, Y.; Lei, X.; Liao, L.; Fu, T.; Yuan, Y.; Huang, X.; Zou, L.; Liu, Y.; Ruan, R.; et al. Chemical composition and evaluation of antioxidant activities, antimicrobial, and anti-melanogenesis effect of the essential oils extracted from Dalbergia pinnata (Lour.) Prain. J. Ethnopharmacol. 2020, 254, 112731. [Google Scholar] [CrossRef]
- Ferdousi, F.; Kondo, S.; Sasaki, K.; Uchida, Y.; Ohkohchi, N.; Zheng, Y.-W.; Isoda, H. Microarray analysis of verbenalin-treated human amniotic epithelial cells reveals therapeutic potential for Alzheimer’s Disease. Aging 2020, 12, 5516–5538. [Google Scholar] [CrossRef] [PubMed]
- Mattar, J.M.; Majchrzak, M.; Iannucci, J.; Bartman, S.; Robinson, J.K.; Grammas, P. Sex Differences in Metabolic Indices and Chronic Neuroinflammation in Response to Prolonged High-Fat Diet in ApoE4 Knock-In Mice. Int. J. Mol. Sci. 2022, 23, 3921. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s, A. 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Katzmarski, N.; Ziegler-Waldkirch, S.; Scheffler, N.; Witt, C.; Abou-Ajram, C.; Nuscher, B.; Prinz, M.; Haass, C.; Meyer-Luehmann, M. Abeta oligomers trigger and accelerate Abeta seeding. Brain Pathol. 2020, 30, 36–45. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Maccioni, R.B.; Rojo, L.E.; Fernandez, J.A.; Kuljis, R.O. The role of neuroimmunomodulation in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2009, 1153, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.D.; Cohen, L.S.; Corbo, C.; Morozova, V.; ElIdrissi, A.; Phillips, G.; Kleiman, F.E. Hyperphosphorylation of Tau Associates with Changes in Its Function Beyond Microtubule Stability. Front. Cell. Neurosci. 2018, 12, 338. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015, 11, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Qiu, Q.; Zhang, H.; Chu, L.; Du, Y.; Zhang, J.; Zhou, C.; Liang, F.; Shi, S.; Wang, S.; et al. Concordance between the assessment of Abeta42, T-tau, and P-T181-tau in peripheral blood neuronal-derived exosomes and cerebrospinal fluid. Alzheimers Dement. 2019, 15, 1071–1080. [Google Scholar] [CrossRef]
- Wesseling, H.; Mair, W.; Kumar, M.; Schlaffner, C.N.; Tang, S.; Beerepoot, P.; Fatou, B.; Guise, A.J.; Cheng, L.; Takeda, S.; et al. Tau PTM Profiles Identify Patient Heterogeneity and Stages of Alzheimer’s Disease. Cell 2020, 183, 1699–1713. [Google Scholar] [CrossRef]
- Cortés, N.; Andrade, V.; Guzmán-Martínez, L.; Estrella, M.; Maccioni, R.B. Neuroimmune Tau Mechanisms: Their Role in the Progression of Neuronal Degeneration. Int. J. Mol. Sci. 2018, 19, 956. [Google Scholar] [CrossRef]
- Combs, C.K.; Karlo, J.C.; Kao, S.C.; Landreth, G.E. beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 2001, 21, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Blasko, I.; Veerhuis, R.; Stampfer-Kountchev, M.; Saurwein-Teissl, M.; Eikelenboom, P.; Grubeck-Loebenstein, B. Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1-40 and Abeta1-42 by human astrocytes. Neurobiol. Dis. 2000, 7, 682–689. [Google Scholar] [CrossRef]
- Liao, Y.F.; Wang, B.J.; Cheng, H.T.; Kuo, L.H.; Wolfe, M.S. Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J. Biol. Chem. 2004, 279, 49523–49532. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Zhong, Z.; Lindholm, K.; Berning, L.; Lee, W.; Lemere, C.; Staufenbiel, M.; Li, R.; Shen, Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J. Cell Biol. 2007, 178, 829–841. [Google Scholar] [CrossRef]
