Simple Summary
Abnormal hemodynamics and cardiac and pulmonary development intertwine to form the most important features of children with congenital heart diseases (CHDs). Here, we review the varieties of hemodynamic abnormalities that exist in children with CHDs and the recently developed neonatal rodent models of CHDs, which have inspired and brought us new ideas and inspirations in cardiomyocyte proliferation and maturation, as well as in alveolar development.
Abstract
Hemodynamics is the eternal theme of the circulatory system. Abnormal hemodynamics and cardiac and pulmonary development intertwine to form the most important features of children with congenital heart diseases (CHDs), thus determining these children’s long-term quality of life. Here, we review the varieties of hemodynamic abnormalities that exist in children with CHDs, the recently developed neonatal rodent models of CHDs, and the inspirations these models have brought us in the areas of cardiomyocyte proliferation and maturation, as well as in alveolar development. Furthermore, current limitations, future directions, and clinical decision making based on these inspirations are highlighted. Understanding how CHD-associated hemodynamic scenarios shape postnatal heart and lung development may provide a novel path to improving the long-term quality of life of children with CHDs, transplantation of stem cell-derived cardiomyocytes, and cardiac regeneration.
1. Introduction
Since Harvey’s discovery of the circulatory system in 1628, it has been recognized that the primary function of the circulatory system is to provide organs, including the heart and lungs, with well-oxygenated blood and nutrients, as well as to transport all types of harmful metabolites [1,2]. The hemodynamics resulting from blood flowing in the circulatory system inevitably becomes the eternal theme of the circulatory system [1,2]. Insufficient embryonic volume load is one of the main causes of left ventricular dysplasia, while adult volume overload (VO) or pressure overload (PO) lead to cardiac failure [3,4,5,6,7]. However, pediatric hearts and lungs are in a stage of active development. For example, 90% of alveoli form between the ages of 0 and 7 years, and there is an extensive cardiomyocyte maturation transition [8,9]. Hemodynamic modifications of embryonic or adult hearts and lungs are well established [3,4,5,6,7] but leave a vacuum to be filled in our understanding of the hemodynamic shaping of pediatric hearts and lungs.
Congenital heart diseases (CHDs) represent the most prevalent birth defect in the world [10,11]. With the progress in pediatric cardiac surgery, cardiology, and cardiac intensive care, most simple CHDs can be corrected, and the overall mortality shows a significant downward trend. However, the mortalities of complex CHDs, such as hypoplastic left heart syndrome, Ebstein anomaly (EA), and tetralogy of Fallot (TOF), are still very high. Most of these cannot be physiologically corrected, leaving residual hemodynamic abnormalities, such as cardiac volume overload (VO), pressure overload (PO), pulmonary congestion and reduced pulmonary blood flow (RPF); therefore, hemodynamic abnormalities dominate postnatal cardiac and pulmonary development in children with CHDs [12,13,14,15,16,17]. As a result, the long-term life quality of children with complex CHDs is poor, with significantly reduced exercise capacity, impaired cardiac performance, and some children ultimately requiring heart or lung transplantation [18,19].
In addition, cardiovascular therapies that are very effective in adults have limited effectiveness or even cause harm when treating infants and young children [20,21]. A possible reason underlying this phenomenon is that the extremely small size of a neonatal mouse heart challenges the creation of neonatal surgical mouse models of cardiovascular diseases. Consequently, clinicians or scientists generally use adult mouse models to explore mechanisms and pathophysiology and to obtain targets for pediatric cardiovascular diseases.
Here, we have reviewed the abnormal hemodynamics in children with CHDs and the recently developed neonatal surgeries for producing CHD-hemodynamic-associated animal models, which include neonatal aortic and inferior vena cava fistula (nACF) surgery, neonatal pulmonary artery banding (nPAB) surgery, neonatal pulmonary vein banding (nPVB) surgery, and neonatal transverse aortic constriction (nTAC) surgery [22,23,24,25,26,27,28,29,30,31,32,33]. nACF produces atrial and ventricular VO and pulmonary congestion; nPAB produces right ventricular (RV) PO and RPF; nPVB produces pulmonary vein stenosis (PVS), which is one of the most challenging issues of pediatric CHDs [34]; and nTAC produces left ventricular (LV) PO. These neonatal surgeries greatly inspire our understanding of the hemodynamic melody of postnatal cardiac and pulmonary development, such as cardiomyocyte proliferation and maturation and also pulmonary dysplasia.
