
Inflammation as a Sex-Specific Mediator in the Relationship between Maternal and Offspring Obesity in C57Bl/6J Mice

 , , and
, , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Blood and Tissue Collection
2.3. Plasma Analyses
2.4. Hypothalamic Gene Expression Analyses
2.5. In Vivo Physiology Analyses
2.6. Statistical Analysis and Data Presentation
3. Results
3.1. Dam Characteristics
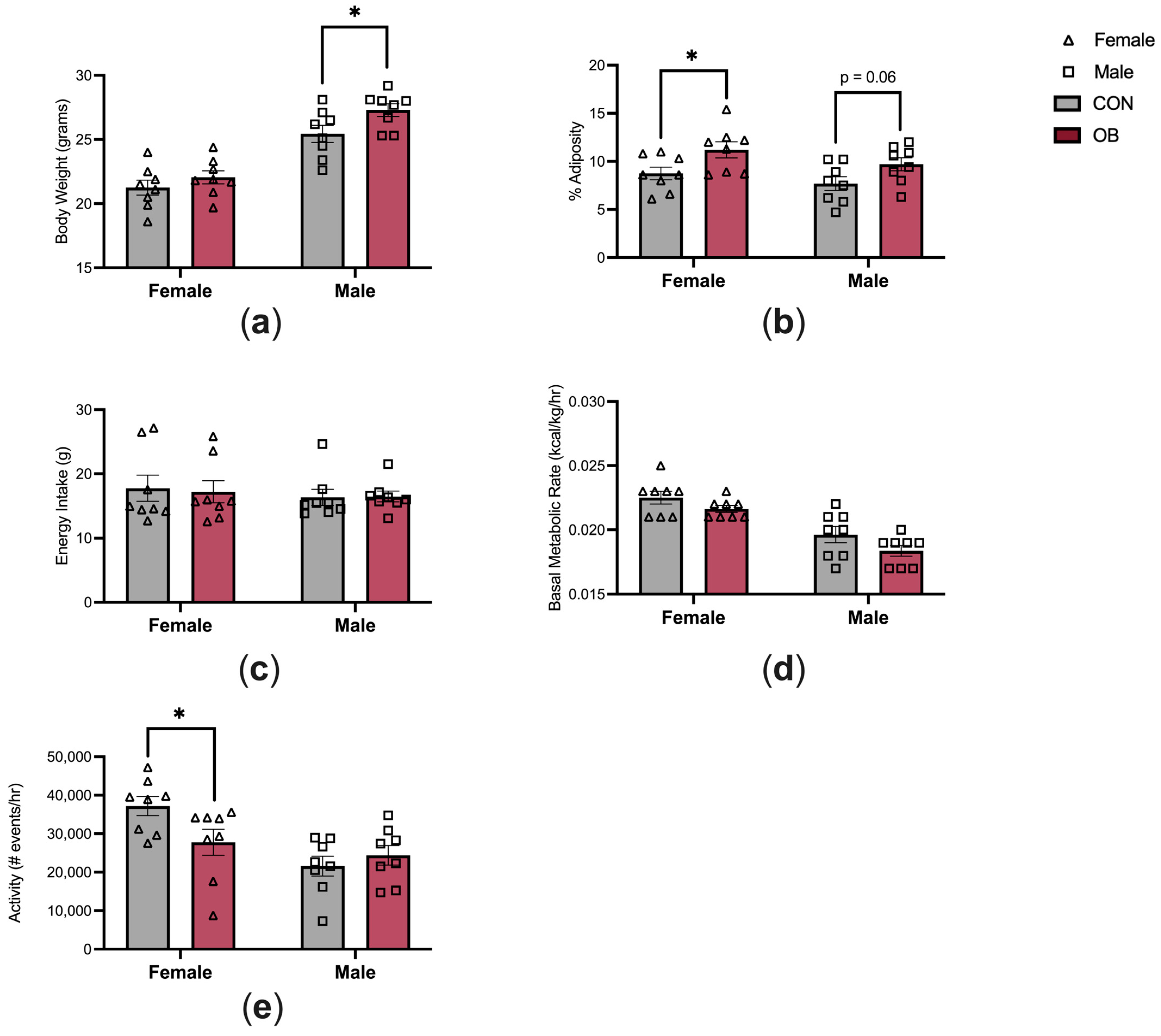
3.2. Offspring Phenotype
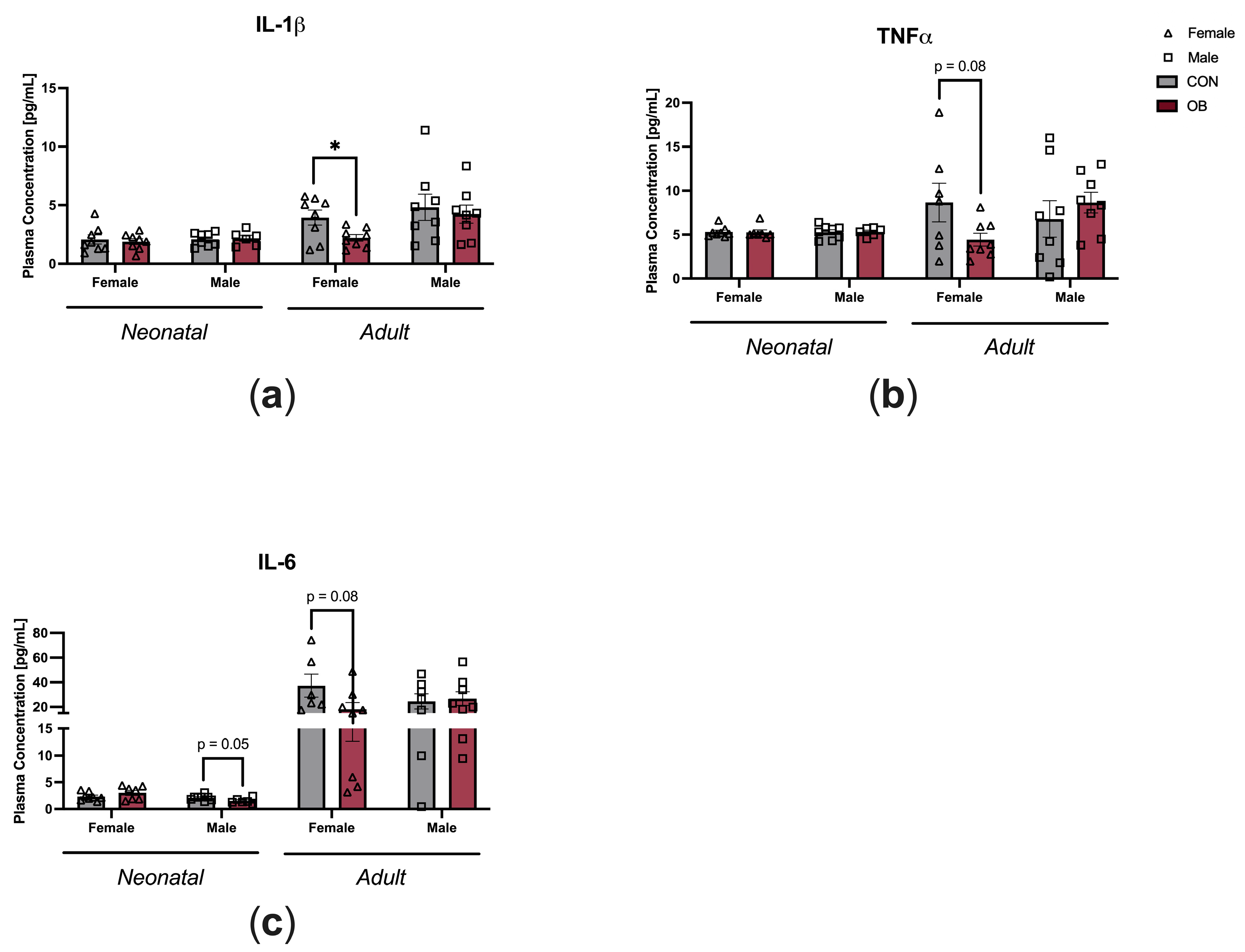
3.3. Offspring Inflammation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Catalano, P.M.; Shankar, K. Obesity and pregnancy: Mechanisms of short term and long term adverse consequences for mother and child. BMJ 2017, 356, j1. [Google Scholar] [CrossRef] [PubMed]
- Sagi-Dain, L. Obesity in Pregnancy: ACOG Practice Bulletin, Number 230. Obstet. Gynecol. 2021, 138, 489. [Google Scholar] [CrossRef] [PubMed]
- Schoonejans, J.M.; Ozanne, S.E. Developmental programming by maternal obesity: Lessons from animal models. Diabet. Med. 2021, 38, e14694. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.J.; Powell, T.L.; Barrett, E.S.; Hardy, D.B. Developmental origins of metabolic diseases. Physiol. Rev. 2021, 101, 739–795. [Google Scholar] [CrossRef] [PubMed]
- Dhana, K.; Haines, J.; Liu, G.; Zhang, C.; Wang, X.; Field, A.E.; Chavarro, J.E.; Sun, Q. Association between maternal adherence to healthy lifestyle practices and risk of obesity in offspring: Results from two prospective cohort studies of mother-child pairs in the United States. BMJ 2018, 362, k2486. [Google Scholar] [CrossRef] [PubMed]
- Kulhanek, D.; Abrahante Llorens, J.E.; Buckley, L.; Tkac, I.; Rao, R.; Paulsen, M.E. Female and male C57BL/6J offspring exposed to maternal obesogenic diet develop altered hypothalamic energy metabolism in adulthood. Am. J. Physiol. Endocrinol. Metab. 2022, 323, E448–E466. [Google Scholar] [CrossRef] [PubMed]
- Segovia, S.A.; Vickers, M.H.; Reynolds, C.M. The impact of maternal obesity on inflammatory processes and consequences for later offspring health outcomes. J. Dev. Orig. Health Dis. 2017, 8, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Kulhanek, D.; Rao, R.B.; Paulsen, M.E. Excess sucrose intake during pregnancy programs fetal brain glucocorticoid receptor expression in female but not male C57Bl/6J mice. Obes. Sci. Pract. 2021, 7, 462–472. [Google Scholar] [CrossRef]
