
Hemodynamic Melody of Postnatal Cardiac and Pulmonary Development in Children with Congenital Heart Diseases


Abstract
Simple Summary
Abstract
1. Introduction
2. Abnormal Hemodynamics in Children with CHD
2.1. Abnormal Hemodynamics in Ventricles
2.2. Abnormal Hemodynamics in Lungs
3. Immature Hearts and Lungs in Children
3.1. Immature Hearts in Children
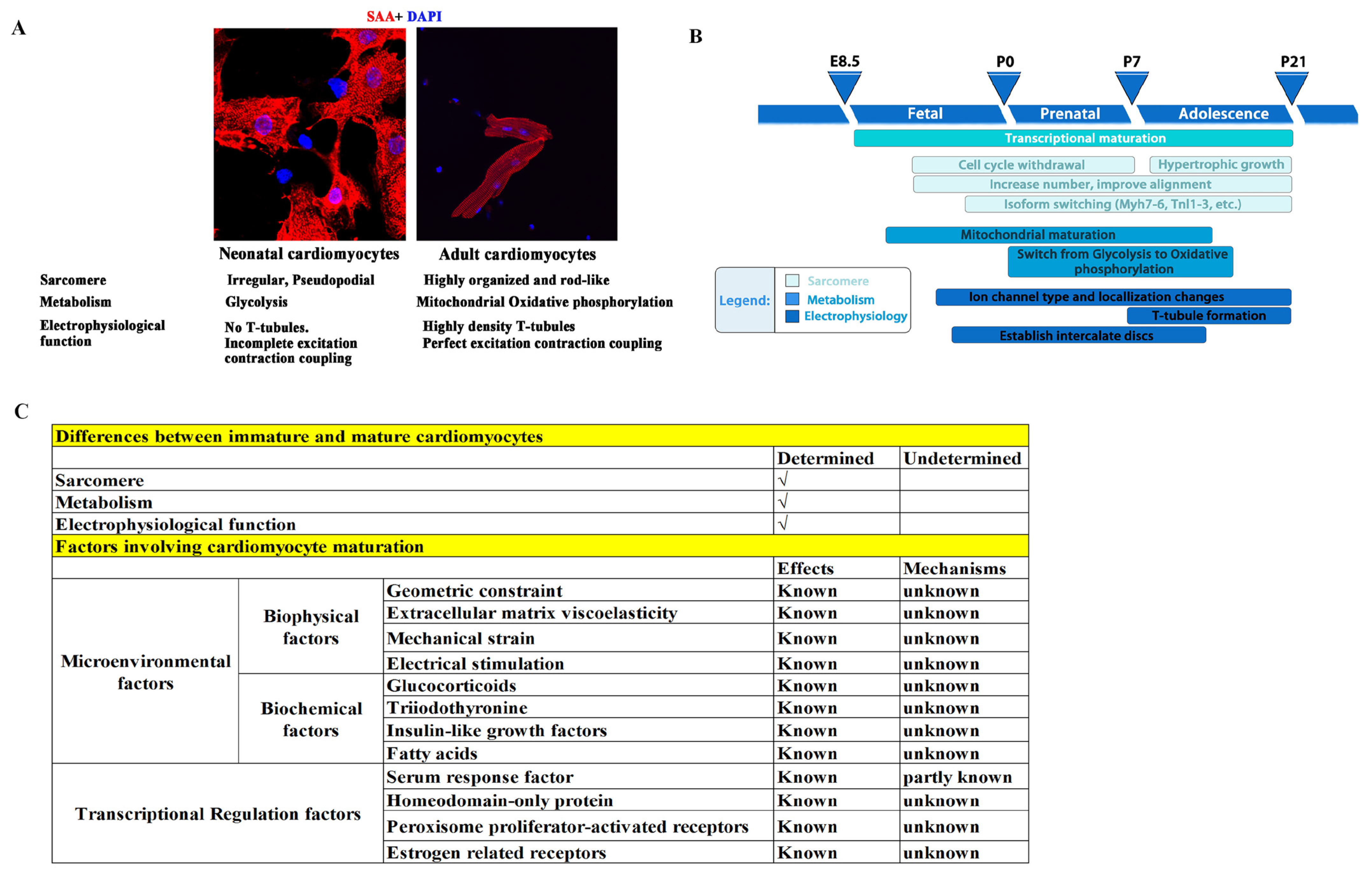
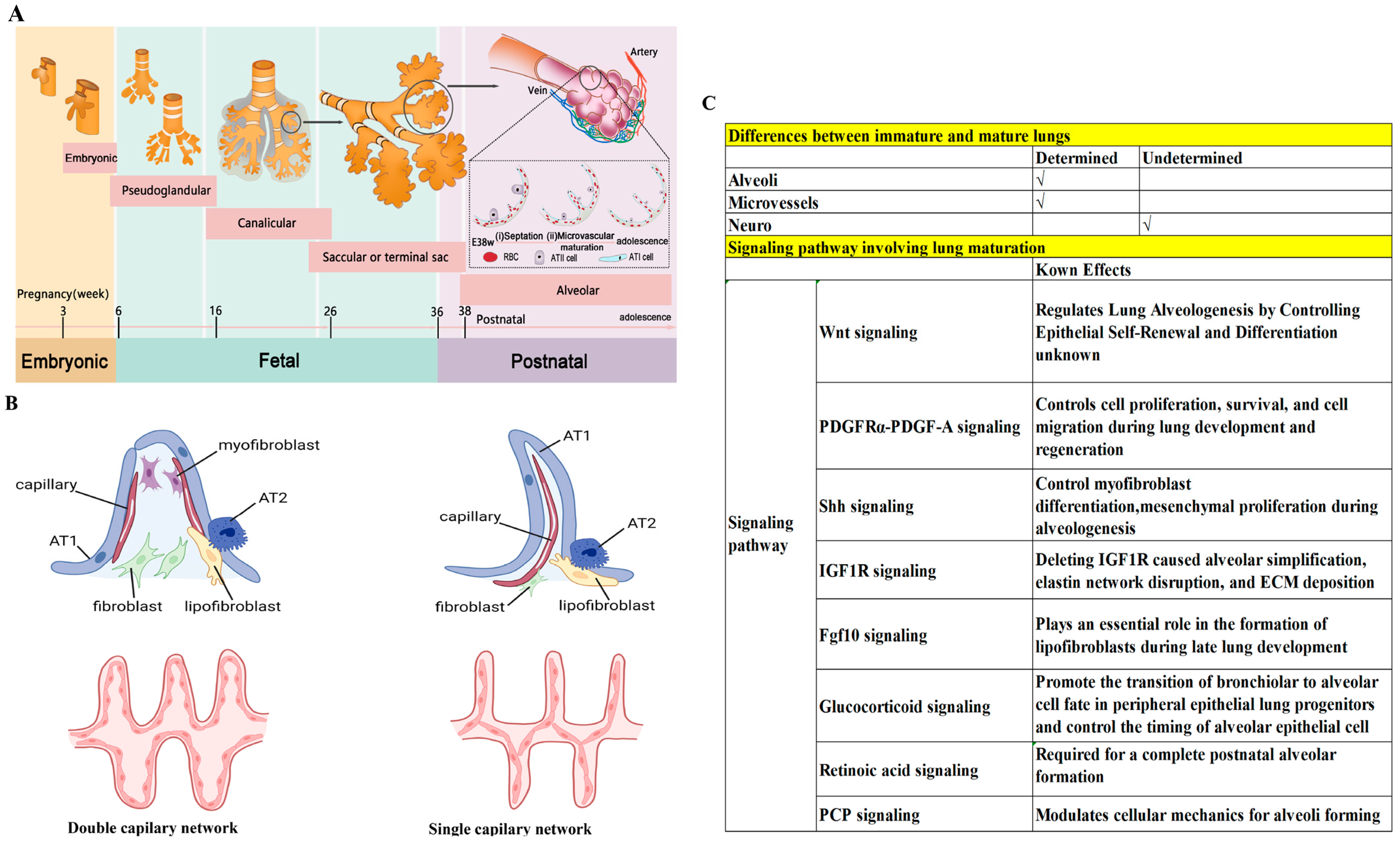
3.2. Immature Lungs in Children
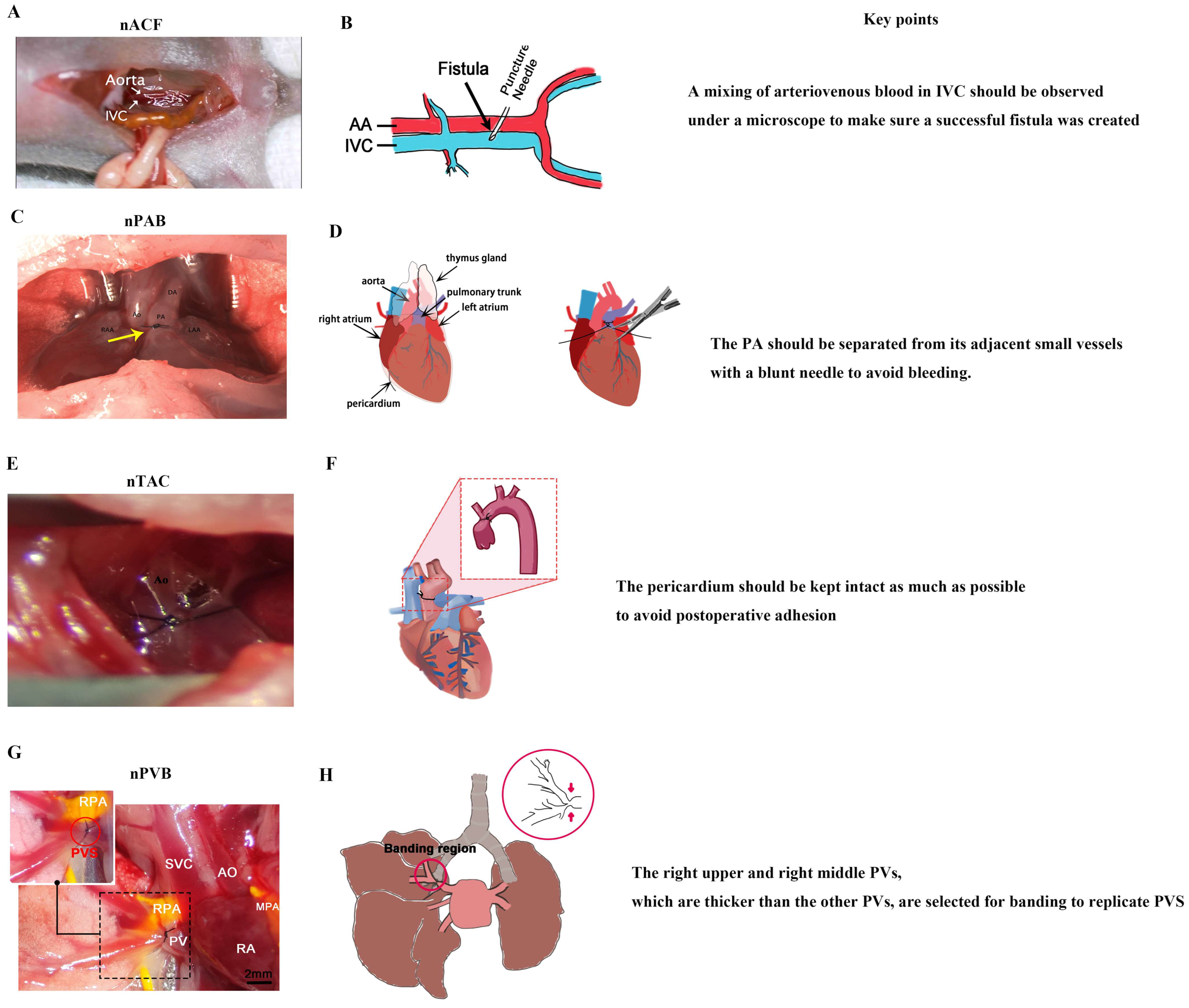
4. Creation of Neonatal Surgical Rodent Models of CHDs
4.1. Challenges in Constructing Neonatal Surgical Rodent Models of CHDs
4.2. Key Points of Constructing Neonatal Surgical Rodent Models of CHDs
4.2.1. nACF Surgery
4.2.2. nPAB Surgery
4.2.3. nTAC Surgery
4.2.4. nPVB Surgery
5. Hemodynamic Melody of Pediatric Heart and Lung Development
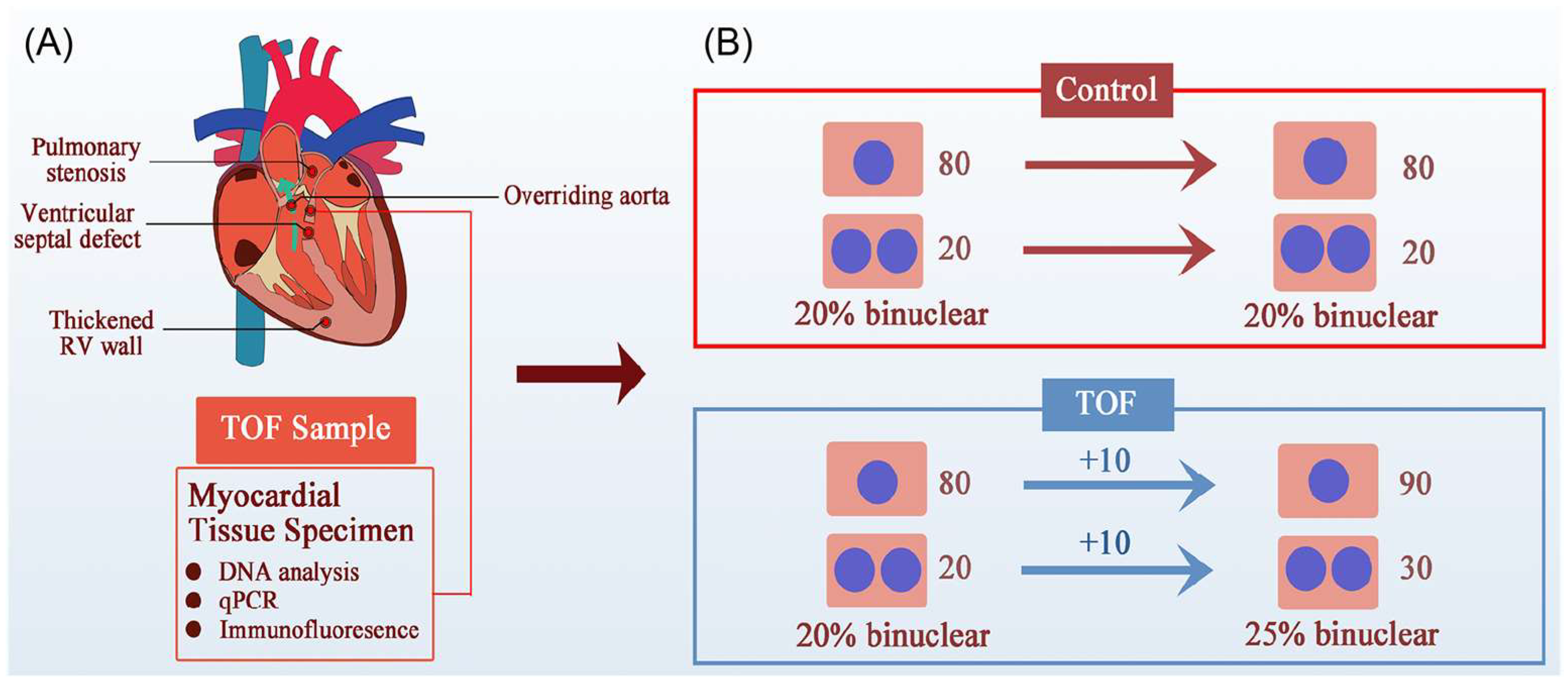
5.1. Cardiomyocyte Proliferation
5.2. Cardiomyocyte Maturation
5.3. Lung Development
6. Summary and Prospects
6.1. Clinical Decision Making
6.2. Limitations and Future Directions
7. Conclusions
8. Searching Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aird, W.C. Discovery of the cardiovascular system: From Galen to William Harvey. J. Thromb. Haemost. 2011, 9 (Suppl. 1), 118–129. [Google Scholar] [CrossRef] [PubMed]
- Bynum, B.; Bynum, H. William Harvey’s demonstration rod. Lancet 2015, 386, 1933. [Google Scholar] [CrossRef]
- Dewan, S.; Krishnamurthy, A.; Kole, D.; Conca, G.; Kerckhoffs, R.; Puchalski, M.D.; Omens, J.H.; Sun, H.; Nigam, V.; McCulloch, A.D. Model of Human Fetal Growth in Hypoplastic Left Heart Syndrome: Reduced Ventricular Growth Due to Decreased Ventricular Filling and Altered Shape. Front. Pediatr. 2017, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Feit, L.R.; Copel, J.A.; Kleinman, C.S. Foramen ovale size in the normal and abnormal human fetal heart: An indicator of transatrial flow physiology. Ultrasound Obstet. Gynecol. 1991, 1, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Tanai, E.; Frantz, S. Pathophysiology of Heart Failure. Compr. Physiol. 2015, 6, 187–214. [Google Scholar] [CrossRef] [PubMed]
- Thandavarayan, R.A.; Chitturi, K.R.; Guha, A. Pathophysiology of Acute and Chronic Right Heart Failure. Cardiol. Clin. 2020, 38, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Nagalingam, R.S.; Chattopadhyaya, S.; Al-Hattab, D.S.; Cheung, D.Y.C.; Schwartz, L.Y.; Jana, S.; Aroutiounova, N.; Ledingham, D.A.; Moffatt, T.L.; Landry, N.M.; et al. Scleraxis and fibrosis in the pressure-overloaded heart. Eur. Heart J. 2022, 43, 4739–4750. [Google Scholar] [CrossRef] [PubMed]
- Burri, P.H. Fetal and postnatal development of the lung. Annu. Rev. Physiol. 1984, 46, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Whitsett, J.A.; Wert, S.E.; Trapnell, B.C. Genetic disorders influencing lung formation and function at birth. Hum. Mol. Genet. 2004, 13, R207–R215. [Google Scholar] [CrossRef]
- Pedra, C.A.; Haddad, J.; Pedra, S.F.; Peirone, A.; Pilla, C.B.; Marin-Neto, J.A. Paediatric and congenital heart disease in South America: An overview. Heart 2009, 95, 1385–1392. [Google Scholar] [CrossRef]
- Fernandes, P.S.; Magalhães, L.R.; Pezzini, T.R.; de Sousa Santos, E.F.; Calderon, M.G. Congenital heart diseases trends in São Paulo State, Brazil: A national live birth data bank analysis. World J. Pediatr. 2022, 18, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Galdos, F.X.; Guo, Y.; Paige, S.L.; VanDusen, N.J.; Wu, S.M.; Pu, W.T. Cardiac Regeneration: Lessons from Development. Circ. Res. 2017, 120, 941–959. [Google Scholar] [CrossRef] [PubMed]
- GBD 2017 Congenital Heart Disease Collaborators. Global, Regional, and National Burden of Congenital Heart Disease, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet Child Adolesc. Health 2020, 4, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Beaton, A.Z.; Boehme, A.K.; Buxton, A.E.; et al. Heart Disease and Stroke Statistics-2023 Update: A Report from the American Heart Association. Circulation 2023, 147, e93–e621. [Google Scholar] [CrossRef] [PubMed]
- Corporan, D.; Segura, A.; Padala, M. Ultrastructural Adaptation of the Cardiomyocyte to Chronic Mitral Regurgitation. Front. Cardiovasc. Med. 2021, 8, 714774. [Google Scholar] [CrossRef] [PubMed]
- Offen, S.M.; Baker, D.; Puranik, R.; Celermajer, D.S. Right ventricular volume and its relationship to functional tricuspid regurgitation. Int. J. Cardiol. Heart Vasc. 2021, 38, 100940. [Google Scholar] [CrossRef] [PubMed]
