
PseUpred-ELPSO Is an Ensemble Learning Predictor with Particle Swarm Optimizer for Improving the Prediction of RNA Pseudouridine Sites



Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Benchmark Datasets
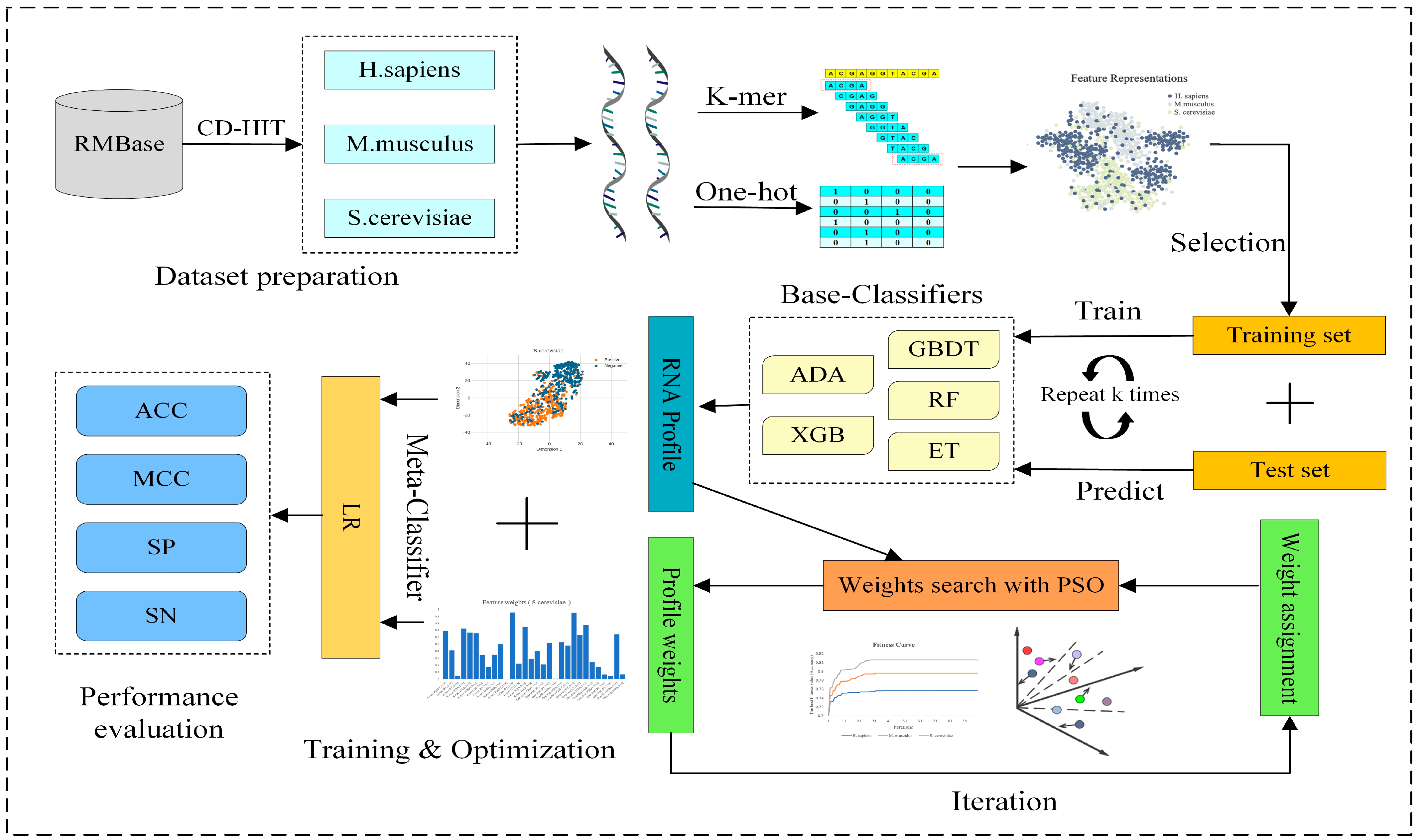
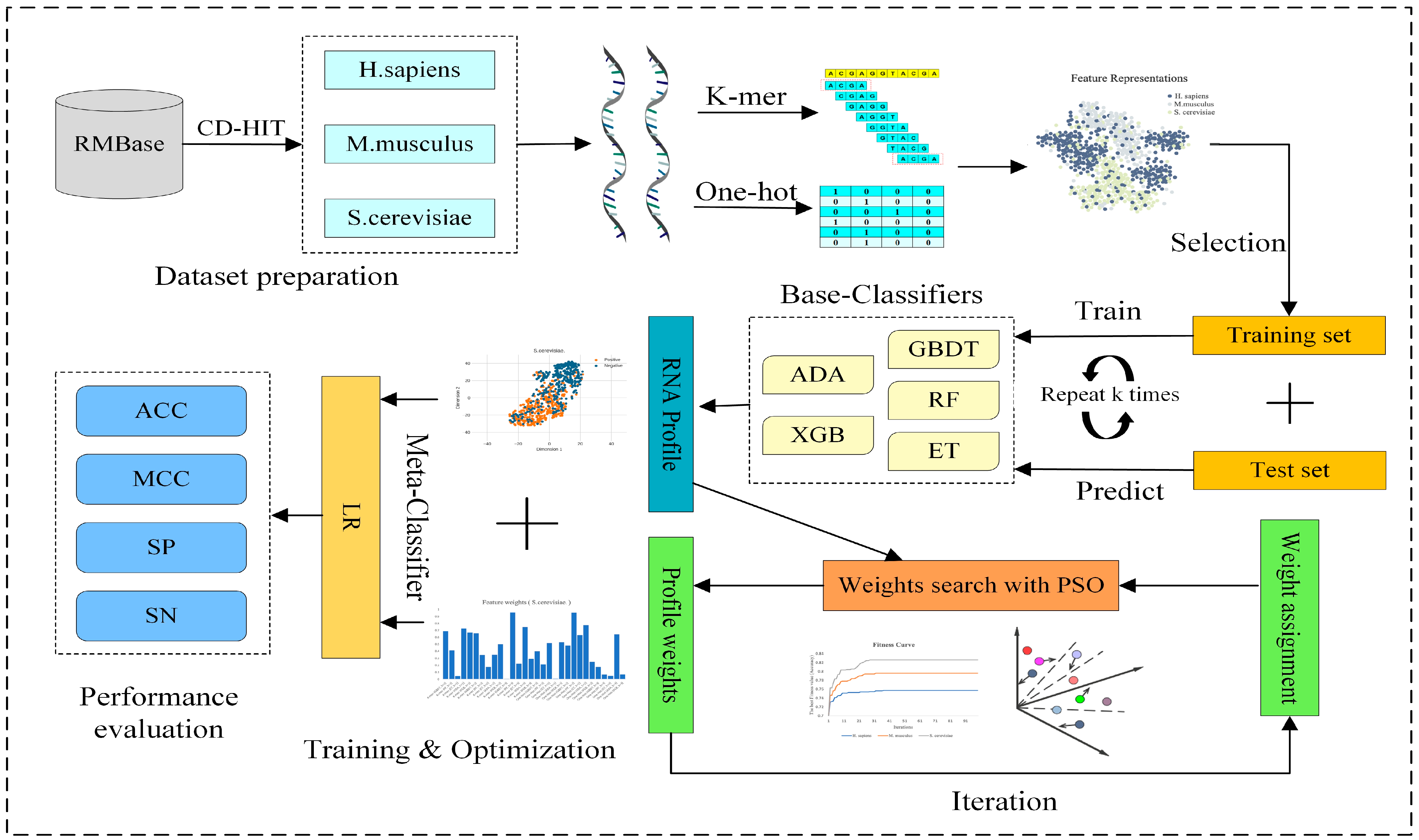
2.2. Overview of PseUpred-ELPSO
2.3. Feature Representation
2.3.1. One-Hot Encoding
2.3.2. K-mer Nucleotide Frequency (K-mer)
2.4. Machine Learning
2.4.1. Adaptive Boosting (ADA)
2.4.2. Gradient Boosting Decision Tree (GBDT)
2.4.3. Extreme Gradient Boosting (XGB)
2.4.4. Random Forest (RF)
2.4.5. Extra Trees (ET)
2.5. Stacking Strategy Combined with Particle Swarm Optimization (PSO)
2.5.1. Stacking Strategy
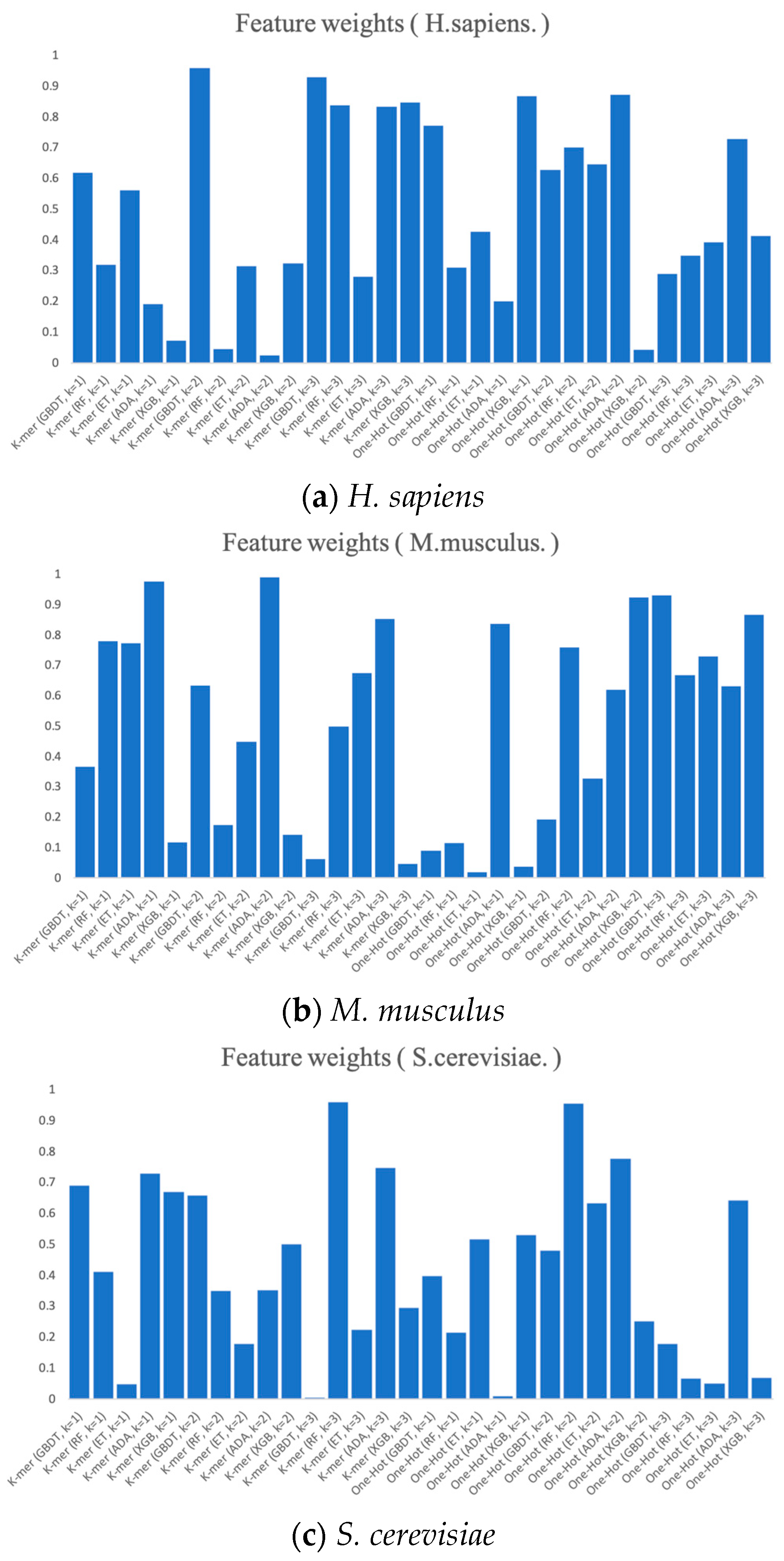
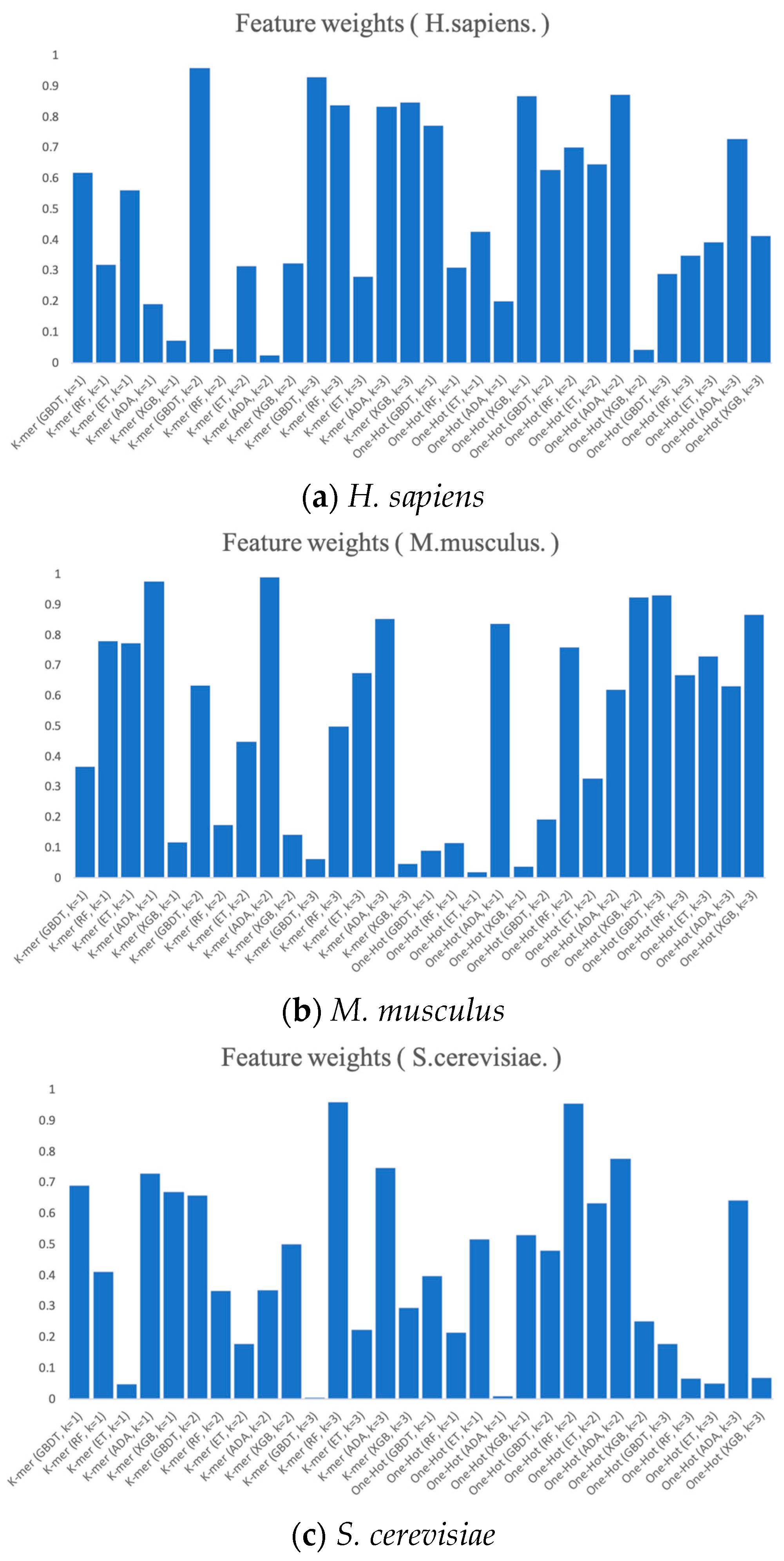
2.5.2. Feature Selection
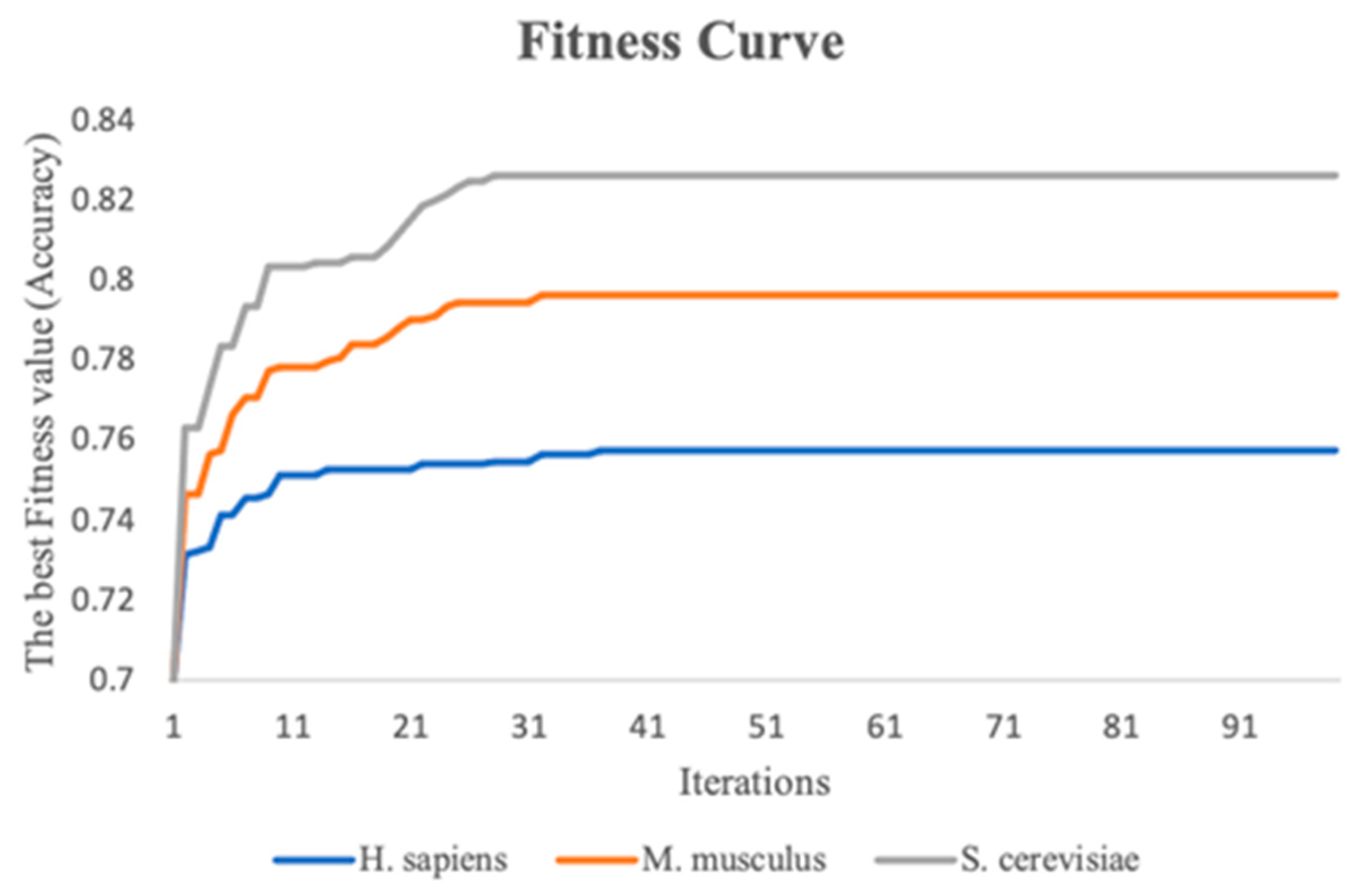
2.5.3. Particle Swarm Optimization (PSO)
2.5.4. PSO to Optimize the Weights of RNA Profile
2.6. Model Evaluation
3. Results
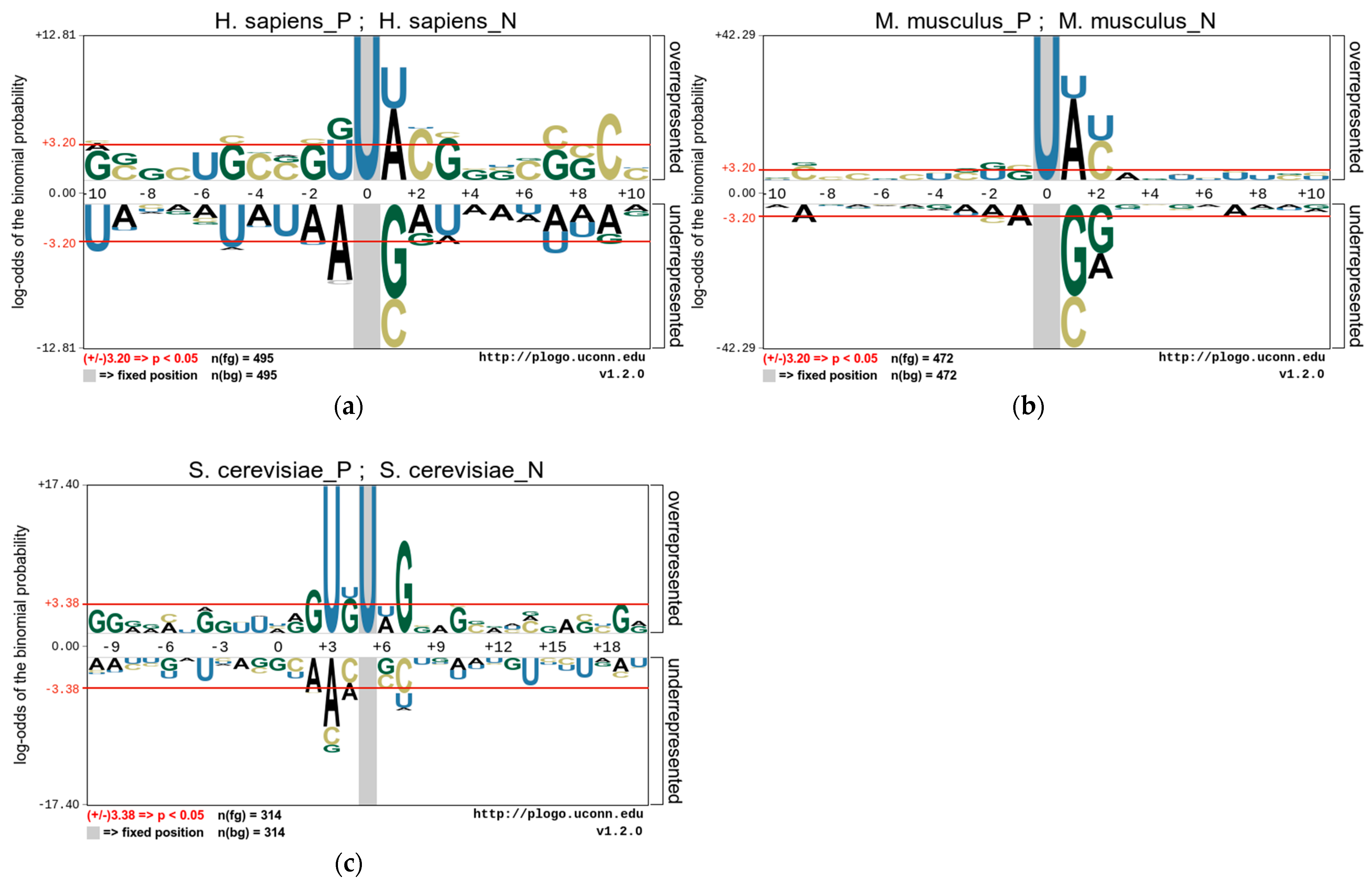
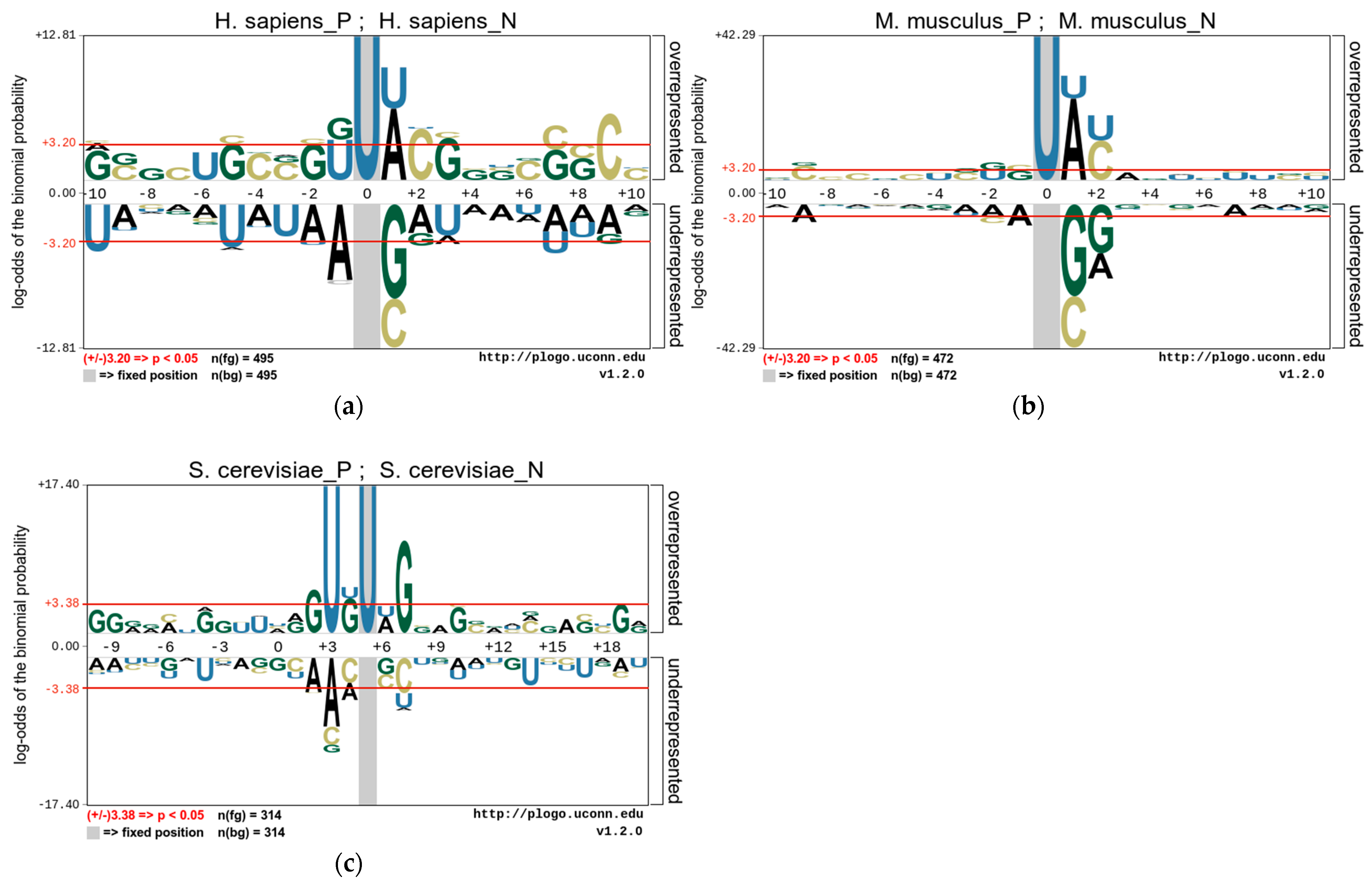
3.1. Nucleotide Composition Preference Analysis
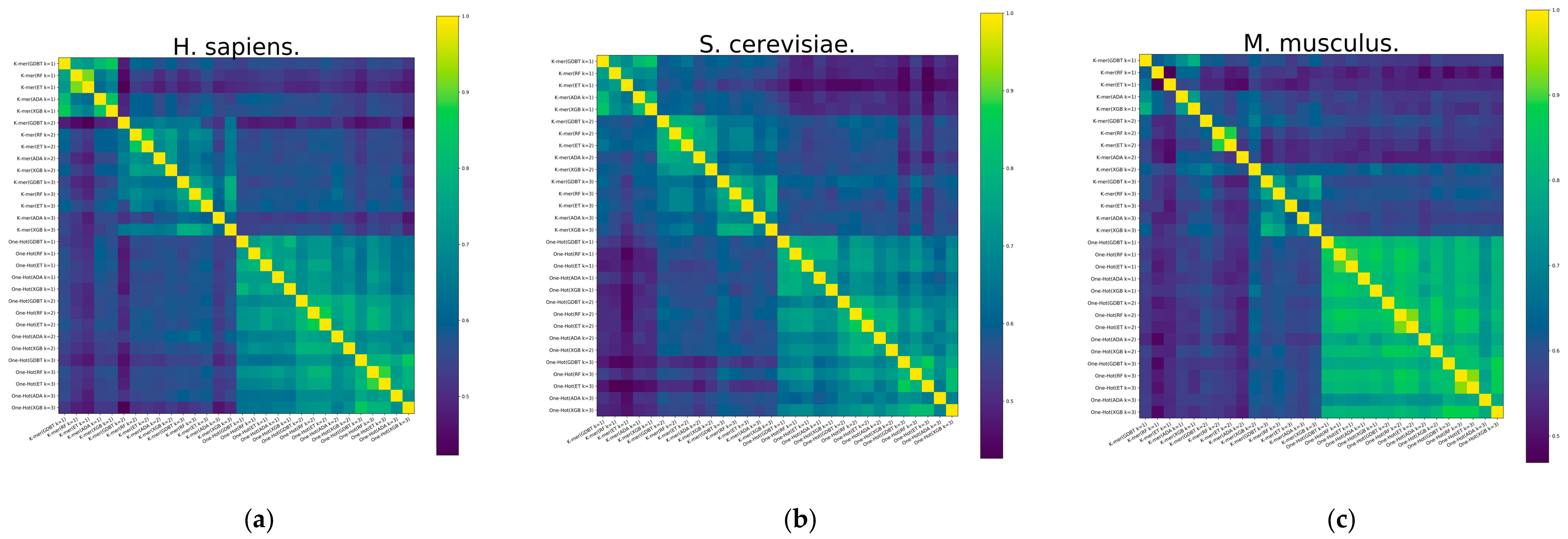
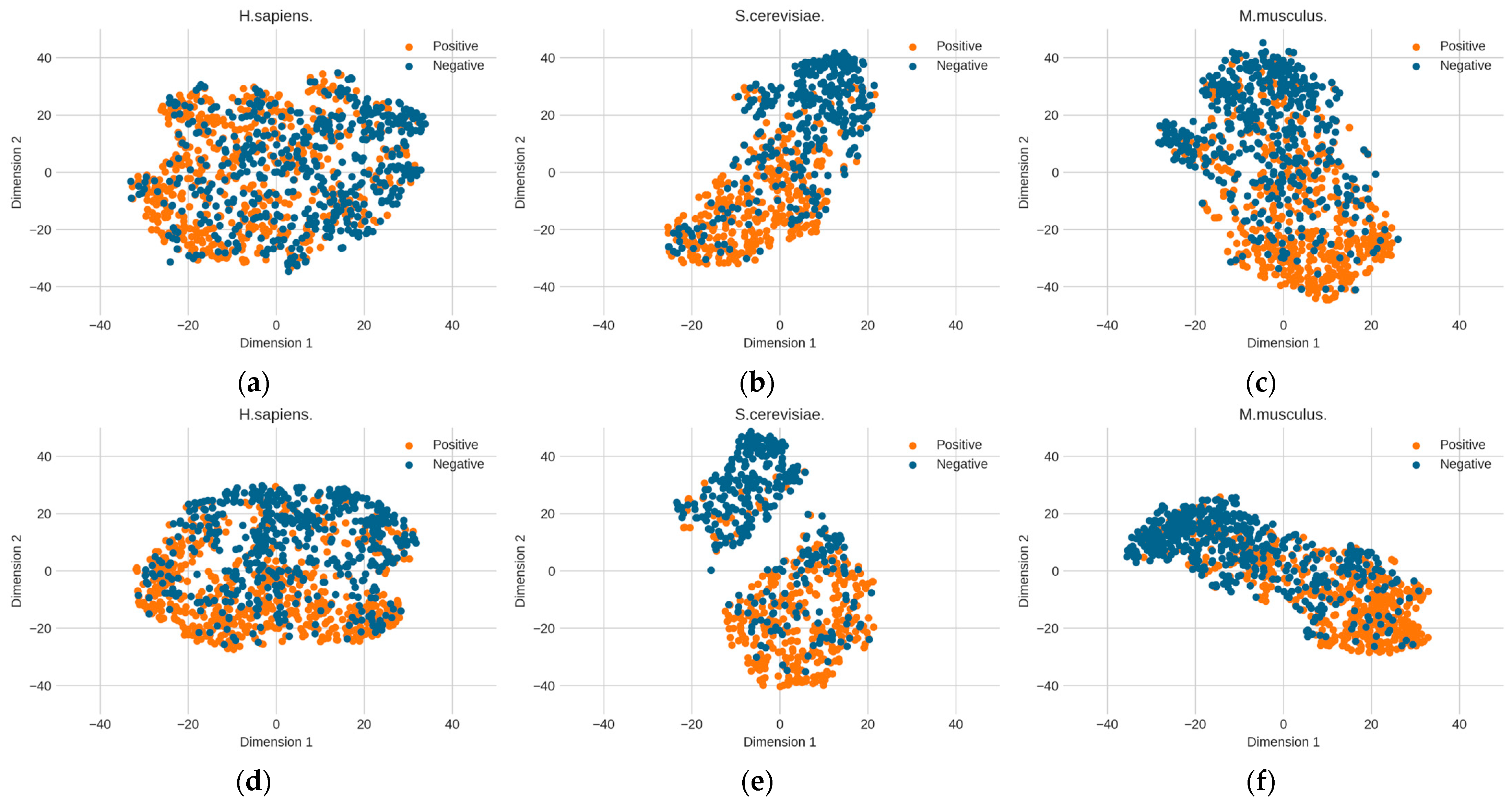
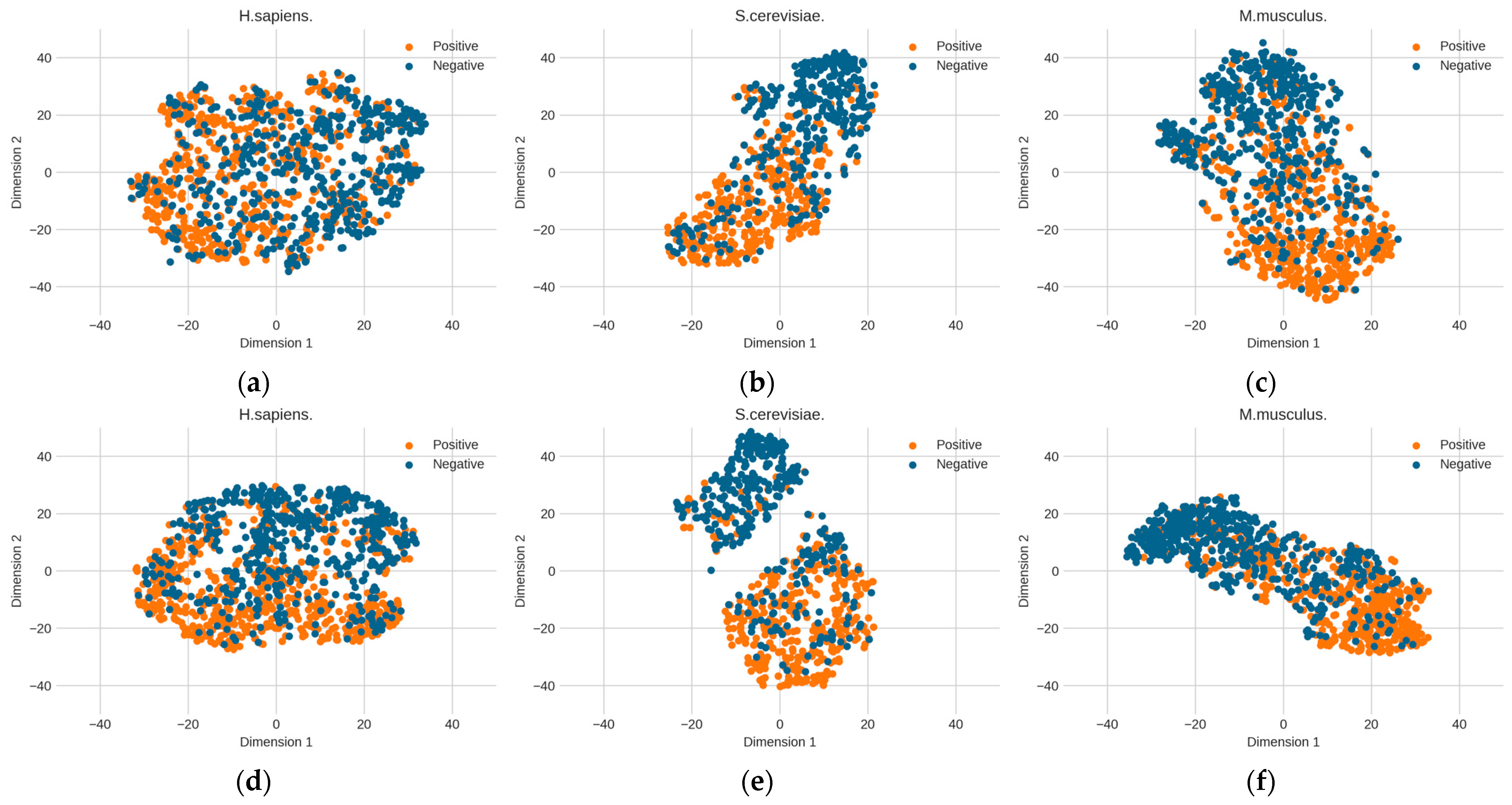
3.2. RNA Profile Analysis
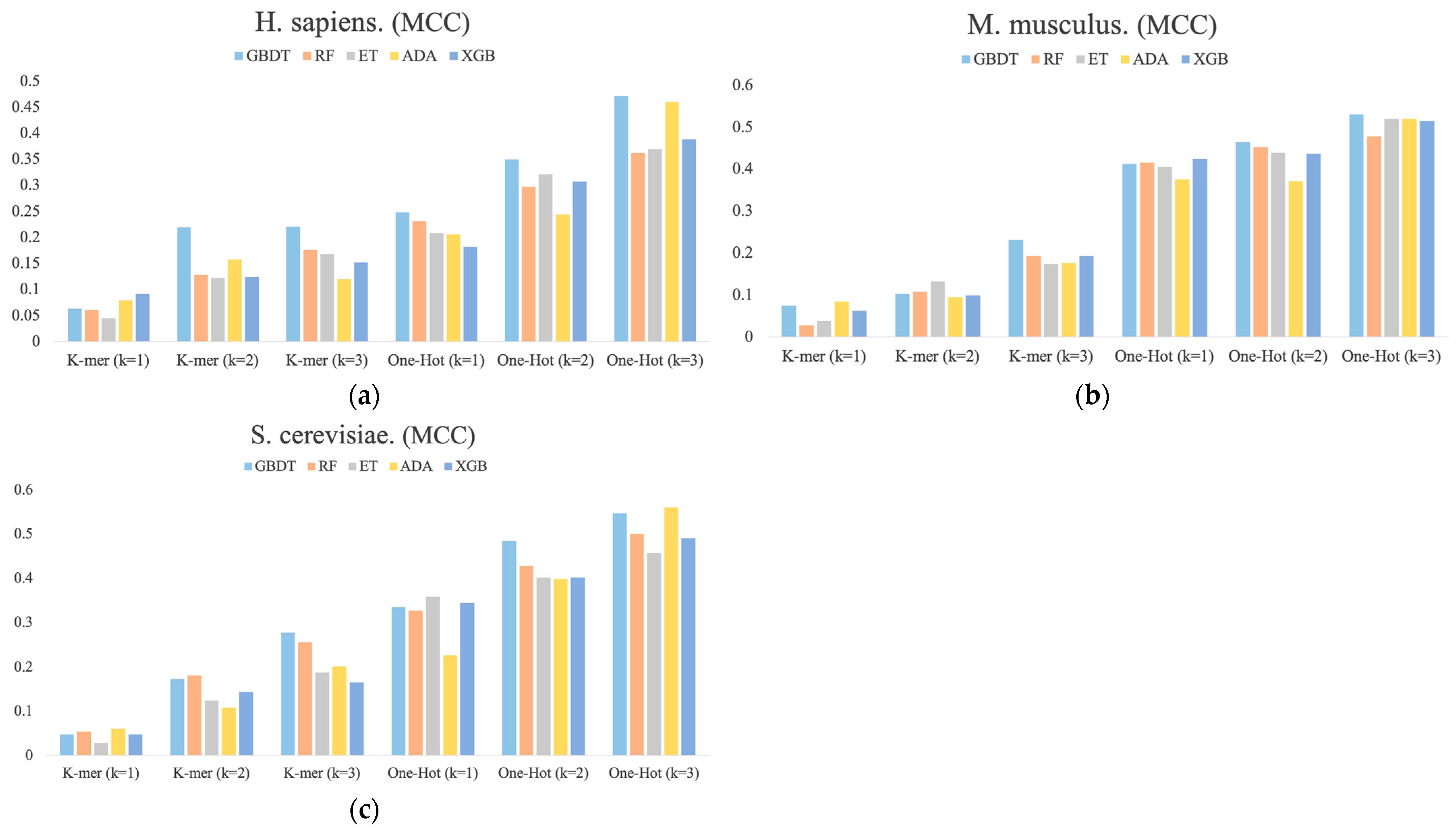
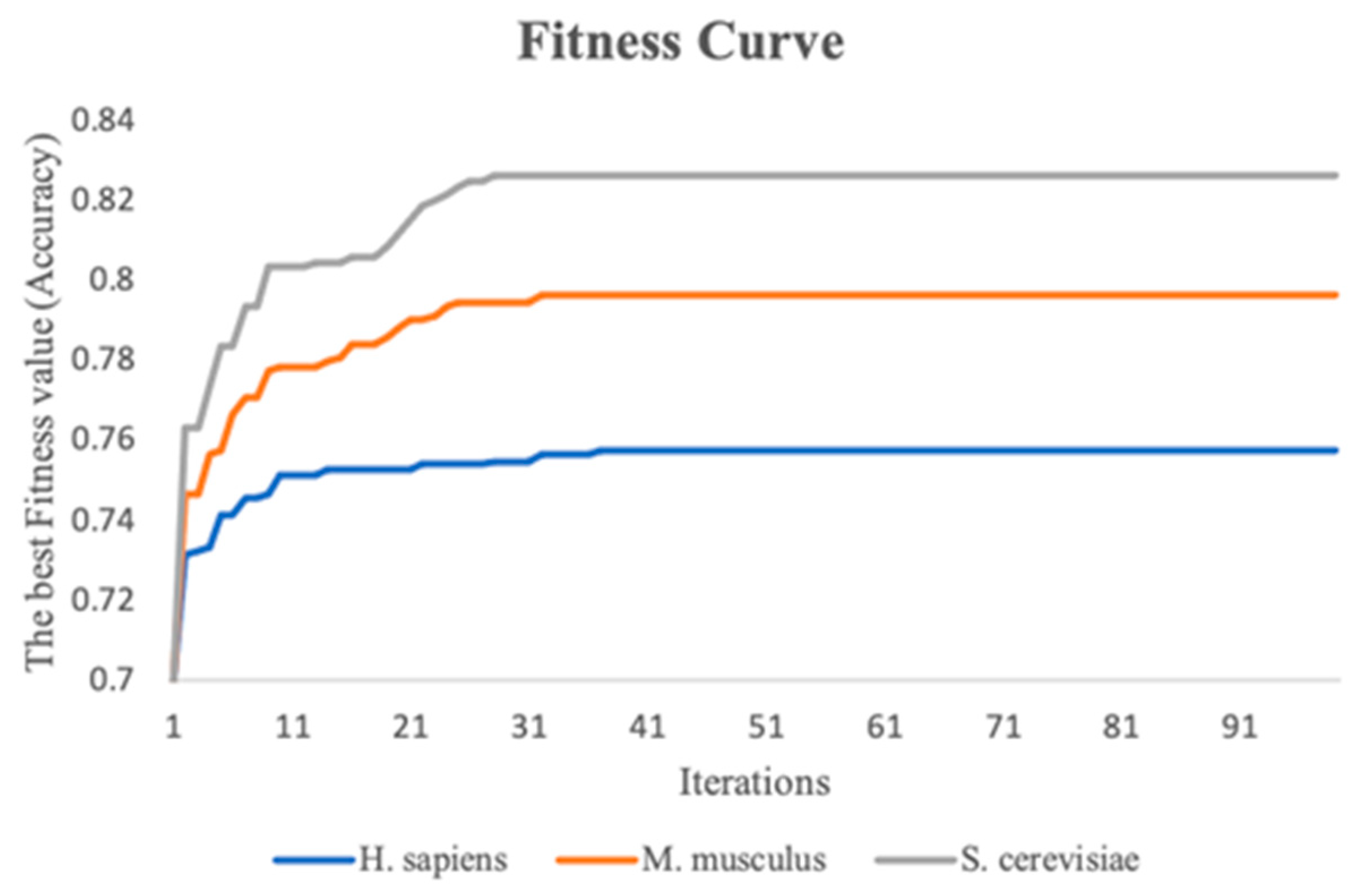
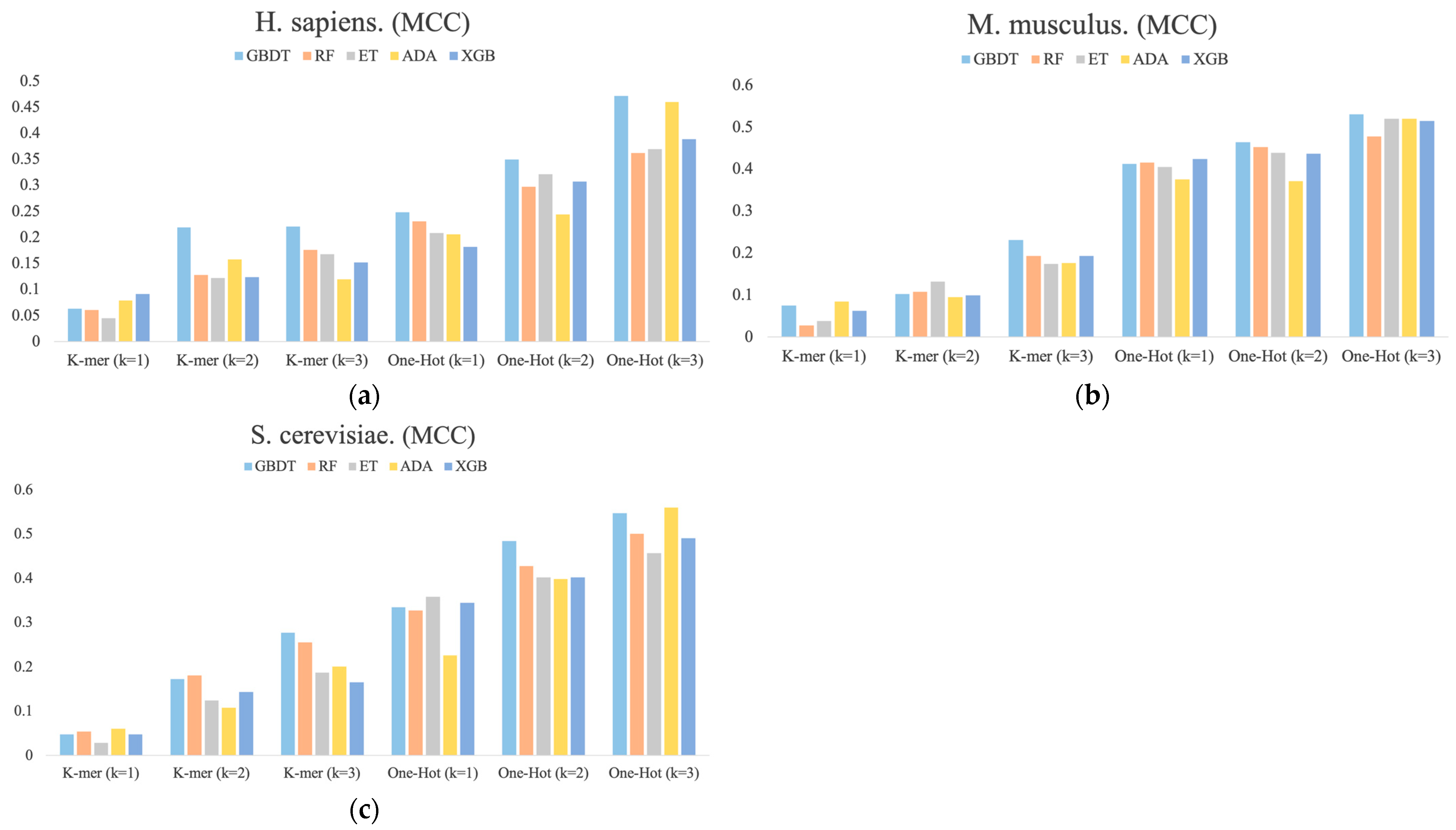
3.3. The Stacking Strategy Combined with PSO Improved the Performance
3.4. Comparison with State-of-the-Art Predictors
3.5. Comparative Analysis on Independent Datasets
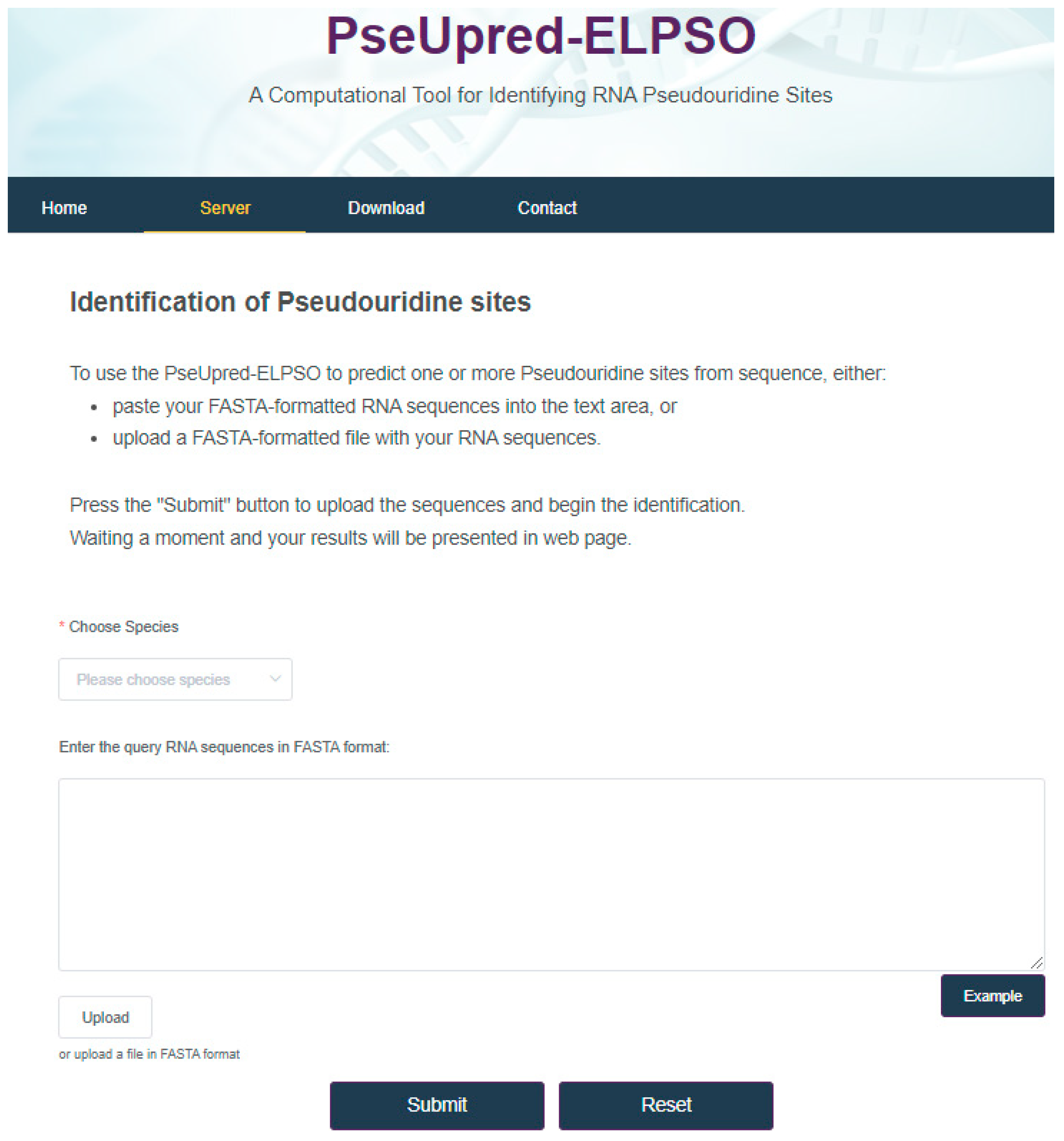