2. Abnormal Hemodynamics in Children with CHD
2.1. Abnormal Hemodynamics in Ventricles
CHD usually leads to ventricular VO and PO, specifically RV VO, LV VO, RV PO, and LV PO. Table 1 summarizes each type of abnormal hemodynamics and its corresponding CHDs.

Table 1.
Abnormal hemodynamics classifications and corresponding congenital heart diseases (CHDs).
There are three types of CHDs that can result in RV VO. The first type is CHD with a left-to-right shunt, which includes atrial septal defect (ASD), patent ductus arteriosus (PDA), partial anomalous pulmonary vein drainage (PAPVD), atrioventricular septal defect (AVSD), and ventricular septal defect (VSD) [17,35,36,37,38]. The second type is right-heart-valve regurgitation-associated CHDs, which include tricuspid regurgitation (TR), EA, pulmonary regurgitation, pulmonary valve insufficiency (PVI), and TOF after patch enlargement of the RV outflow tract correction [39,40,41,42,43]. The third includes advanced pulmonary hypertension (PH), increased returning blood (e.g., in thyrotoxicosis), and functional univentricular conditions (e.g., hypoplastic left heart syndrome and Fontan surgery) [44,45,46].
Right-to-left shunting and left-heart-valvular regurgitation can lead to LV VO. Right-to-left shunting-associated CHDs include ASD with tricuspid stenosis (TS) or RV outflow obstruction (RVOT) [47]. These defects result in a high pressure in the right atrium compared to the left atrium, leading to a right-to-left shunt. Left-heart-valvular regurgitation includes congenital mitral regurgitation (CMR), acquired mitral regurgitation (rheumatic or calcific mitral valve), aortic valve calcification, and bicuspid aortic valve (BAV) [48,49,50]. In addition, increased venous return can lead to LV VO.
PH, left cardiac defects that cause PH at later stages, and RVOT obstructive diseases may cause RV PO. CHDs that cause PH include PVS, large VSD, and PDA [34,35,38,51]. Large VSD and PDA lead to increased pulmonary blood flow, resulting in pulmonary congestion and small vessel remodeling, which in turn cause PH. CHD with a left cardiac defect that causes PH includes mitral or aortic stenosis (MS or AS) and coarctation/interruption of the aortic arch (CAA/IAA) [49,50,52]. CHDs with RVOT obstruction include TOF, isolated pulmonary artery obstruction or embolism, pulmonary artery stenosis (PAS), and pulmonary valve stenosis (PS) [53,54,55,56].
Hypertension and left ventricular outflow tract (LVOT) obstruction lead to LV PO. Hypertension is uncommon in children. CHD with LVOT obstruction includes BAV, CAA/IAA, mitral or aortic stenosis, and hypertrophic cardiomyopathy [49,50,52,57].
It is evident from Table 1 that various hemodynamic abnormalities, including both ventricular VO and PO, usually coexist in the same type of CHD. In addition, these hemodynamic abnormalities are closely associated with disease progression, and sometimes one type of abnormal hemodynamic abnormality dominates a particular stage of disease progression. The extents of VO and PO often determine the performance of the RV and LV, as well as surgical approaches and the prognosis of children with CHDs. Therefore, gaining a comprehensive understanding of how VO and PO shape postnatal cardiac development may help establish a theoretical foundation for enhancing the quality of life in children with CHD.
2.2. Abnormal Hemodynamics in Lungs
A left-to-right shunt or increased venous return will induce increased pulmonary blood flow (IPF), which leads to pulmonary congestion, ultimately resulting in pulmonary hypertension and congestive heart failure. RVOT obstructions, such as TOF and PS, result in RPF, which leads to pulmonary dysplasia, reduced exercise endurance, and renders patients less susceptible to COVID-19 infection [25,29,58,59]. A deeper understanding of the mechanisms by which IPF and RPF regulate postnatal lung development may help improve lung function and exercise capacity in children with CHD.