- Otani, H.; Kagaya, Y.; Yamane, Y.; Chida, M.; Ito, K.; Namiuchi, S.; Shiba, N.; Koseki, Y.; Ninomiya, M.; Ikeda, J.; et al. Long-term right ventricular volume overload increases myocardial fluorodeoxyglucose uptake in the interventricular septum in patients with atrial septal defect. Circulation 2000, 101, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.W.; Fisher, S.A.; Wehman, B.; Mehra, M.R. Adults with congenital heart disease and heart transplantation: Optimizing outcomes. J. Heart Lung Transplant. 2014, 33, 873–877. [Google Scholar] [CrossRef]
- Palomo-López, N.; Escalona-Rodríguez, S.; Martín-Villén, L.; Herruzo-Avilés, Á.; Hinojosa-Pérez, R.; Escoresca-Ortega, A.; Porras-López, M.; Corcia-Palomo, Y.; Adsuar-Gómez, A. Transplantation in Congenital Heart Disease: A Challenge. Transplant. Proc. 2020, 52, 577–579. [Google Scholar] [CrossRef]
- van der Bom, T.; Winter, M.M.; Bouma, B.J.; Groenink, M.; Vliegen, H.W.; Pieper, P.G.; van Dijk, A.P.; Sieswerda, G.T.; Roos-Hesselink, J.W.; Zwinderman, A.H.; et al. Effect of valsartan on systemic right ventricular function: A double-blind, randomized, placebo-controlled pilot trial. Circulation 2013, 127, 322–330. [Google Scholar] [CrossRef]
- Mathur, K.; Hsu, D.T.; Lamour, J.M.; Aydin, S.I. Safety of Enalapril in Infants: Data from the Pediatric Heart Network Infant Single Ventricle Trial. J. Pediatr. 2020, 227, 218–223. [Google Scholar] [CrossRef]
- Hu, Y.; Li, D.; Zhou, C.; Xiao, Y.; Sun, S.; Jiang, C.; Chen, L.; Liu, J.; Zhang, H.; Li, F.; et al. Molecular Changes in Prepubertal Left Ventricular Development under Experimental Volume Overload. Front. Cardiovasc. Med. 2022, 9, 850248. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Hu, Y.; Xiao, Y.; Wang, S.; Jiang, C.; Liu, J.; Zhang, H.; Hong, H.; Li, F.; Ye, L. Postnatal Right Ventricular Developmental Track Changed by Volume Overload. J. Am. Heart Assoc. 2021, 10, e020854. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Jiang, C.; Zhao, L.; Sun, S.; Xiao, Y.; Ye, L.; Sun, Q.; Li, J. Metabolic maturation during postnatal right ventricular development switches to heart-contraction regulation due to volume overload. J. Cardiol. 2022, 79, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Li, D.B.; Xu, X.X.; Hu, Y.Q.; Cui, Q.; Xiao, Y.Y.; Sun, S.J.; Chen, L.J.; Ye, L.C.; Sun, Q. Congenital heart disease-associated pulmonary dysplasia and its underlying mechanisms. Am. J. Physiol. Lung Cell Mol. Physiol. 2023, 324, L89–L101. [Google Scholar] [CrossRef]
- Zhou, C.; Li, D.; Cui, Q.; Sun, Q.; Hu, Y.; Xiao, Y.; Jiang, C.; Qiu, L.; Zhang, H.; Ye, L.; et al. Ability of the Right Ventricle to Serve as a Systemic Ventricle in Response to the Volume Overload at the Neonatal Stage. Biology 2022, 11, 1831. [Google Scholar] [CrossRef]
- Zhou, C.; Sun, S.; Hu, M.; Xiao, Y.; Yu, X.; Ye, L.; Qiu, L. Downregulated developmental processes in the postnatal right ventricle under the influence of a volume overload. Cell Death Discov. 2021, 7, 208. [Google Scholar] [CrossRef]
- Li, D.; Qiu, L.; Hong, H.; Chen, H.; Zhao, P.; Xiao, Y.; Zhang, H.; Sun, Q.; Ye, L. A neonatal rat model of pulmonary vein stenosis. Cell Biosci. 2023, 13, 112. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, J.; Fang, Y.; Hu, Y.; Xiao, Y.; Cui, Q.; Jiang, C.; Sun, S.; Chen, H.; Ye, L.; et al. Impaired cell-cell communication and axon guidance because of pulmonary hypoperfusion during postnatal alveolar development. Respir. Res. 2023, 24, 12. [Google Scholar] [CrossRef]
- Wang, S.; Ye, L.; Hong, H.; Tang, C.; Li, M.; Zhang, Z.; Liu, J. A neonatal rat model of increased right ventricular afterload by pulmonary artery banding. J. Thorac. Cardiovasc. Surg. 2017, 154, 1734–1739. [Google Scholar] [CrossRef]
- Ye, L.; Wang, S.; Xiao, Y.; Jiang, C.; Huang, Y.; Chen, H.; Zhang, H.; Zhang, H.; Liu, J.; Xu, Z.; et al. Pressure Overload Greatly Promotes Neonatal Right Ventricular Cardiomyocyte Proliferation: A New Model for the Study of Heart Regeneration. J. Am. Heart Assoc. 2020, 9, e015574. [Google Scholar] [CrossRef] [PubMed]
- Malek Mohammadi, M.; Abouissa, A.; Heineke, J. A surgical mouse model of neonatal pressure overload by transverse aortic constriction. Nat. Protoc. 2021, 16, 775–790. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhu, H.; Wang, S.; Xu, X.; Ye, L. Establishment and Confirmation of a Postnatal Right Ventricular Volume Overload Mouse Model. J. Vis. Exp. 2023, 196, e65372. [Google Scholar] [CrossRef] [PubMed]
- McLennan, D.I.; Solano, E.C.R.; Handler, S.S.; Lincoln, J.; Mitchell, M.E.; Kirkpatrick, E.C. Pulmonary Vein Stenosis: Moving from Past Pessimism to Future Optimism. Front. Pediatr. 2021, 9, 747812. [Google Scholar] [CrossRef] [PubMed]