- Kulhanek, D.; Weigel, R.; Paulsen, M.E. Maternal High-Fat-High-Carbohydrate Diet-Induced Obesity Is Associated with Increased Appetite in Peripubertal Male but Not Female C57Bl/6J Mice. Nutrients 2020, 12, 2919. [Google Scholar] [CrossRef]
- Menting, M.D.; Mintjens, S.; van de Beek, C.; Frick, C.J.; Ozanne, S.E.; Limpens, J.; Roseboom, T.J.; Hooijmans, C.R.; van Deutekom, A.W.; Painter, R.C. Maternal obesity in pregnancy impacts offspring cardiometabolic health: Systematic review and meta-analysis of animal studies. Obes. Rev. 2019, 20, 675–685. [Google Scholar] [CrossRef]
- Juul, S.E.; Comstock, B.A.; Heagerty, P.J.; Mayock, D.E.; Goodman, A.M.; Hauge, S.; Gonzalez, F.; Wu, Y.W. High-Dose Erythropoietin for Asphyxia and Encephalopathy (HEAL): A Randomized Controlled Trial—Background, Aims, and Study Protocol. Neonatology 2018, 113, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Juul, S.E.; Comstock, B.A.; Wadhawan, R.; Mayock, D.E.; Courtney, S.E.; Robinson, T.; Ahmad, K.A.; Bendel-Stenzel, E.; Baserga, M.; LaGamma, E.F.; et al. A Randomized Trial of Erythropoietin for Neuroprotection in Preterm Infants. N. Engl. J. Med. 2020, 382, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Gali Ramamoorthy, T.; Begum, G.; Harno, E.; White, A. Developmental programming of hypothalamic neuronal circuits: Impact on energy balance control. Front. Neurosci. 2015, 9, 126. [Google Scholar] [CrossRef] [PubMed]
- Ziko, I.; De Luca, S.; Dinan, T.; Barwood, J.M.; Sominsky, L.; Cai, G.; Kenny, R.; Stokes, L.; Jenkins, T.A.; Spencer, S.J. Neonatal overfeeding alters hypothalamic microglial profiles and central responses to immune challenge long-term. Brain Behav. Immun. 2014, 41, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Skowronski, A.A.; Leibel, R.L.; LeDuc, C.A. Neurodevelopmental Programming of Adiposity: Contributions to Obesity Risk. Endocr. Rev. 2024, 45, 253–280. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Valdearcos, M.; Douglass, J.D.; Robblee, M.M.; Dorfman, M.D.; Stifler, D.R.; Bennett, M.L.; Gerritse, I.; Fasnacht, R.; Barres, B.A.; Thaler, J.P.; et al. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab. 2017, 26, 185–197.e3. [Google Scholar] [CrossRef] [PubMed]
- Valdearcos, M.; Robblee, M.M.; Benjamin, D.I.; Nomura, D.K.; Xu, A.W.; Koliwad, S.K. Microglia Dictate the Impact of Saturated Fat Consumption on Hypothalamic Inflammation and Neuronal Function. Cell Rep. 2014, 9, 2124–2138. [Google Scholar] [CrossRef] [PubMed]