PVS is another deadly CHD [28,34]. Stenosis of the pulmonary vein leads to the remodeling of small pulmonary vessels, which in turn leads to PH, and ultimately right heart failure occurs. Despite advancements in surgical techniques, interventional procedures, and postoperative monitoring, the prognosis for children with PVS remains unfavorable, with 60% of children with PVS dying within 2 years after diagnosis [34]. Moreover, due to stenosis, there may be oozing blood upstream of the pulmonary vessels, giving rise to hemoptysis and pulmonary infections, which further deteriorate the prognosis of children with PVS. Understanding how and why PVS occurs is crucial to treating children with PVS.
3. Immature Hearts and Lungs in Children
3.1. Immature Hearts in Children
Unlike adult mature hearts and lungs, children’s hearts and lungs are immature and at an active stage of development [8,12].
To meet the physiological demands of pumping blood at adulthood, there are three main characteristics that immature cardiomyocytes (CMs) need to develop (Figure 1A,B) [8,60,61,62,63,64,65,66]: (1) sarcomere maturation, which includes sarcomere component maturation (an Myh7 to Myh6 and TnI1 to TnI3 switch), and arrangement maturation (disordered and irregular arrangement switched to a rod-like and orderly arrangement); (2) metabolism maturation, which is a shift from anaerobic glycolysis to oxidative phosphorylation due to mitochondrial maturation, with an increase in the number of mitochondrial and their ridges, as well as a closer proximity of mitochondria to sarcomeres; and (3) electrophysiological maturation, in which transverse tubules (T-tubules) form with a gradually increased density and integrity and are accompanied by a significant improvement in calcium handling and excitation–contraction coupling. Failure of CM maturation results in arrhythmias and heart failure.
In terms of sarcomere maturation, several sarcomere components switch from a fetal to an adult isoform: (1) Myosin heavy chain (Myh) switches from fetal MYH7 to adult MYH6 in mice. In contrast, MYH7 is the predominant isoform in the adult heart of humans, and is it already established by 5 weeks of gestation [67,68]. (2) Myosin light chain (MYL) switches from fetal MYL7 (MLC2a) to adult MYL2 (MLC2v) in ventricles. In addition, MYL7 expression becomes restricted to atrial CMs in adults [69,70]. (3) Troponin switches from fetal TNNI1 to adult TNNI3 [71].
In terms of metabolism maturation, in addition to the mitochondria switch, metabolic transcriptional regulators undergo active changes, which include upregulations of genes involved in fatty acid metabolism, oxidative phosphorylation, and mitochondrial biogenesis, such as Ppargc1a/b, Ppara, Nrf1/2, and Esrra/b/g, and downregulations of glycolytic genes [72,73]. Glycolytic enzymes switch from fetal HK1 (hexokinase 1) to HK2 (hexokinase 2) [74], and COX (cytochrome c oxidase) subunit 8 switches from fetal COX8A to adult COX8B [75].
In terms of electrophysiological maturation, in addition to T-tubule formation, there are also important switches: (1) The resting membrane potential changes from less negative (approximately −50 to −60 mV) to more negative (approximately −85 mV) because of the higher expressions of the potassium channels (Kir) Kir2.1 and Kir2.2, encoded by genes KCNJ2 and KCNJ [76]. (2) The upstroke velocity action potential switches from slow (approximately 15–30 V/s) to fast (approximately 70–242 V/s) due to the higher expressions of SCN5A and other sodium channels [77]. (3) The plateau phase of the action potential switches from short to long due to the higher expression of the Cav1.2 core component CACNA1C and alternative splicing of its auxiliary subunit CACNB2 [78].
However, the mechanisms that govern CM maturation remain elusive. It is now generally believed that microenvironmental factors and intracellular transcriptional regulation, as well as post transcriptional modifications, play important roles. For example, mature primary cultured CMs gradually lose their maturity, and immature CMs transferred to adult hearts can mature [79,80], indicating that the microenvironment surrounding CMs plays an important role in the regulation of maturation. In addition, geometric constraints, extracellular matrix viscoelasticity, mechanical strain, and electrical stimulation are currently used to promote CM maturation in vitro [81,82,83,84], thus providing evidence that microenvironmental factors play a role in cardiomyocyte maturation. However, due to the largely unknown mechanisms by which microenvironmental factors regulate CM maturation, the maturity of CMs obtained through environmental factor stimulation is much lower than that of mature adult cardiomyocytes. Furthermore, biochemical and transcriptional factors have also been suggested to play critical roles in CM maturation [85,86,87,88,89,90,91,92]. We summarize the CM maturation factors that have been reported and those that remain unknown in Figure 1C.