- Abu-Shaweesh, J.M.; Almidani, E. PDA: Does it matter? Int. J. Pediatr. Adolesc. Med. 2020, 7, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Manjunath, S.C.; Usha, M.K. Isolated Partial Anomalous Pulmonary Venous Connection: Development of Volume Overload and Elevated Estimated Pulmonary Pressure in Adults. J. Clin. Imaging Sci. 2019, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Calkoen, E.E.; Hazekamp, M.G.; Blom, N.A.; Elders, B.B.; Gittenberger-de Groot, A.C.; Haak, M.C.; Bartelings, M.M.; Roest, A.A.; Jongbloed, M.R. Atrioventricular septal defect: From embryonic development to long-term follow-up. Int. J. Cardiol. 2016, 202, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Grosse-Brockhoff, F.; Loogen, F. Ventricular septal defect. Circulation 1968, 38 (Suppl. 1), 13–20. [Google Scholar] [CrossRef] [PubMed]
- Hahn, R.T. Tricuspid Regurgitation. N. Engl. J. Med. 2023, 388, 1876–1891. [Google Scholar] [CrossRef]
- Holst, K.A.; Connolly, H.M.; Dearani, J.A. Ebstein’s Anomaly. Methodist. Debakey Cardiovasc. J. 2019, 15, 138–144. [Google Scholar] [CrossRef]
- Bouzas, B.; Kilner, P.J.; Gatzoulis, M.A. Pulmonary regurgitation: Not a benign lesion. Eur. Heart J. 2005, 26, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Tatewaki, H.; Shiose, A. Pulmonary valve replacement after repaired Tetralogy of Fallot. Gen. Thorac. Cardiovasc. Surg. 2018, 66, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Said, S.M.; Mainwaring, R.D.; Ma, M.; Tacy, T.A.; Hanley, F.L. Pulmonary Valve Repair for Patients with Acquired Pulmonary Valve Insufficiency. Ann. Thorac. Surg. 2016, 101, 2294–2301. [Google Scholar] [CrossRef] [PubMed]
- Sanz, J.; Sánchez-Quintana, D.; Bossone, E.; Bogaard, H.J.; Naeije, R. Anatomy, Function, and Dysfunction of the Right Ventricle: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 1463–1482. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Stan, M.N. Thyrotoxicosis: Diagnosis and Management. Mayo Clin. Proc. 2019, 94, 1048–1064. [Google Scholar] [CrossRef]
- Metcalf, M.K.; Rychik, J. Outcomes in Hypoplastic Left Heart Syndrome. Pediatr. Clin. N. Am. 2020, 67, 945–962. [Google Scholar] [CrossRef]
- Cohen, M.L.; Spray, T.; Gutierrez, F.; Barzilai, B.; Bauwens, D. Congenital tricuspid valve stenosis with atrial septal defect and left anterior fascicular block. Clin. Cardiol. 1990, 13, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Enriquez-Sarano, M.; Akins, C.W.; Vahanian, A. Mitral regurgitation. Lancet 2009, 373, 1382–1394. [Google Scholar] [CrossRef] [PubMed]
- Cramariuc, D.; Bahlmann, E.; Gerdts, E. Grading of Aortic Stenosis: Is it More Complicated in Women? Eur. Cardiol. 2022, 17, e21. [Google Scholar] [CrossRef]
- Bravo-Jaimes, K.; Prakash, S.K. Genetics in bicuspid aortic valve disease: Where are we? Prog. Cardiovasc. Dis. 2020, 63, 398–406. [Google Scholar] [CrossRef]
- Mandras, S.A.; Mehta, H.S.; Vaidya, A. Pulmonary Hypertension: A Brief Guide for Clinicians. Mayo Clin. Proc. 2020, 95, 1978–1988. [Google Scholar] [CrossRef]
- Chetan, D.; Mertens, L.L. Challenges in diagnosis and management of coarctation of the aorta. Curr. Opin. Cardiol. 2022, 37, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Dickey, J.; Phelan, C. Unrepaired Tetralogy of Fallot in Adulthood. N. Engl. J. Med. 2020, 382, e97. [Google Scholar] [CrossRef] [PubMed]
- Rali, P.M.; Criner, G.J. Submassive Pulmonary Embolism. Am. J. Respir. Crit. Care Med. 2018, 198, 588–598. [Google Scholar] [CrossRef]
- Patel, A.B.; Ratnayaka, K.; Bergersen, L. A review: Percutaneous pulmonary artery stenosis therapy: State-of-the-art and look to the future. Cardiol. Young 2019, 29, 93–99. [Google Scholar] [CrossRef]
- Marchini, F.; Meossi, S.; Passarini, G.; Campo, G.; Pavasini, R. Pulmonary Valve Stenosis: From Diagnosis to Current Management Techniques and Future Prospects. Vasc. Health Risk Manag. 2023, 19, 379–390. [Google Scholar] [CrossRef]
- Maron, B.J.; Maron, M.S. Hypertrophic cardiomyopathy. Lancet 2013, 381, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Abassi, H.; Gavotto, A.; Picot, M.C.; Bertet, H.; Matecki, S.; Guillaumont, S.; Moniotte, S.; Auquier, P.; Moreau, J.; Amedro, P. Impaired pulmonary function and its association with clinical outcomes, exercise capacity and quality of life in children with congenital heart disease. Int. J. Cardiol. 2019, 285, 86–92. [Google Scholar] [CrossRef]
- Diller, G.P.; Dimopoulos, K.; Okonko, D.; Li, W.; Babu-Narayan, S.V.; Broberg, C.S.; Johansson, B.; Bouzas, B.; Mullen, M.J.; Poole-Wilson, P.A.; et al. Exercise intolerance in adult congenital heart disease: Comparative severity, correlates, and prognostic implication. Circulation 2005, 112, 828–835. [Google Scholar] [CrossRef]