- De Bona Schraiber, R.; De Mello, A.H.; Garcez, M.L.; De Bem Silveira, G.; Zacaron, R.P.; De Souza Goldim, M.P.; Budni, J.; Silveira, P.C.L.; Petronilho, F.; Ferreira, G.K.; et al. Diet-induced obesity causes hypothalamic neurochemistry alterations in Swiss mice. Metab. Brain Dis. 2019, 34, 565–573. [Google Scholar] [CrossRef]
- Seong, J.; Kang, J.Y.; Sun, J.S.; Kim, K.W. Hypothalamic inflammation and obesity: A mechanistic review. Arch. Pharmacal Res. 2019, 42, 383–392. [Google Scholar] [CrossRef]
- Bae-Gartz, I.; Janoschek, R.; Kloppe, C.S.; Vohlen, C.; Roels, F.; Oberthur, A.; Alejandre Alcazar, M.A.; Lippach, G.; Muether, P.S.; Dinger, K.; et al. Running Exercise in Obese Pregnancies Prevents IL-6 Trans-signaling in Male Offspring. Med. Sci. Sports Exerc. 2016, 48, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Rother, E.; Kuschewski, R.; Alcazar, M.A.; Oberthuer, A.; Bae-Gartz, I.; Vohlen, C.; Roth, B.; Dotsch, J. Hypothalamic JNK1 and IKKbeta activation and impaired early postnatal glucose metabolism after maternal perinatal high-fat feeding. Endocrinology 2012, 153, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Bae-Gartz, I.; Janoschek, R.; Breuer, S.; Schmitz, L.; Hoffmann, T.; Ferrari, N.; Branik, L.; Oberthuer, A.; Kloppe, C.S.; Appel, S.; et al. Maternal Obesity Alters Neurotrophin-Associated MAPK Signaling in the Hypothalamus of Male Mouse Offspring. Front. Neurosci. 2019, 13, 962. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, S.; Luo, X.; Zhang, C. Emerging role of hypothalamus in the metabolic regulation in the offspring of maternal obesity. Front. Nutr. 2023, 10, 1094616. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Glendining, K.A.; Grattan, D.R.; Jasoni, C.L. Maternal obesity leads to increased proliferation and numbers of astrocytes in the developing fetal and neonatal mouse hypothalamus. Int. J. Dev. Neurosci. 2016, 53, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Gisslen, T.; Ennis, K.; Bhandari, V.; Rao, R. Recurrent hypoinsulinemic hyperglycemia in neonatal rats increases PARP-1 and NF-kappaB expression and leads to microglial activation in the cerebral cortex. Pediatr. Res. 2015, 78, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Gisslen, T.; Singh, G.; Georgieff, M.K. Fetal inflammation is associated with persistent systemic and hippocampal inflammation and dysregulation of hippocampal glutamatergic homeostasis. Pediatr. Res. 2019, 85, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Segura, B.J.; Georgieff, M.K.; Gisslen, T. Fetal inflammation induces acute immune tolerance in the neonatal rat hippocampus. J. Neuroinflammation 2021, 18, 69. [Google Scholar] [CrossRef]