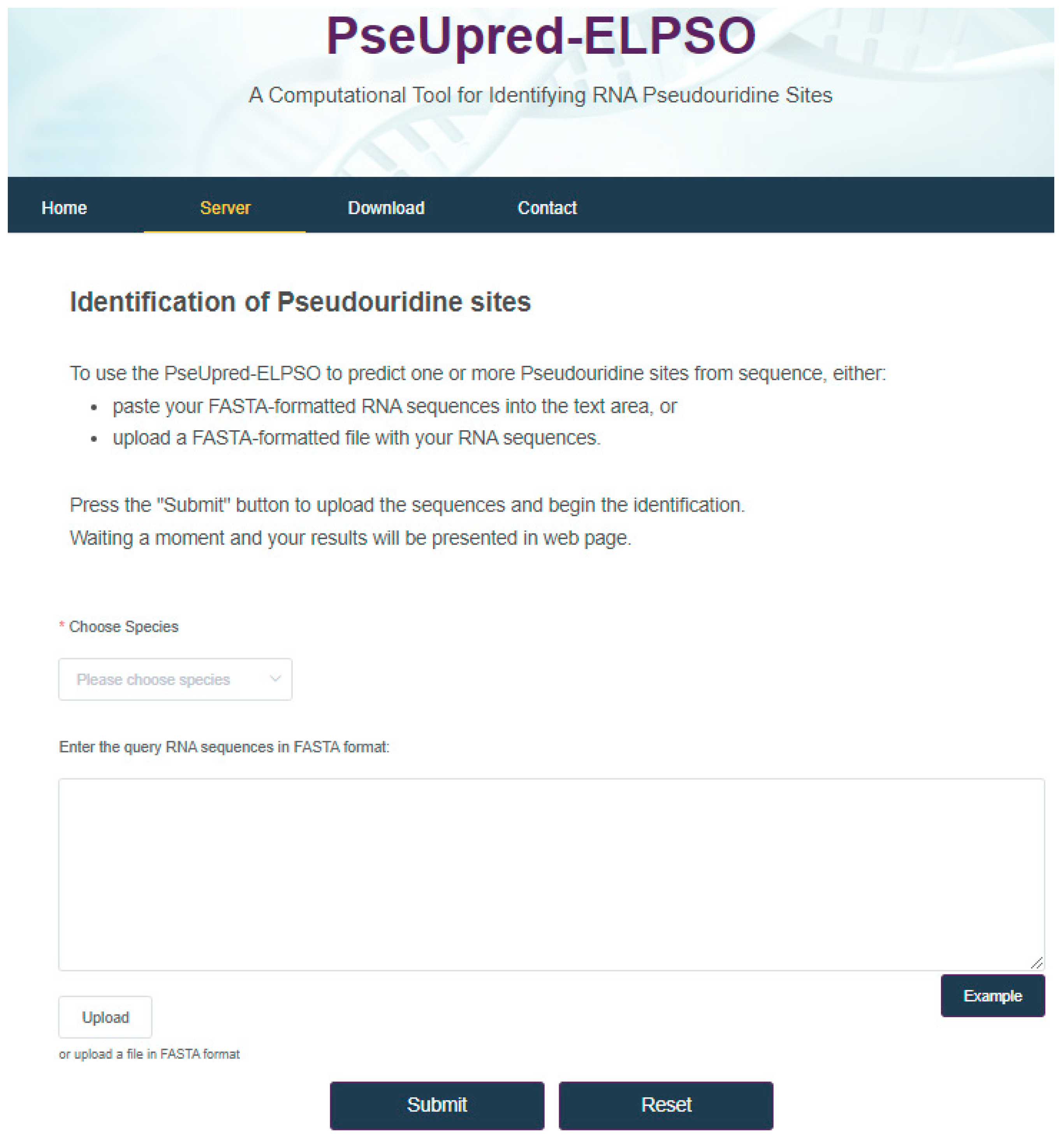
4. Web Server for PseUpred-ELPSO
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ge, J.; Yu, Y.T. RNA pseudouridylation: New insights into an old modification. Trends Biochem. Sci. 2013, 38, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.R.; Veltri, C.A.; Nielsen, L. An RNA Model System for Investigation of Pseudouridine Stabilization of the Codon-Anticodon Interaction in TRNA Lys, TRNA His and TRNA Tyr. J. Biomol. Struct. Dyn. 1998, 15, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- de Crécy-Lagard, V.; Hutinet, G.; Cediel-Becerra, J.D.D.; Yuan, Y.; Zallot, R.; Chevrette, M.G.; Ratnayake, R.M.M.N.; Jaroch, M.; Quaiyum, S.; Bruner, S. Biosynthesis and function of 7-deazaguanine derivatives in bacteria and phages. Microbiol. Mol. Biol. Rev. 2024, 29, e00199-23. [Google Scholar] [CrossRef] [PubMed]
- Langdon, E.M.; Qiu, Y.; Ghanbari Niaki, A.; McLaughlin, G.A.; Weidmann, C.A.; Gerbich, T.M.; Smith, J.A.; Crutchley, J.M.; Termini, C.M.; Weeks, K.M.; et al. mRNA structure determines specificity of a polyQ-driven phase separation. Science 2018, 360, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, A.F.; Riordan, D.P.; Brown, P.O. Transcriptome-Wide Mapping of Pseudouridines: Pseudouridine Synthases Modify Specific MRNAs in S. cerevisiae. PLoS ONE 2014, 9, 0110799. [Google Scholar] [CrossRef] [PubMed]
- Carlile, T.M.; Rojas-Duran, M.F.; Zinshteyn, B.; Shin, H.; Bartoli, K.M.; Gilbert, W.V. Pseudouridine Profiling Reveals Regulated MRNA Pseudouridylation in Yeast and Human Cells. Nature 2014, 515, 143–146. [Google Scholar] [CrossRef]