- Talman, V.; Teppo, J.; Pöhö, P.; Movahedi, P.; Vaikkinen, A.; Karhu, S.T.; Trošt, K.; Suvitaival, T.; Heikkonen, J.; Pahikkala, T.; et al. Molecular Atlas of Postnatal Mouse Heart Development. J. Am. Heart Assoc. 2018, 7, e010378. [Google Scholar] [CrossRef]
- Guo, Y.; Pu, W.T. Cardiomyocyte Maturation: New Phase in Development. Circ. Res. 2020, 126, 1086–1106. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, K.N.; Kikuchi, R.; Ngoh, G.A.; Coughlan, K.A.; Dominguez, I.; Stanley, W.C.; Walsh, K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ. Res. 2012, 111, 1012–1026. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Novotova, M.; Fortin, D.; Garnier, A.; Ventura-Clapier, R.; Veksler, V.; Joubert, F. Postnatal development of mouse heart: Formation of energetic microdomains. J. Physiol. 2010, 588 Pt 13, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.O.; Chiang, D.Y.; Wang, W.; Beavers, D.L.; Dixit, S.S.; Skapura, D.G.; Landstrom, A.P.; Song, L.S.; Ackerman, M.J.; Wehrens, X.H. Junctophilin-2 is necessary for T-tubule maturation during mouse heart development. Cardiovasc. Res. 2013, 100, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Yang, H.; Zhang, S.S.; Cho, H.C.; Kalashnikova, M.; Sun, B.; Zhang, H.; Bhargava, A.; Grabe, M.; Olgin, J.; et al. Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat. Med. 2014, 20, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Ong, L.P.; Bargehr, J.; Knight-Schrijver, V.R.; Lee, J.; Colzani, M.; Bayraktar, S.; Bernard, W.G.; Marchiano, S.; Bertero, A.; Murry, C.E.; et al. Epicardially secreted fibronectin drives cardiomyocyte maturation in 3D-engineered heart tissues. Stem Cell Rep. 2023, 18, 936–951. [Google Scholar] [CrossRef] [PubMed]
- Reiser, P.J.; Portman, M.A.; Ning, X.H.; Schomisch Moravec, C. Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1814–H1820. [Google Scholar] [CrossRef]
- Cui, Y.; Zheng, Y.; Liu, X.; Yan, L.; Fan, X.; Yong, J.; Hu, Y.; Dong, J.; Li, Q.; Wu, X.; et al. Single-Cell Transcriptome Analysis Maps the Developmental Track of the Human Heart. Cell Rep. 2019, 26, 1934–1950.e5. [Google Scholar] [CrossRef] [PubMed]
- Kubalak, S.W.; Miller-Hance, W.C.; O’Brien, T.X.; Dyson, E.; Chien, K.R. Chamber specification of atrial myosin light chain-2 expression precedes septation during murine cardiogenesis. J. Biol. Chem. 1994, 269, 16961–16970. [Google Scholar] [CrossRef]
- O’Brien, T.X.; Lee, K.J.; Chien, K.R. Positional specification of ventricular myosin light chain 2 expression in the primitive murine heart tube. Proc. Natl. Acad. Sci. USA 1993, 90, 5157–5161. [Google Scholar] [CrossRef]
- Bedada, F.B.; Chan, S.S.; Metzger, S.K.; Zhang, L.; Zhang, J.; Garry, D.J.; Kamp, T.J.; Kyba, M.; Metzger, J.M. Acquisition of a quantitative, stoichiometrically conserved ratiometric marker of maturation status in stem cell-derived cardiac myocytes. Stem Cell Rep. 2014, 3, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., 2nd; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Uosaki, H.; Cahan, P.; Lee, D.I.; Wang, S.; Miyamoto, M.; Fernandez, L.; Kass, D.A.; Kwon, C. Transcriptional Landscape of Cardiomyocyte Maturation. Cell Rep. 2015, 13, 1705–1716. [Google Scholar] [CrossRef] [PubMed]
- Calmettes, G.; John, S.A.; Weiss, J.N.; Ribalet, B. Hexokinase-mitochondrial interactions regulate glucose metabolism differentially in adult and neonatal cardiac myocytes. J. Gen. Physiol. 2013, 142, 425–436. [Google Scholar] [CrossRef] [PubMed]
- DeLaughter, D.M.; Bick, A.G.; Wakimoto, H.; McKean, D.; Gorham, J.M.; Kathiriya, I.S.; Hinson, J.T.; Homsy, J.; Gray, J.; Pu, W.; et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell 2016, 39, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Goversen, B.; van der Heyden, M.A.G.; van Veen, T.A.B.; de Boer, T.P. The immature electrophysiological phenotype of iPSC-CMs still hampers in vitro drug screening: Special focus on IK1. Pharmacol. Ther. 2018, 183, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Haufe, V.; Camacho, J.A.; Dumaine, R.; Günther, B.; Bollensdorff, C.; von Banchet, G.S.; Benndorf, K.; Zimmer, T. Expression pattern of neuronal and skeletal muscle voltage-gated Na+ channels in the developing mouse heart. J. Physiol. 2005, 564 Pt 3, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Link, S.; Meissner, M.; Held, B.; Beck, A.; Weissgerber, P.; Freichel, M.; Flockerzi, V. Diversity and developmental expression of L-type calcium channel beta2 proteins and their influence on calcium current in murine heart. J. Biol. Chem. 2009, 284, 30129–30137. [Google Scholar] [CrossRef] [PubMed]
- Ellingsen, O.; Davidoff, A.J.; Prasad, S.K.; Berger, H.J.; Springhorn, J.P.; Marsh, J.D.; Kelly, R.A.; Smith, T.W. Adult rat ventricular myocytes cultured in defined medium: Phenotype and electromechanical function. Am. J. Physiol. 1993, 265 Pt 2, H747–H754. [Google Scholar] [CrossRef]