- Rosario, F.J.; Kanai, Y.; Powell, T.L.; Jansson, T. Increased placental nutrient transport in a novel mouse model of maternal obesity with fetal overgrowth. Obesity 2015, 23, 1663–1670. [Google Scholar] [CrossRef]
- Hahn, W.S.; Kuzmicic, J.; Burrill, J.S.; Donoghue, M.A.; Foncea, R.; Jensen, M.D.; Lavandero, S.; Arriaga, E.A.; Bernlohr, D.A. Proinflammatory cytokines differentially regulate adipocyte mitochondrial metabolism, oxidative stress, and dynamics. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1033–E1045. [Google Scholar] [CrossRef]
- Beilharz, J.E.; Maniam, J.; Morris, M.J. Short-term exposure to a diet high in fat and sugar, or liquid sugar, selectively impairs hippocampal-dependent memory, with differential impacts on inflammation. Behav. Brain Res. 2016, 306, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bobbo, V.C.; Engel, D.F.; Jara, C.P.; Mendes, N.F.; Haddad-Tovolli, R.; Prado, T.P.; Sidarta-Oliveira, D.; Morari, J.; Velloso, L.A.; Araujo, E.P. Interleukin-6 actions in the hypothalamus protects against obesity and is involved in the regulation of neurogenesis. J. Neuroinflammation 2021, 18, 192. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.A. Role of interleukins in obesity: Implications for metabolic disease. Trends Endocrinol. Metab. 2014, 25, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Kammoun, H.L.; Kraakman, M.J.; Febbraio, M.A. Adipose tissue inflammation in glucose metabolism. Rev. Endocr. Metab. Disord. 2014, 15, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Young, S.L.; Grattan, D.R.; Jasoni, C.L. Obesity during pregnancy disrupts placental morphology, cell proliferation, and inflammation in a sex-specific manner across gestation in the mouse. Biol. Reprod. 2014, 90, 130. [Google Scholar] [CrossRef]
- Liang, X.; Yang, Q.; Fu, X.; Rogers, C.J.; Wang, B.; Pan, H.; Zhu, M.J.; Nathanielsz, P.W.; Du, M. Maternal obesity epigenetically alters visceral fat progenitor cell properties in male offspring mice. J. Physiol. 2016, 594, 4453–4466. [Google Scholar] [CrossRef] [PubMed]
- Sethi, J.K.; Hotamisligil, G.S. Metabolic Messengers: Tumour necrosis factor. Nat. Metab. 2021, 3, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Bouret, S.G. Nutritional programming of hypothalamic development: Critical periods and windows of opportunity. Int. J. Obes. Suppl. 2012, 2, S19–S24. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef]