- Schwartz, S.; Bernstein, D.A.; Mumbach, M.R.; Jovanovic, M.; Herbst, R.H.; León-Ricardo, B.X.; Engreitz, J.M.; Guttman, M.; Satija, R.; Lander, E.S.; et al. Transcriptome-Wide Mapping Reveals Widespread Dynamic-Regulated Pseudouridylation of NcRNA and MRNA. Cell 2014, 159, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, P.; Ma, S.; Song, J.; Bai, J.; Sun, F.; Yi, C. Chemical Pulldown Reveals Dynamic Pseudouridylation of the Mammalian Transcriptome. Nat. Chem. Biol. 2015, 11, 592–597. [Google Scholar] [CrossRef]
- Li, Y.-H.; Zhang, G.; Cui, Q. PPUS: A Web Server to Predict PUS-Specific Pseudouridine Sites. Bioinformatics 2015, 31, 3362–3364. [Google Scholar] [CrossRef]
- Chen, W.; Tang, H.; Ye, J.; Lin, H.; Chou, K.-C. IRNA-PseU: Identifying RNA Pseudouridine Sites. Mol. Ther. Nucleic Acids 2016, 5, e332. [Google Scholar]
- He, J.; Fang, T.; Zhang, Z.; Huang, B.; Zhu, X.; Xiong, Y. PseUI: Pseudouridine Sites Identification Based on RNA Sequence Information. BMC Bioinform. 2018, 19, 306. [Google Scholar] [CrossRef] [PubMed]
- Tahir, M.; Tayara, H.; Chong, K.T. IPseU-CNN: Identifying RNA Pseudouridine Sites Using Convolutional Neural Networks. Mol. Ther.-Nucleic Acids 2019, 16, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Chen, W.; Lin, H. XG-PseU: An EXtreme Gradient Boosting Based Method for Identifying Pseudouridine Sites. Mol. Genet. Genom. 2020, 295, 13–21. [Google Scholar] [CrossRef]