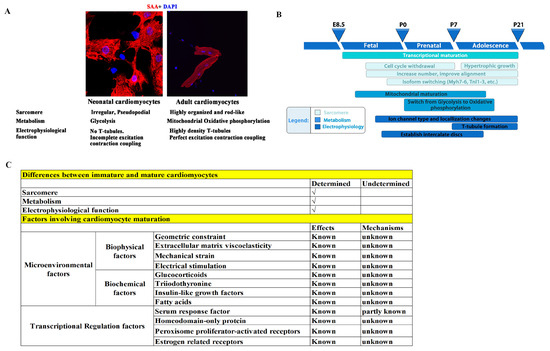
Figure 1.
Illustration of the three main characteristics that immature CMs require for development. (A) Neonatal and adult CMs have differences in sarcomere, metabolism, and electrophysiological functions. (B) Three main characteristics that immature CMs must develop, and the presumed key time points of these maturation events in rodents. Redrawn from [12]. (C) Summary of cardiomyocyte maturation factors that have been reported and those that remain unknown [67,68,69,70,71,72,73,74,75,76,77,78,81,82,83,84,85,86,87,88,89,90,91,92].
Figure 1.
Illustration of the three main characteristics that immature CMs require for development. (A) Neonatal and adult CMs have differences in sarcomere, metabolism, and electrophysiological functions. (B) Three main characteristics that immature CMs must develop, and the presumed key time points of these maturation events in rodents. Redrawn from [12]. (C) Summary of cardiomyocyte maturation factors that have been reported and those that remain unknown [67,68,69,70,71,72,73,74,75,76,77,78,81,82,83,84,85,86,87,88,89,90,91,92].
3.2. Immature Lungs in Children
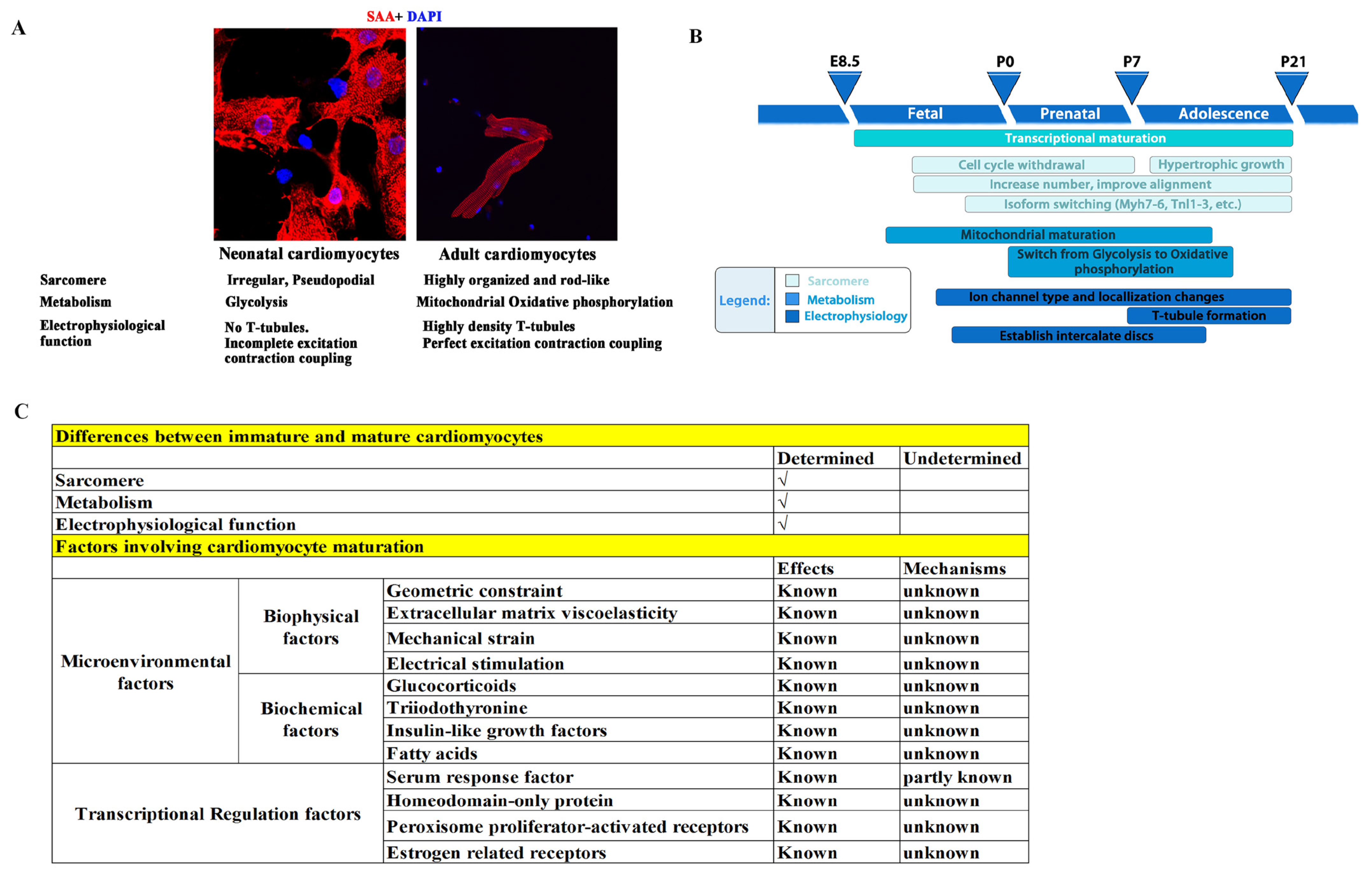
There are five developmental stages of human lungs (Figure 2A) [93,94,95,96]: (1) The embryonic stage (pulmonary bud stage)—occurring between the third and sixth weeks of gestation, this stage marks the appearance of precursor cells and epidermal cells known as pulmonary buds. (2) The pseudoglandular stage (bronchial stage)—taking place between the sixth