- Cho, G.S.; Lee, D.I.; Tampakakis, E.; Murphy, S.; Andersen, P.; Uosaki, H.; Chelko, S.; Chakir, K.; Hong, I.; Seo, K.; et al. Neonatal Transplantation Confers Maturation of PSC-Derived Cardiomyocytes Conducive to Modeling Cardiomyopathy. Cell Rep. 2017, 18, 571–582. [Google Scholar] [CrossRef]
- Heidi Au, H.T.; Cui, B.; Chu, Z.E.; Veres, T.; Radisic, M. Cell culture chips for simultaneous application of topographical and electrical cues enhance phenotype of cardiomyocytes. Lab Chip 2009, 9, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Jacot, J.G.; McCulloch, A.D.; Omens, J.H. Substrate stiffness affects the functional maturation of neonatal rat ventricular myocytes. Biophys. J. 2008, 95, 3479–3487. [Google Scholar] [CrossRef] [PubMed]
- Mihic, A.; Li, J.; Miyagi, Y.; Gagliardi, M.; Li, S.H.; Zu, J.; Weisel, R.D.; Keller, G.; Li, R.K. The effect of cyclic stretch on maturation and 3D tissue formation of human embryonic stem cell-derived cardiomyocytes. Biomaterials 2014, 35, 2798–2808. [Google Scholar] [CrossRef] [PubMed]
- Radisic, M.; Park, H.; Shing, H.; Consi, T.; Schoen, F.J.; Langer, R.; Freed, L.E.; Vunjak-Novakovic, G. Functional assembly of engineered myocardium by electrical stimulation of cardiac myocytes cultured on scaffolds. Proc. Natl. Acad. Sci. USA 2004, 101, 18129–18134. [Google Scholar] [CrossRef] [PubMed]
- Rog-Zielinska, E.A.; Richardson, R.V.; Denvir, M.A.; Chapman, K.E. Glucocorticoids and foetal heart maturation; implications for prematurity and foetal programming. J. Mol. Endocrinol. 2014, 52, R125–R135. [Google Scholar] [CrossRef]
- Krüger, M.; Sachse, C.; Zimmermann, W.H.; Eschenhagen, T.; Klede, S.; Linke, W.A. Thyroid hormone regulates developmental titin isoform transitions via the phosphatidylinositol-3-kinase/AKT pathway. Circ. Res. 2008, 102, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Butler, J.H. Parturition-related changes in insulin-like growth factors-I and -II in the perinatal lamb. J. Endocrinol. 1983, 99, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Rodriguez, M.L.; Leonard, A.; Sun, L.; Fischer, K.A.; Wang, Y.; Ritterhoff, J.; Zhao, L.; Kolwicz, S.C., Jr.; Pabon, L.; et al. Fatty Acids Enhance the Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2019, 13, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Jardin, B.D.; Zhou, P.; Sethi, I.; Akerberg, B.N.; Toepfer, C.N.; Ai, Y.; Li, Y.; Ma, Q.; Guatimosim, S.; et al. Hierarchical and stage-specific regulation of murine cardiomyocyte maturation by serum response factor. Nat. Commun. 2018, 9, 3837. [Google Scholar] [CrossRef]
- Friedman, C.E.; Nguyen, Q.; Lukowski, S.W.; Helfer, A.; Chiu, H.S.; Miklas, J.; Levy, S.; Suo, S.; Han, J.J.; Osteil, P.; et al. Single-Cell Transcriptomic Analysis of Cardiac Differentiation from Human PSCs Reveals HOPX-Dependent Cardiomyocyte Maturation. Cell Stem Cell 2018, 23, 586–598.e8. [Google Scholar] [CrossRef]
- Lee, W.S.; Kim, J. Peroxisome Proliferator-Activated Receptors and the Heart: Lessons from the Past and Future Directions. PPAR Res. 2015, 2015, 271983. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; McDonald, C.; Petrenko, N.B.; Leblanc, M.; Wang, T.; Giguere, V.; Evans, R.M.; Patel, V.V.; Pei, L. Estrogen-related receptor α (ERRα) and ERRγ are essential coordinators of cardiac metabolism and function. Mol. Cell Biol. 2015, 35, 1281–1298. [Google Scholar] [CrossRef]
- Thébaud, B.; Goss, K.N.; Laughon, M.; Whitsett, J.A.; Abman, S.H.; Steinhorn, R.H.; Aschner, J.L.; Davis, P.G.; McGrath-Morrow, S.A.; Soll, R.F.; et al. Bronchopulmonary dysplasia. Nat. Rev. Dis. Primers 2019, 5, 78. [Google Scholar] [CrossRef] [PubMed]
- Salaets, T.; Aertgeerts, M.; Gie, A.; Vignero, J.; de Winter, D.; Regin, Y.; Jimenez, J.; Vande Velde, G.; Allegaert, K.; Deprest, J.; et al. Preterm birth impairs postnatal lung development in the neonatal rabbit model. Respir. Res. 2020, 21, 59. [Google Scholar] [CrossRef] [PubMed]
- Jobe, A.H. Animal Models, Learning Lessons to Prevent and Treat Neonatal Chronic Lung Disease. Front. Med. 2015, 2, 49. [Google Scholar] [CrossRef]
- Zepp, J.A.; Morley, M.P.; Loebel, C.; Kremp, M.M.; Chaudhry, F.N.; Basil, M.C.; Leach, J.P.; Liberti, D.C.; Niethamer, T.K.; Ying, Y.; et al. Genomic, epigenomic, and biophysical cues controlling the emergence of the lung alveolus. Science 2021, 371, eabc3172. [Google Scholar] [CrossRef]
- Frank, B.S.; Ivy, D.D. Pediatric Pulmonary Arterial Hypertension. Pediatr. Clin. N. Am. 2020, 67, 903–921. [Google Scholar] [CrossRef] [PubMed]