- Bi, Y.; Xiang, D.; Ge, Z.; Li, F.; Jia, C.; Song, J. An Interpretable Prediction Model for Identifying N7-Methylguanosine Sites Based on XGBoost and SHAP. Mol. Ther.-Nucleic Acids 2020, 22, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Zhang, J.; Ding, H.; Zou, Q. RF-PseU: A Random Forest Predictor for RNA Pseudouridine Sites. Front. Bioeng. Biotechnol. 2020, 8, 134. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.J.; Li, J.H.; Liu, S.; Wu, J.; Zhou, H.; Qu, L.H.; Yang, J.H. RMBase: A resource for decoding the landscape of RNA modifications from high-throughput sequencing data. Nucleic Acids Res. 2016, 44, D259–D265. [Google Scholar] [CrossRef]
- Xuan, J.; Chen, L.; Chen, Z.; Pang, J.; Huang, J.; Lin, J.; Zheng, L.; Li, B.; Qu, L.; Yang, J.R. MBase v3.0: Decode the landscape, mechanisms and functions of RNA modifications. Nucleic Acids Res. 2024, 52, D273–D284. [Google Scholar] [CrossRef]
- Freund, Y.; Schapire, R.E. A decision-theoretic generalization of on-line learning and an application to boosting. J. Comput. Syst. Sci. 1997, 55, 119–139. [Google Scholar] [CrossRef]
- Friedman, J.H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD’ 16), San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Geurts, P.; Ernst, D.; Wehenkel, L. Extremely randomized trees. Mach. Learn. 2006, 63, 3–22. [Google Scholar] [CrossRef]
- Wang, H.; Liu, H.; Huang, T.; Li, G.; Zhang, L.; Sun, Y. EMDLP: Ensemble multiscale deep learning model for RNA methylation site prediction. BMC Bioinform. 2022, 23, 221. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Su, W.; Lou, L.; Qiu, W.; Xiao, X.; Xu, Z. DLm6Am: A Deep-Learning-Based Tool for Identifying N6,2′-O-Dimethyladenosine Sites in RNA Sequences. Int. J. Mol. Sci. 2022, 23, 11026. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Wei, Z.; Sun, M. EMDL_m6Am: Identifying N6,2’-O-dimethyladenosine sites based on stacking ensemble deep learning. BMC Bioinform. 2023, 24, 397. [Google Scholar] [CrossRef]
- Zhou, B.; Ding, M.; Feng, J.; Ji, B.; Huang, P.; Zhang, J.; Yu, X.; Cao, Z.; Yang, Y.; Zhou, Y.; et al. EVlncRNA-Dpred: Improved prediction of experimentally validated lncRNAs by deep learning. Brief. Bioinform. 2023, 24, bbac583. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Shao, Y.; Zhang, Y.; Zhou, W.; Pang, S. A deep learning method for drug-target affinity prediction based on sequence interaction information mining. PeerJ 2023, 11, e16625. [Google Scholar] [CrossRef]
- Avila Santos, A.P.; de Almeida, B.L.; Bonidia, R.P.; Stadler, P.F.; Stefanic, P.; Mandic-Mulec, I.; Rocha, U.; Sanches, D.S.; de Carvalho, A.C. BioDeepfuse: A hybrid deep learning approach with integrated feature extraction techniques for enhanced non-coding RNA classification. RNA Biol. 2024, 21, 1–12. [Google Scholar] [CrossRef]
- Aylward, A.J.; Petrus, S.; Mamerto, A.; Hartwick, N.T.; Michael, T.P. PanKmer: K-mer-based and reference-free pangenome analysis. Bioinformatics 2023, 39, btad621. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liu, M.; Zhang, P.; Zhang, H. iEnhancer-SKNN: A stacking ensemble learning-based method for enhancer identification and classification using sequence information. Brief. Funct. Genom. 2023, 22, 302–311. [Google Scholar] [CrossRef]