- Tarkowski, E.; Andreasen, N.; Tarkowski, A.K.; Blennow, K. Intrathecal inflammation precedes development of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.W.; Barnum, S.R.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimers Disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.G.; Zhu, S.G.; Jones, R.A.; Griffin, W.S.; Mrak, R.E. Interleukin-1 promotes expression and phosphorylation of neurofilament and tau proteins in vivo. Exp. Neurol. 2000, 163, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S.T. Interleukin-1 Mediates Pathological Effects of Microglia on Tau Phosphorylation and on Synaptophysin Synthesis in Cortical Neurons through a p38-MAPK Pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Rothaug, M.; Becker-Pauly, C.; Rose-John, S. The role of interleukin-6 signaling in nervous tissue. Biochim. Biophys. Acta 2016, 1863, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, P.; Jansen-West, K.R.; Beccard, A.; Ceballos-Diaz, C.; Levites, Y.; Verbeeck, C.; Zubair, A.C.; Dickson, D.W.; Golde, T.E.; Das, P. Massive gliosis induced by interleukin-6 suppresses Aβ deposition in vivo: Evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010, 24, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Ringheim, G.E.; Szczepanik, A.M.; Petko, W.; Burgher, K.L.; Zhu, S.Z.; Chao, C.C. Enhancement of beta-amyloid precursor protein transcription and expression by the soluble interleukin-6 receptor/interleukin-6 complex. Brain Res. Mol. Brain Res. 1998, 55, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, R.A.; Orellana, D.I.; Gonzalez-Billault, C.; Maccioni, R.B. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 2004, 295, 245–257. [Google Scholar] [CrossRef]
- Xiang, Z.; Ho, L.; Valdellon, J.; Borchelt, D.R.; Kelley, K.A.; Spielman, L.; Aisen, P.S.; Pasinetti, G.M. Cyclooxygenase (COX)-2 and cell cycle activity in a transgenic mouse model of Alzheimer’s Disease neuropathology. Neurobiol. Aging 2002, 23, 327–334. [Google Scholar] [CrossRef]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- D’Autréaux, B.; Tolédano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Jyrkkanen, H.K.; Levonen, A.L. Activation of stress signaling pathways by electrophilic oxidized and nitrated lipids. Free Radic. Biol. Med. 2012, 52, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Young, I.S.; Woodside, J.V. Antioxidants in health and disease. J. Clin. Pathol. 2001, 54, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.P.; Suescun, E.A.; Yang, S.Y. Effect of age-related lipid peroxidation on membrane fluidity and phospholipase A2: Modulation by dietary restriction. Mech. Aging Dev. 1992, 65, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Aruoma, O.I.; Neergheen, V.S.; Bahorun, T.; Jen, L.S. Free Radicals, Antioxidants and Diabetes: Embryopathy, Retinopathy, Neuropathy, Nephropathy and Cardiovascular Complications. Neuroembryol. Aging 2007, 4, 117–137. [Google Scholar] [CrossRef]
- Rammal, H.; Bouayed, J.; Soulimani, R. A direct relationship between aggressive behavior in the resident/intruder test and cell oxidative status in adult male mice. Eur. J. Pharmacol. 2010, 627, 173–176. [Google Scholar] [CrossRef]
- Kurutaş, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2015, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Sauer, H.; Wartenberg, M.; Hescheler, J. Reactive Oxygen Species as Intracellular Messengers During Cell Growth and Differentiation. Cell. Physiol. Biochem. 2001, 11, 173–186. [Google Scholar] [CrossRef]
- Grivennikova, V.G.; Vinogradov, A.D. Generation of superoxide by the mitochondrial Complex I. Biochim. Biophys. Acta 2006, 1757, 553–561. [Google Scholar] [CrossRef]
- Singh, P.P.; Mahadi, F.; Roy, A.; Sharma, P. Reactive oxygen species, reactive nitrogen species and antioxidants in etiopathogenesis of diabetes mellitus type-2. Indian J. Clin. Biochem. 2009, 24, 324–342. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. 1984, 219, 1–14. [Google Scholar] [CrossRef]
- Floyd, R.A.; Hensley, K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol. Aging 2002, 23, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Perry, G.; Pappolla, M.A.; Friedland, R.P.; Hirai, K.; Chiba, S.; Smith, M.A. Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome. J. Neuropathol. Exp. Neurol. 2000, 59, 1011–1017. [Google Scholar] [CrossRef]