and sixteenth weeks of gestation, this stage is characterized by bronchial formation. The pulmonary buds proliferate and migrate to the visceral mesenchyme, forming airways and peripheral acinar buds, and eventually developing into alveoli. (3) The tubular phase (alveolar tubular phase)—this stage occurs from 16 to 26 weeks of gestation and involves the differentiation of intra-airway epithelial cells, including basal cells, cupped cells, ciliated cells, and other secretory cells. (4) The vesicular phase (terminal vesicular phase)—spanning from 26 to 36 weeks of gestation, the differentiation of peripheral glandular alveoli characterizes this phase, including cubic type 2 alveolar (AT2) cells and squamous type 1 alveolar (AT1) cells. At this stage, alveoli gradually expand and are covered by AT1 cells, and AT2 cells differentiate to produce alveolar surfactant. (5) The alveolar phase—this phase, which extends from 36 weeks of gestation to adolescence, is the main stage of alveolar formation, with over 90% of alveoli forming at ages 0–7 years. Bronchopulmonary dysplasia, a major complication of prematurity, occurs when the immature lungs fail to develop due to various damaging factors such as hyperoxia, toxicity, and inflammation. Therefore, the lungs of children are immature and in a critical stage of alveolar development, while adult lungs have completed their development. Consequently, many drugs effective in treating adult lung diseases have limited efficacy in addressing lung diseases in infants and children [97,98].