- Mishra, A.; Pokhrel, P.; Hoque, M.T. StackDPPred: A Stacking Based Prediction of DNA-Binding Protein from Sequence. Bioinformatics 2019, 35, 433–441. [Google Scholar] [CrossRef]
- Wu, H.; Wu, Y.; Jiang, Y.; Zhou, B.; Zhou, H.; Chen, Z.; Xiong, Y.; Liu, Q.; Zhang, H. scHiCStackL: A stacking ensemble learning-based method for single-cell Hi-C classification using cell embedding. Brief. Bioinform. 2022, 23, bbab396. [Google Scholar] [CrossRef]
- Le, D.H.; Pham, V.H.; Nguyen, T.T. An ensemble learning-based method for prediction of novel disease-microRNA associations. In Proceedings of the 2017 9th International Conference on Knowledge and Systems Engineering (KSE), Hue, Vietnam, 19–21 October 2017; pp. 7–12. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Wang, D.; Tan, D.; Liu, L. Particle Swarm Optimization Algorithm: An Overview. Soft Comput. 2018, 22, 387–408. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, S.; Ji, G. A Comprehensive Survey on Particle Swarm Optimization Algorithm and Its Applications. Math. Probl. Eng. 2015, 2015, 931256. [Google Scholar] [CrossRef]
- Rokbani, N.; Abraham, A.; Alimi, A.M. Fuzzy Ant Supervised by PSO and Simplified Ant Supervised PSO Applied to TSP. In Proceedings of the 13th International Conference on Hybrid Intelligent Systems (HIS 2013), Salamanca, Spain, 11–13 September 2013; IEEE: Gammarth, Tunisia, 2013; pp. 251–255. [Google Scholar]
- O’Shea, J.P.; Chou, M.F.; Quader, S.A.; Ryan, J.K.; Church, G.M.; Schwartz, D. pLogo: A probabilistic approach to visualizing sequence motifs. Nat. Methods 2013, 10, 1211–1212. [Google Scholar] [CrossRef]