- Lvy, D.D.; Abman, S.H.; Barst, R.J.; Berger, R.M.; Bonnet, D.; Fleming, T.R.; Haworth, S.G.; Raj, J.U.; Rosenzweig, E.B.; Schulze Neick, I.; et al. Pediatric pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62 (Suppl. 25), D117–D126. [Google Scholar] [CrossRef]
- Herring, M.J.; Putney, L.F.; Wyatt, G.; Finkbeiner, W.E.; Hyde, D.M. Growth of alveoli during postnatal development in humans based on stereological estimation. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L338–L344. [Google Scholar] [CrossRef]
- Ochs, M.; Nyengaard, J.R.; Jung, A.; Knudsen, L.; Voigt, M.; Wahlers, T.; Richter, J.; Gundersen, H.J. The number of alveoli in the human lung. Am. J. Respir. Crit. Care Med. 2004, 169, 120–124. [Google Scholar] [CrossRef]
- Rippa, A.L.; Alpeeva, E.V.; Vasiliev, A.V.; Vorotelyak, E.A. Alveologenesis: What Governs Secondary Septa Formation. Int. J. Mol. Sci. 2021, 22, 12107. [Google Scholar] [CrossRef]
- Mund, S.I.; Stampanoni, M.; Schittny, J.C. Developmental alveolarization of the mouse lung. Dev. Dyn. 2008, 237, 2108–2116. [Google Scholar] [CrossRef]
- Beauchemin, K.J.; Wells, J.M.; Kho, A.T.; Philip, V.M.; Kamir, D.; Kohane, I.S.; Graber, J.H.; Bult, C.J. Temporal dynamics of the developing lung transcriptome in three common inbred strains of laboratory mice reveals multiple stages of postnatal alveolar development. PeerJ 2016, 4, e2318. [Google Scholar] [CrossRef]
- Frank, D.B.; Peng, T.; Zepp, J.A.; Snitow, M.; Vincent, T.L.; Penkala, I.J.; Cui, Z.; Herriges, M.J.; Morley, M.P.; Zhou, S.; et al. Emergence of a Wave of Wnt Signaling that Regulates Lung Alveologenesis by Controlling Epithelial Self-Renewal and Differentiation. Cell Rep. 2016, 17, 2312–2325. [Google Scholar] [CrossRef]
- Lee, J.H.; Tammela, T.; Hofree, M.; Choi, J.; Marjanovic, N.D.; Han, S.; Canner, D.; Wu, K.; Paschini, M.; Bhang, D.H.; et al. Anatomically and Functionally Distinct Lung Mesenchymal Populations Marked by Lgr5 and Lgr6. Cell 2017, 170, 1149–1163.e12. [Google Scholar] [CrossRef] [PubMed]
- Kugler, M.C.; Loomis, C.A.; Zhao, Z.; Cushman, J.C.; Liu, L.; Munger, J.S. Sonic Hedgehog Signaling Regulates Myofibroblast Function during Alveolar Septum Formation in Murine Postnatal Lung. Am. J. Respir Cell Mol. Biol. 2017, 57, 280–293. [Google Scholar] [CrossRef]
- He, H.; Snowball, J.; Sun, F.; Na, C.L.; Whitsett, J.A. IGF1R controls mechanosignaling in myofibroblasts required for pulmonary alveologenesis. JCI Insight 2021, 6, e144863. [Google Scholar] [CrossRef] [PubMed]
- Al Alam, D.; El Agha, E.; Sakurai, R.; Kheirollahi, V.; Moiseenko, A.; Danopoulos, S.; Shrestha, A.; Schmoldt, C.; Quantius, J.; Herold, S.; et al. Evidence for the involvement of fibroblast growth factor 10 in lipofibroblast formation during embryonic lung development. Development 2015, 142, 4139–4150. [Google Scholar] [CrossRef] [PubMed]
- Alanis, D.M.; Chang, D.R.; Akiyama, H.; Krasnow, M.A.; Chen, J. Two nested developmental waves demarcate a compartment boundary in the mouse lung. Nat. Commun. 2014, 5, 3923. [Google Scholar] [CrossRef]
- McGowan, S.; Jackson, S.K.; Jenkins-Moore, M.; Dai, H.H.; Chambon, P.; Snyder, J.M. Mice bearing deletions of retinoic acid receptors demonstrate reduced lung elastin and alveolar numbers. Am. J. Respir Cell Mol. Biol. 2000, 23, 162–167. [Google Scholar] [CrossRef]
- Zhang, K.; Yao, E.; Lin, C.; Chou, Y.T.; Wong, J.; Li, J.; Wolters, P.J.; Chuang, P.T. A mammalian Wnt5a-Ror2-Vangl2 axis controls the cytoskeleton and confers cellular properties required for alveologenesis. eLife 2020, 9, e53688. [Google Scholar] [CrossRef] [PubMed]
- Marino, B.S.; Cassedy, A.; Brown, K.L.; Franklin, R.; Gaynor, J.W.; Cvetkovic, M.; Laker, S.; Levinson, K.; MacGloin, H.; Mahony, L.; et al. Long-Term Quality of Life in Congenital Heart Disease Surgical Survivors: Multicenter Retrospective Study of Surgical and ICU Explanatory Factors. Pediatr. Crit. Care Med. 2023, 24, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, L.; Yang, T.; Huang, P.; Wang, L.; Zhao, L.; Zhang, S.; Ye, Z.; Chen, L.; Zheng, Z.; et al. Congenital Heart Disease and Risk of Cardiovascular Disease: A Meta-Analysis of Cohort Studies. J. Am. Heart Assoc. 2019, 8, e012030. [Google Scholar] [CrossRef]
- Shi, B.; Zhang, X.; Song, Z.; Dai, Z.; Luo, K.; Chen, B.; Zhou, Z.; Cui, Y.; Feng, B.; Zhu, Z.; et al. Targeting gut microbiota-derived kynurenine to predict and protect the remodeling of the pressure-overloaded young heart. Sci. Adv. 2023, 9, eadg7417. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Li, D.; Xiao, Y.; Sun, S.; Ye, L.; Hu, Y.; Hong, H. Volume overload impedes the maturation of sarcomeres and T-tubules in the right atria: A potential cause of atrial arrhythmia following delayed atrial septal defect closure. Front. Physiol. 2023, 14, 1237187. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Hong, H.; Li, M.; Xu, X.; Wang, S.; Xiao, Y.; Zheng, S.; Wang, Z.; Yan, Y.; Chen, H.; et al. A surgical mouse model of neonatal right ventricular outflow tract obstruction by pulmonary artery banding. J. Heart Lung Transplant. 2024, 43, 496–507. [Google Scholar] [CrossRef]