- Cero, C.; Razzoli, M.; Han, R.; Sahu, B.S.; Patricelli, J.; Guo, Z.; Zaidman, N.A.; Miles, J.M.; O’Grady, S.M.; Bartolomucci, A. The neuropeptide TLQP-21 opposes obesity via C3aR1-mediated enhancement of adrenergic-induced lipolysis. Mol. Metab. 2017, 6, 148–158. [Google Scholar] [CrossRef]
- Nie, Y.; Gavin, T.P.; Kuang, S. Measurement of Resting Energy Metabolism in Mice Using Oxymax Open Circuit Indirect Calorimeter. Bio Protoc. 2015, 5, e1602. [Google Scholar] [CrossRef] [PubMed]
- Ellulu, M.S.; Khaza’ai, H.; Rahmat, A.; Patimah, I.; Abed, Y. Obesity can predict and promote systemic inflammation in healthy adults. Int. J. Cardiol. 2016, 215, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Wondmkun, Y.T. Obesity, Insulin Resistance, and Type 2 Diabetes: Associations and Therapeutic Implications. Diabetes Metab. Syndr. Obes. 2020, 13, 3611–3616. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, K.; Rohde, T.; Asp, S.; Schjerling, P.; Pedersen, B.K. Pro- and anti-inflammatory cytokine balance in strenuous exercise in humans. J. Physiol. 1999, 515 Pt 1, 287–291. [Google Scholar] [CrossRef]
- Nash, D.; Hughes, M.G.; Butcher, L.; Aicheler, R.; Smith, P.; Cullen, T.; Webb, R. IL-6 signaling in acute exercise and chronic training: Potential consequences for health and athletic performance. Scand. J. Med. Sci. Sports 2023, 33, 4–19. [Google Scholar] [CrossRef] [PubMed]
- McNay, D.E.G.; Briançon, N.; Kokoeva, M.V.; Maratos-Flier, E.; Flier, J.S. Remodeling of the arcuate nucleus energy-balance circuit is inhibited in obese mice. J. Clin. Investig. 2012, 122, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Bach, E.; Nielsen, R.R.; Vendelbo, M.H.; Moller, A.B.; Jessen, N.; Buhl, M.; Hafstrøm, T.K.; Holm, L.; Pedersen, S.B.; Pilegaard, H.; et al. Direct effects of TNF-alpha on local fuel metabolism and cytokine levels in the placebo-controlled, bilaterally infused human leg: Increased insulin sensitivity, increased net protein breakdown, and increased IL-6 release. Diabetes 2013, 62, 4023–4029. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Patsalos, O.; Dalton, B.; Leppanen, J.; Ibrahim, M.A.A.; Himmerich, H. Impact of TNF-alpha Inhibitors on Body Weight and BMI: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2020, 11, 481. [Google Scholar] [CrossRef]
- Choi, J.S. Effects of Maternal and Post-Weaning High-Fat Diet on Leptin Resistance and Hypothalamic Appetite Genes in Sprague Dawley Rat Offspring. Clin. Nutr. Res. 2018, 7, 276–290. [Google Scholar] [CrossRef] [PubMed]