- Gonzalez, F.J. Role of cytochromes P450 in chemical toxicity and oxidative stress: Studies with CYP2E1. Mutat. Res. 2005, 569, 101–110. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A.; Linn, S. DNA damage and oxygen radical toxicity. Science 1988, 240, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Calingasan, N.Y.; Yu, F.; Mauck, W.M.; Toidze, M.; Almeida, C.G.; Takahashi, R.H.; Carlson, G.A.; Flint Beal, M.; Lin, M.T.; et al. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J. Neurochem. 2004, 89, 1308–1312. [Google Scholar] [CrossRef]
- Melov, S.; Adlard, P.A.; Morten, K.; Johnson, F.; Golden, T.R.; Hinerfeld, D.; Schilling, B.; Mavros, C.; Masters, C.L.; Volitakis, I.; et al. Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS ONE 2007, 2, e536. [Google Scholar] [CrossRef]
- Esposito, L.; Raber, J.; Kekonius, L.; Yan, F.; Yu, G.Q.; Bien-Ly, N.; Puolivali, J.; Scearce-Levie, K.; Masliah, E.; Mucke, L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J. Neurosci. 2006, 26, 5167–5179. [Google Scholar] [CrossRef] [PubMed]
- An, F.; Bai, Y.; Xuan, X.; Bian, M.; Zhang, G.; Wei, C. 1,8-Cineole Ameliorates Advanced Glycation End Products-Induced Alzheimer’s Disease-like Pathology In Vitro and In Vivo. Molecules 2022, 27, 3913. [Google Scholar] [CrossRef] [PubMed]
- Hernández, F.; Barreda, E.G.d.; Fuster-Matanzo, A.; Lucas, J.J.; Ávila, J. GSK3: A possible link between beta amyloid peptide and tau protein. Exp. Neurol. 2010, 223, 322–325. [Google Scholar] [CrossRef]
- Engel, T.; Hernandez, F.; Avila, J.; Lucas, J.J. Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J. Neurosci. 2006, 26, 5083–5090. [Google Scholar] [CrossRef] [PubMed]
- Sayas, C.L.; Avila, J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.; Perry, E.K.; McKeith, I.G. Key Papers in Geriatric Psychiatry. Series Editor: Alistair Burns Correlation of Cholinergic Abnormalities with Senile Plaques and Mental Test Scores in Senile Dementia. E. Perry, B. Tomlinson, G. Blessed, K. Bergmann, P. Gibson and R. Perry, British Medical Journal (1978)2, 1457–1459. Int. J. Geriatr. Psychiatry 1996, 11, 765–771. [Google Scholar]
- Whitehouse, P.J.; Price, D.L.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer disease: Evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann. Neurol. 1981, 10, 122–126. [Google Scholar] [CrossRef]
- Wilcock, G.K.; Esiri, M.M.; Bowen, D.M.; Smith, C.C. Alzheimer’s disease. Correlation of cortical choline acetyltransferase activity with the severity of dementia and histological abnormalities. J. Neurol. Sci. 1982, 57, 407–417. [Google Scholar] [CrossRef]
- Muir, J.L. Acetylcholine, aging, and Alzheimer’s disease. Pharmacol. Biochem. Behav. 1997, 56, 687–696. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef]
- Francis, P.T. The interplay of neurotransmitters in Alzheimer’s disease. CNS Spectr. 2005, 10, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Kihara, T.; Shimohama, S. Alzheimer’s disease and acetylcholine receptors. Acta Neurobiol. Exp. 2004, 64, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Sberna, G.; Saez-Valero, J.; Beyreuther, K.; Masters, C.L.; Small, D.H. The amyloid beta-protein of Alzheimer’s disease increases acetylcholinesterase expression by increasing intracellular calcium in embryonal carcinoma P19 cells. J. Neurochem. 1997, 69, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; Reid, G.A.; Martin, E. Biochemical and histochemical comparison of cholinesterases in normal and Alzheimer brain tissues. Curr. Alzheimer Res. 2010, 7, 386–400. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M.; Geula, C. Butyrylcholinesterase reactivity differentiates the amyloid plaques of aging from those of dementia. Ann. Neurol. 1994, 36, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, I.R.; Maxwell, S.P.; Reid, G.A.; Cash, M.K.; DeBay, D.R.; Darvesh, S. Quantification of Butyrylcholinesterase Activity as a Sensitive and Specific Biomarker of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Guillozet, A.L.; Smiley, J.F.; Mash, D.C.; Mesulam, M.M. Butyrylcholinesterase in the life cycle of amyloid plaques. Ann. Neurol. 1997, 42, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; Cash, M.K.; Reid, G.A.; Martin, E.; Mitnitski, A.; Geula, C. Butyrylcholinesterase Is Associated with β-Amyloid Plaques in the Transgenic APPSWE/PSEN1dE9 Mouse Model of Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2012, 71, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.A.; Darvesh, S. Butyrylcholinesterase-knockout reduces brain deposition of fibrillar beta-amyloid in an Alzheimer mouse model. Neuroscience 2015, 298, 424–435. [Google Scholar] [CrossRef]
- Darvesh, S.; Reid, G.A. Reduced fibrillar beta-amyloid in subcortical structures in a butyrylcholinesterase-knockout Alzheimer disease mouse model. Chem. Biol. Interact. 2016, 259, 307–312. [Google Scholar] [CrossRef]
- He, X.; Qin, S.; Yu, G.; Zhang, S.; Yi, F. Study on the Effect of Dalbergia pinnata (Lour.) Prain Essential Oil on Electroencephalography upon Stimulation with Different Auditory Effects. Molecules 2024, 29, 1584. [Google Scholar] [CrossRef]
- Arjmandi-Rad, S.; Vestergaard Nieland, J.D.; Goozee, K.G.; Vaseghi, S. The effects of different acetylcholinesterase inhibitors on EEG patterns in patients with Alzheimer’s disease: A systematic review. Neurol. Sci. 2024, 45, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Scuteri, D.; Morrone, L.A.; Rombola, L.; Avato, P.R.; Bilia, A.R.; Corasaniti, M.T.; Sakurada, S.; Sakurada, T.; Bagetta, G. Aromatherapy and Aromatic Plants for the Treatment of Behavioural and Psychological Symptoms of Dementia in Patients with Alzheimer’s Disease: Clinical Evidence and Possible Mechanisms. Evid. Based Complement. Altern. Med. 2017, 2017, 9416305. [Google Scholar] [CrossRef]
- Koyama, S.; Heinbockel, T. The Effects of Essential Oils and Terpenes in Relation to Their Routes of Intake and Application. Int. J. Mol. Sci. 2020, 21, 1558. [Google Scholar] [CrossRef]
- Zhao, Z.-Y.; Zhang, Y.; Zhang, Y.-H.; Wei, X.-Z.; Wang, H.; Zhang, M.; Yang, Z.-J.; Zhang, C.-H. The protective underlying mechanisms of Schisandrin on SH-SY5Y cell model of Alzheimer’s disease. J. Toxicol. Environ. Health Part A 2019, 82, 1019–1026. [Google Scholar] [CrossRef]
- Meraz-Rios, M.A.; Toral-Rios, D.; Franco-Bocanegra, D.; Villeda-Hernandez, J.; Campos-Pena, V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef]
- Yang, X.; Chang, Y.; Wei, W. Emerging role of targeting macrophages in rheumatoid arthritis: Focus on polarization, metabolism and apoptosis. Cell Prolif. 2020, 53, e12854. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.-W.; Du, S.-S.; Fang, Y.; Zhang, W. Targeting TYK2 for Fighting Diseases: Recent Advance of TYK2 Inhibitors. Curr. Med. Chem. 2023, 31, 2900–2920. [Google Scholar]
- Wang, L.; Liang, C.; Li, F.; Guan, D.; Wu, X.; Fu, X.; Lu, A.; Zhang, G. PARP1 in Carcinomas and PARP1 Inhibitors as Antineoplastic Drugs. Int. J. Mol. Sci. 2017, 18, 2111. [Google Scholar] [CrossRef]
- Boluda, J.C.H.; Gómez, M.H.; Pérez, A.R. Inhibidores de JAK2. Med. Clin. 2016, 147, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, A. Dementia in 2014. Towards early diagnosis in Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 69–70. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Wang, L.; Yu, H.; Chen, D.; Zhu, W.; Sun, C. Pharmacological Effects of Polyphenol Phytochemicals on the JAK-STAT Signaling Pathway. Front. Pharmacol. 2021, 12, 716672. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Name | Website |