- van der Maaten, L.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Type | ACC | MCC | SN | SP |
|---|---|---|---|---|---|
| H. sapiens | Stack | 0.745 | 0.49 | 0.758 | 0.734 |
| Stack (PSO) | 0.757 | 0.52 | 0.775 | 0.742 | |
| S. cerevisiae | Stack | 0.795 | 0.59 | 0.820 | 0.773 |
| Stack (PSO) | 0.826 | 0.66 | 0.860 | 0.799 | |
| M. musculus | Stack | 0.776 | 0.55 | 0.774 | 0.779 |
| Stack (PSO) | 0.797 | 0.59 | 0.790 | 0.803 |
| Species | Predictor | ACC | MCC | SN | SP |
|---|---|---|---|---|---|
| S. cerevisiae | PseUI | 0.641 | 0.29 | 0.647 | 0.643 |
| IRna-PseU | 0.645 | 0.29 | 0.647 | 0.643 | |
| XG-PseU | 0.682 | 0.37 | 0.668 | 0.695 | |
| iPseU-CNN | 0.682 | 0.37 | 0.664 | 0.705 | |
| RF-PseU | 0.748 | 0.49 | 0.772 | 0.724 | |
| PseUpred-ELPSO | 0.826 | 0.66 | 0.860 | 0.799 | |
| H. sapiens | IRna-PseU | 0.604 | 0.21 | 0.610 | 0.598 |
| PseUI | 0.642 | 0.28 | 0.649 | 0.636 | |
| RF-PseU | 0.643 | 0.29 | 0.661 | 0.626 | |
| XG-PseU | 0.661 | 0.32 | 0.635 | 0.687 | |
| iPseU-CNN | 0.660 | 0.34 | 0.650 | 0.680 | |
| PseUpred-ELPSO | 0.757 | 0.52 | 0.775 | 0.742 | |
| M. musculus | IRna-PseU | 0.691 | 0.38 | 0.733 | 0.648 |
| PseUI | 0.704 | 0.41 | 0.799 | 0.703 | |
| iPseU-CNN | 0.718 | 0.44 | 0.748 | 0.691 | |
| XG-PseU | 0.720 | 0.45 | 0.765 | 0.676 | |
| RF-PseU | 0.748 | 0.50 | 0.731 | 0.765 | |
| PseUpred-ELPSO | 0.797 | 0.59 | 0.790 | 0.803 |
| Species | Predictor | ACC | MCC | SN | SP |
|---|---|---|---|---|---|