- Du, J.; Zheng, L.; Gao, P.; Yang, H.; Yang, W.J.; Guo, F.; Liang, R.; Feng, M.; Wang, Z.; Zhang, Z.; et al. A small-molecule cocktail promotes mammalian cardiomyocyte proliferation and heart regeneration. Cell Stem Cell 2022, 29, 545–558.e13. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, L.; Liu, H.; Duan, R.; Zhou, H.; Zhang, F.; He, X.; Lu, D.; Xiong, K.; Xiong, M.; et al. LRP6 downregulation promotes cardiomyocyte proliferation and heart regeneration. Cell Res. 2021, 31, 450–462. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, C.H.; Ammanamanchi, N.; Suresh, S.; Lewarchik, C.; Rao, K.; Uys, G.M.; Han, L.; Abrial, M.; Yimlamai, D.; et al. Control of cytokinesis by β-adrenergic receptors indicates an approach for regulating cardiomyocyte endowment. Sci. Transl. Med. 2019, 11, eaaw6419. [Google Scholar] [CrossRef]
- Yutzey, K.E. Cytokinesis, Beta-Blockers, and Congenital Heart Disease. N. Engl. J. Med. 2020, 382, 291–293. [Google Scholar] [CrossRef]
- Gu, J.; Chen, X.; Jin, Y.; Liu, M.; Xu, Q.; Liu, X.; Luo, Z.; Ling, S.; Liu, N.; Liu, S. A Neonatal Mouse Model for Pressure Overload: Myocardial Response Corresponds to Severity. Front. Cardiovasc. Med. 2021, 8, 660246. [Google Scholar] [CrossRef] [PubMed]
- Malek Mohammadi, M.; Abouissa, A.; Azizah, I.; Xie, Y.; Cordero, J.; Shirvani, A.; Gigina, A.; Engelhardt, M.; Trogisch, F.A.; Geffers, R.; et al. Induction of cardiomyocyte proliferation and angiogenesis protects neonatal mice from pressure overload-associated maladaptation. JCI Insight 2019, 5, e128336. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Wang, S.; Wang, Y.; Yang, J.; Bao, N.; Liu, J.; Zhang, Z. Neonatal Heart Responds to Pressure Overload with Differential Alterations in Various Cardiomyocyte Maturation Programs That Accommodate Simultaneous Hypertrophy and Hyperplasia. Front. Cell Dev. Biol. 2020, 8, 596960. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pu, W.; He, L.; Li, Y.; Zhao, H.; Li, Y.; Liu, K.; Huang, X.; Weng, W.; Wang, Q.D.; et al. Cell proliferation fate mapping reveals regional cardiomyocyte cell-cycle activity in subendocardial muscle of left ventricle. Nat. Commun. 2021, 12, 5784. [Google Scholar] [CrossRef] [PubMed]
- Bossers, G.P.L.; Günthel, M.; van der Feen, D.E.; Hagdorn, Q.A.J.; Koop, A.C.; van Duijvenboden, K.; Barnett, P.; Borgdorff, M.A.J.; Christoffels, V.M.; Silljé, H.H.W.; et al. Neuregulin-1 enhances cell-cycle activity, delays cardiac fibrosis, and improves cardiac performance in rat pups with right ventricular pressure load. J. Thorac. Cardiovasc. Surg. 2022, 164, e493–e510. [Google Scholar] [CrossRef]
- Cui, Q.; Sun, S.; Zhu, H.; Xiao, Y.; Jiang, C.; Zhang, H.; Liu, J.; Ye, L.; Shen, J. Volume Overload Initiates an Immune Response in the Right Ventricle at the Neonatal Stage. Front. Cardiovasc. Med. 2021, 8, 772336. [Google Scholar] [CrossRef] [PubMed]
- Canseco, D.C.; Kimura, W.; Garg, S.; Mukherjee, S.; Bhattacharya, S.; Abdisalaam, S.; Das, S.; Asaithamby, A.; Mammen, P.P.; Sadek, H.A. Human ventricular unloading induces cardiomyocyte proliferation. J. Am. Coll. Cardiol. 2015, 65, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Davoodianidalik, M.; Punzmann, H.; Kellay, H.; Xia, H.; Shats, M.; Francois, N. Fluctuation-Induced Interaction in Turbulent Flows. Phys. Rev. Lett. 2022, 128, 024503. [Google Scholar] [CrossRef] [PubMed]
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A tense situation: Forcing tumour progression. Nat. Rev. Cancer 2009, 9, 108–122. [Google Scholar] [CrossRef]
- Mehta, V.; Pang, K.L.; Rozbesky, D.; Nather, K.; Keen, A.; Lachowski, D.; Kong, Y.; Karia, D.; Ameismeier, M.; Huang, J.; et al. The guidance receptor plexin D1 is a mechanosensor in endothelial cells. Nature 2020, 578, 290–295. [Google Scholar] [CrossRef]
- Karbassi, E.; Fenix, A.; Marchiano, S.; Muraoka, N.; Nakamura, K.; Yang, X.; Murry, C.E. Cardiomyocyte maturation: Advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020, 17, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, M.; Hao, K.; Lei, W.; Tang, M.; Hu, S. Cardiomyocyte Maturation-the Road is not Obstructed. Stem Cell Rev. Rep. 2022, 18, 2966–2981. [Google Scholar] [CrossRef]