|---|---|---|
| Database | TCMSP | https://www.tcmsp-e.com/#/database (accessed on 4 December 2023) |
| PubChem | https://pubchem.ncbi.nlm.nih.gov (accessed on 4 December 2023) | |
| Uniprot | https://www.uniprot.org (accessed on 10 December 2023) | |
| Swiss Target Prediction | http://www.swisstargetprediction.ch (accessed on 10 December 2023) | |
| GeneCards | https://www.genecards.org (accessed on 15 December 2023) | |
| STRING | https://string-db.org/ (accessed on 15 December 2023) | |
| KEGG | https://www.kegg.jp/ (accessed on 15 December 2023) | |
| AlphaFold | https://alphafold.ebi.ac.uk (accessed on 17 December 2023) | |
| Analysis platform | CB-Dock2 | https://cadd.labshare.cn/cb-dock2/index.php (accessed on 19 December 2023) |
| Hiplot | https://hiplot.com.cn/home/index.html (accessed on 19 December 2023) |
| NO. | Key Targets | Name | Uniprot ID | Degree |
|---|---|---|---|---|
| 1 | CCND1 | Cyclin D1 | P24385 | 19 |
| 2 | STAT3 | Signal Transducer And Activator Of Transcription 3 | P40763 | 15 |
| 3 | EP300 | E1A Binding Protein P300 | Q09472 | 13 |
| 4 | CREBBP | CREB Binding Protein | Q92793 | 12 |
| 5 | HDAC1 | Histone Deacetylase 1 | Q13547 | 12 |
| 6 | CDK2 | Cyclin Dependent Kinase 2 | P24941 | 12 |
| 7 | PARP1 | Poly (ADP-Ribose) Polymerase 1 | P09874 | 11 |
| 8 | CASP3 | Caspase 3 | P42574 | 10 |
| 9 | CCNB1 | Cyclin B1 | P14635 | 10 |
| 10 | CDK1 | Cyclin Dependent Kinase 1 | P06493 | 10 |
| 11 | PIK3CA | Phosphatidylinositol-4, 5-Bisphosphate 3-Kinase Catalytic Subunit Alpha | P42336 | 10 |
| 12 | AR | Androgen Receptor | P10275 | 9 |
| 13 | SRC | SRC Proto-Oncogene, Non-Receptor Tyrosine Kinase | P12931 | 9 |
| 14 | CDK4 | Cyclin Dependent Kinase 4 | P11802 | 9 |
| 15 | JAK2 | Janus Kinase 2 | O60674 | 9 |
| 16 | HDAC3 | Histone Deacetylase 3 | O15379 | 8 |
| 17 | HDAC2 | Histone Deacetylase 2 | Q92769 | 8 |
| 18 | CYP19A1 | Cytochrome P450 Family 19 Subfamily A Member 1 | P11511 | 7 |
| 19 | JAK1 | Janus Kinase 1 | P23458 | 7 |
| 20 | PIK3CB | Phosphatidylinositol-4, 5-Bisphosphate 3-Kinase Catalytic Subunit Beta | P42338 | 7 |
| 21 | CYP17A1 | Cytochrome P450 Family 17 Subfamily A Member 1 | P05093 | 6 |
| 22 | KAT2B | Lysine Acetyltransferase 2B | Q92831 | 6 |
| 23 | KDR | Kinase Insert Domain Receptor | P35968 | 6 |
| 24 | TYK2 | Tyrosine Kinase 2 | P29597 | 6 |
| 25 | JAK3 | Janus Kinase 3 | P52333 | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, S.; Fang, J.; He, X.; Yu, G.; Yi, F.; Zhu, G. The Therapeutic Potential of Dalbergia pinnata (Lour.) Prain Essential Oil in Alzheimer’s Disease: EEG Signal Analysis In Vivo, SH-SY5Y Cell Model In Vitro, and Network Pharmacology. Biology 2024, 13, 544. https://doi.org/10.3390/biology13070544
Qin S, Fang J, He X, Yu G, Yi F, Zhu G. The Therapeutic Potential of Dalbergia pinnata (Lour.) Prain Essential Oil in Alzheimer’s Disease: EEG Signal Analysis In Vivo, SH-SY5Y Cell Model In Vitro, and Network Pharmacology. Biology. 2024; 13(7):544. https://doi.org/10.3390/biology13070544
Chicago/Turabian StyleQin, Sheng, Jiayi Fang, Xin He, Genfa Yu, Fengping Yi, and Guangyong Zhu. 2024. "The Therapeutic Potential of Dalbergia pinnata (Lour.) Prain Essential Oil in Alzheimer’s Disease: EEG Signal Analysis In Vivo, SH-SY5Y Cell Model In Vitro, and Network Pharmacology" Biology 13, no. 7: 544. https://doi.org/10.3390/biology13070544
APA StyleQin, S., Fang, J., He, X., Yu, G., Yi, F., & Zhu, G. (2024). The Therapeutic Potential of Dalbergia pinnata (Lour.) Prain Essential Oil in Alzheimer’s Disease: EEG Signal Analysis In Vivo, SH-SY5Y Cell Model In Vitro, and Network Pharmacology. Biology, 13(7), 544. https://doi.org/10.3390/biology13070544