| H. sapiens | IRna-PseU | 0.65 | 0.30 | 0.600 | 0.700 |
| PseUI | 0.655 | 0.31 | 0.630 | 0.680 | |
| iPseU-CNN | 0.690 | 0.40 | 0.777 | 0.608 | |
| XG-PseU | 0.675 | / | / | / | |
| RF-PseU | 0.750 | 0.50 | 0.780 | 0.720 | |
| PseUpred-ELPSO | 0.760 | 0.52 | 0.770 | 0.750 | |
| S. cerevisiae | IRna-PseU | 0.600 | 0.20 | 0.630 | 0.570 |
| PseUI | 0.685 | 0.37 | 0.650 | 0.720 | |
| iPseU-CNN | 0.735 | 0.47 | 0.686 | 0.778 | |
| XG-PseU | 0.710 | / | / | / | |
| RF-PseU | 0.770 | 0.54 | 0.750 | 0.790 | |
| PseUpred-ELPSO | 0.795 | 0.60 | 0.760 | 0.850 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Li, P.; Wang, R.; Gao, X. PseUpred-ELPSO Is an Ensemble Learning Predictor with Particle Swarm Optimizer for Improving the Prediction of RNA Pseudouridine Sites. Biology 2024, 13, 248. https://doi.org/10.3390/biology13040248
Wang X, Li P, Wang R, Gao X. PseUpred-ELPSO Is an Ensemble Learning Predictor with Particle Swarm Optimizer for Improving the Prediction of RNA Pseudouridine Sites. Biology. 2024; 13(4):248. https://doi.org/10.3390/biology13040248
Chicago/Turabian StyleWang, Xiao, Pengfei Li, Rong Wang, and Xu Gao. 2024. "PseUpred-ELPSO Is an Ensemble Learning Predictor with Particle Swarm Optimizer for Improving the Prediction of RNA Pseudouridine Sites" Biology 13, no. 4: 248. https://doi.org/10.3390/biology13040248
APA StyleWang, X., Li, P., Wang, R., & Gao, X. (2024). PseUpred-ELPSO Is an Ensemble Learning Predictor with Particle Swarm Optimizer for Improving the Prediction of RNA Pseudouridine Sites. Biology, 13(4), 248. https://doi.org/10.3390/biology13040248