- Kannan, S.; Kwon, C. Regulation of cardiomyocyte maturation during critical perinatal window. J. Physiol. 2020, 598, 2941–2956. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.T.; Ye, S.; Su, J.; Garg, V. Cardiomyocyte Proliferation and Maturation: Two Sides of the Same Coin for Heart Regeneration. Front. Cell Dev. Biol. 2020, 8, 594226. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.E.; Li, L.; Xia, X.; Fu, W.; Liao, Q.; Lan, C.; Yang, D.; Chen, H.; Yue, R.; Zeng, C.; et al. Dedifferentiation, Proliferation, and Redifferentiation of Adult Mammalian Cardiomyocytes After Ischemic Injury. Circulation 2017, 136, 834–848. [Google Scholar] [CrossRef]
- Krieger, E.V.; Zeppenfeld, K.; DeWitt, E.S.; Duarte, V.E.; Egbe, A.C.; Haeffele, C.; Lin, K.Y.; Robinson, M.R.; Sillman, C.; Upadhyay, S.; et al. Arrhythmias in Repaired Tetralogy of Fallot: A Scientific Statement from the American Heart Association. Circ. Arrhythm. Electrophysiol. 2022, 15, e000084. [Google Scholar] [CrossRef]
- Badagliacca, R.; Papa, S.; Valli, G.; Pezzuto, B.; Poscia, R.; Reali, M.; Manzi, G.; Giannetta, E.; Berardi, D.; Sciomer, S.; et al. Right ventricular dyssynchrony and exercise capacity in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2017, 49, 1601419. [Google Scholar] [CrossRef]
- Van Aerde, N.; Meersseman, P.; Debaveye, Y.; Wilmer, A.; Casaer, M.P.; Gunst, J.; Wauters, J.; Wouters, P.J.; Goetschalckx, K.; Gosselink, R.; et al. Aerobic exercise capacity in long-term survivors of critical illness: Secondary analysis of the post-EPaNIC follow-up study. Intensive Care Med. 2021, 47, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, D.C. Respiratory physiology: Adaptations to high-level exercise. Br. J. Sports Med. 2012, 46, 381–384. [Google Scholar] [CrossRef]
- De Troyer, A.; Yernault, J.C.; Englert, M. Lung hypoplasia in congenital pulmonary valve stenosis. Circulation 1977, 56 Pt 1, 647–651. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Namba, F. An experimental animal model of bronchopulmonary dysplasia: Secondary publication. Pediatr. Int. 2021, 63, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Davis, J.M. Future applications of antioxidants in premature infants. Curr. Opin. Pediatr. 2011, 23, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Mourani, P.M.; Sontag, M.K.; Younoszai, A.; Miller, J.I.; Kinsella, J.P.; Baker, C.D.; Poindexter, B.B.; Ingram, D.A.; Abman, S.H. Early pulmonary vascular disease in preterm infants at risk for bronchopulmonary dysplasia. Am. J. Respir Crit. Care Med. 2015, 191, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Herum, K.M.; Lunde, I.G.; Skrbic, B.; Louch, W.E.; Hasic, A.; Boye, S.; Unger, A.; Brorson, S.H.; Sjaastad, I.; Tønnessen, T.; et al. Syndecan-4 is a key determinant of collagen cross-linking and passive myocardial stiffness in the pressure-overloaded heart. Cardiovasc. Res. 2015, 106, 217–226. [Google Scholar] [CrossRef]
- Yu, C.; Qiu, M.; Zhang, Z.; Song, X.; Du, H.; Peng, H.; Li, Q.; Yang, L.; Xiong, X.; Xia, B.; et al. Transcriptome sequencing reveals genes involved in cadmium-triggered oxidative stress in the chicken heart. Poult. Sci. 2021, 100, 100932. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abnormal Hemodynamics Classification | RV VO | LV VO | RV PO | LV PO |
|---|---|---|---|---|
| CHD | ASD, PDA, PAPVD, VSD, TR, EA, PVI, TOF, AVSD, PAH | ASD+TS/RVOT, MR, MVI, AR, BAV | PVS, VSD, PDA, PAH, MS, AS, TOF, IAA, CAA, PAS, PVS | BAV, IAA, CAA, MS, AS, HCM |
| Surgery | Website | Ref. |
|---|---|---|
| nACF | https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.020854, accessed on 13 August 2021 | [23] |
| nTAC | https://www.nature.com/articles/s41596-020-00434-9, accessed on 16 December 2020 | [32] |
| nPAB | https://www.jtcvs.org/article/S0022-5223(17)31192-3/fulltext#supplementaryMaterial, accessed on 14 June 2017 | [30] |
| nPVB | https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-023-01058-8, accessed on 19 June 2023 | [28] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, S.; Ye, L. Hemodynamic Melody of Postnatal Cardiac and Pulmonary Development in Children with Congenital Heart Diseases. Biology 2024, 13, 234. https://doi.org/10.3390/biology13040234
Zheng S, Ye L. Hemodynamic Melody of Postnatal Cardiac and Pulmonary Development in Children with Congenital Heart Diseases. Biology. 2024; 13(4):234. https://doi.org/10.3390/biology13040234
Chicago/Turabian StyleZheng, Sixie, and Lincai Ye. 2024. "Hemodynamic Melody of Postnatal Cardiac and Pulmonary Development in Children with Congenital Heart Diseases" Biology 13, no. 4: 234. https://doi.org/10.3390/biology13040234
APA StyleZheng, S., & Ye, L. (2024). Hemodynamic Melody of Postnatal Cardiac and Pulmonary Development in Children with Congenital Heart Diseases. Biology, 13(4), 234. https://doi.org/10.3390/biology13040234