- Terrien, J.; Seugnet, I.; Seffou, B.; Herrero, M.J.; Bowers, J.; Chamas, L.; Decherf, S.; Duvernois-Berthet, E.; Djediat, C.; Ducos, B.; et al. Reduced central and peripheral inflammatory responses and increased mitochondrial activity contribute to diet-induced obesity resistance in WSB/EiJ mice. Sci. Rep. 2019, 9, 19696. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, T.; Okuma, Y.; Nomura, Y. Expression of leptin receptors and induction of IL-1beta transcript in glial cells. Biochem. Biophys. Res. Commun. 2000, 273, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Posillico, C.K.; Garcia-Hernandez, R.E.; Tronson, N.C. Sex differences and similarities in the neuroimmune response to central administration of poly I:C. J. Neuroinflam. 2021, 18, 193. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef]
- Morselli, E.; Frank, A.P.; Palmer, B.F.; Rodriguez-Navas, C.; Criollo, A.; Clegg, D.J. A sexually dimorphic hypothalamic response to chronic high-fat diet consumption. Int. J. Obes. 2016, 40, 206–209. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CON (N = 16) | OB (N = 16) | p-Value | |
|---|---|---|---|
| Insulin (ng/mL) | 2.76 ± 0.3 | 2.24 ± 0.3 | 0.26 |
| Leptin (ng/mL) | 4.88 ± 1.3 | 6.67 ± 1.7 | 0.25 |
| Cholesterol (mg/dL) | 102.5 ± 6.5 | 102.4 ± 8.3 | 0.99 |
| NEFA (mEq/L) | 0.85 ± 0.07 | 0.81 ± 0.04 | 0.66 |
| Triglycerides (mg/dL) | 54.3 ± 6.6 | 50.9 ± 6.1 | 0.51 |
| Phospholipids (mg/dL) | 189.5 ± 11.3 | 196.9 ± 12.7 | 0.67 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buckley, L.A.; Kulhanek, D.R.; Bruder, A.; Gisslen, T.; Paulsen, M.E. Inflammation as a Sex-Specific Mediator in the Relationship between Maternal and Offspring Obesity in C57Bl/6J Mice. Biology 2024, 13, 399. https://doi.org/10.3390/biology13060399
Buckley LA, Kulhanek DR, Bruder A, Gisslen T, Paulsen ME. Inflammation as a Sex-Specific Mediator in the Relationship between Maternal and Offspring Obesity in C57Bl/6J Mice. Biology. 2024; 13(6):399. https://doi.org/10.3390/biology13060399
Chicago/Turabian StyleBuckley, Lauren A., Debra R. Kulhanek, Adrienne Bruder, Tate Gisslen, and Megan E. Paulsen. 2024. "Inflammation as a Sex-Specific Mediator in the Relationship between Maternal and Offspring Obesity in C57Bl/6J Mice" Biology 13, no. 6: 399. https://doi.org/10.3390/biology13060399
APA StyleBuckley, L. A., Kulhanek, D. R., Bruder, A., Gisslen, T., & Paulsen, M. E. (2024). Inflammation as a Sex-Specific Mediator in the Relationship between Maternal and Offspring Obesity in C57Bl/6J Mice. Biology, 13(6), 399. https://doi.org/10.3390/biology13060399