Postnatal lung maturation includes two aspects of maturation: (1) Alveoli maturation, in which the number of alveoli in humans increases from 0 to 50 million at birth to >300 million at adulthood [99,100]. (2) Microvascular maturation, in which the double capillary networks in the parenchymal septa at birth are restructured to a single capillary system at adulthood to enhance gas–blood exchange (Figure 2B) [101,102]. Although the significant differences between the lungs of children and those of adults have been clearly established, the mechanisms underlying postnatal lung maturation are far from clear. Transcriptomic analysis has revealed that postnatal lung maturation can be divided into four different stages (stage 1–4), exhibiting a two-phase pattern of angiogenesis, alveolarization, and neurogenesis. For example, in mice, compared to stage 1 (postnatal day 0–3), stage 2 (postnatal day 4–7) has shown a significant upregulation of angiogenic genes; compared to stage 3 (postnatal day 9–12), there was a peak in the expression of angiogenic genes in stage 4 (postnatal day 13–18) [103]. The transcriptomic analysis of postnatal lung development has suggested that there might be a complicated and extensive genomic coordination between vascular, neurogenic, and alveolar processes for lung maturation [103]. A more recent combined genomic, epigenomic, and biophysical analysis has identified that AT1 cells have a distinct signaling hub that integrates Shh/Wnt/Pdgf signaling pathways for early postnatal alveoli formation [96]. Several important pathways regulating postnatal lung maturation have also been suggested [104,105,106,107,108,109,110,111]. However, many important questions need answers. For example, how is the maturation of blood vessels, alveoli, and nerves coordinated and regulated? Why are some premature infants unable to develop mature lungs, despite the strong regenerative ability of alveoli throughout childhood and even adulthood? The answers to these questions will help us to treat the various forms of lung damage inflicted early in life. We illustrate the known lung maturation factors and certain key pathways regulating postnatal lung maturation in Figure 2C.
Figure 2.
Differences between immature and mature lungs. (A) Five developmental stages of human lungs. Note that 90% of alveoli are formed at ages 0–7 years. (B) Different capillary network between immature and mature lungs. Redrawn from [101]. (C) Several pathways involved in lung maturation [99,100,101,102,103,104,105,106,107,108,109,110,111].
Figure 2.
Differences between immature and mature lungs. (A) Five developmental stages of human lungs. Note that 90% of alveoli are formed at ages 0–7 years. (B) Different capillary network between immature and mature lungs. Redrawn from [101]. (C) Several pathways involved in lung maturation [99,100,101,102,103,104,105,106,107,108,109,110,111].
4. Creation of Neonatal Surgical Rodent Models of CHDs
4.1. Challenges in Constructing Neonatal Surgical Rodent Models of CHDs
The lack of neonatal rodent models for cardiac VO, PO, IPF, and RPF limits our understanding of how hemodynamics regulates postnatal heart and lung development [23,28,30,31]. Consequently, managing CHD primarily relies on surgical intervention, and thus the long-term quality of life for children with complex CHD remains poor [112,113]. The challenges of creating neonatal rodent CHD models are as follows: (1) the size of the neonatal rodent heart is extremely small, with limited operation space; (2) neonatal anesthesia is ice cooling, limiting the surgical time to no more than 20 min; and (3) cannibalization may account for additional post-surgical mortality in neonatal surgery. The above three challenges require superb cardiac microsurgical skills.
As the largest pediatric heart center in the world, the heart center of Shanghai Children’s Medical Center has about 4000 open-heart surgeries per year; as a result, surgeons at Shanghai Children’s Medical Center are very skillful in cardiac surgery. Thus, in recent years, they have successfully developed a series of neonatal CHD models (Figure 3), including models of nACF, nPAB, nPVB, and nTAC [23,28,30,31,114].
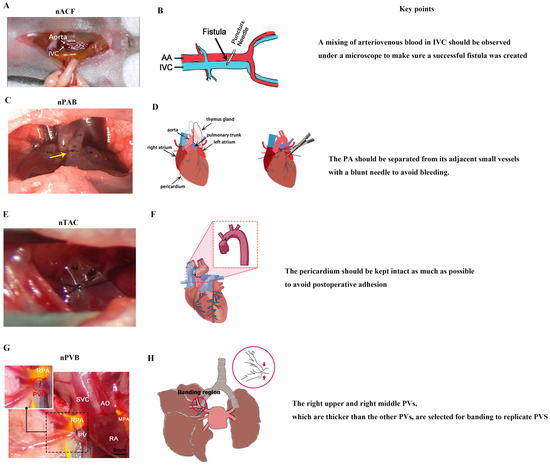
Figure 3.
Neonatal surgical rodent models of CHDs. (A) nACF produces RA VO, RV VO, LA VO, LV VO, and IPF. (B) Surgical diagram of nACF. (C) nPAB produces RV PO and RPF, yellow arrow indicates the banding region. (D) Surgical diagram of nPAB. (E) nTAC produces LVPO. (F) Surgical diagram of nTAC. (G) nPVB produces PVS and IPF. (H) Surgical diagram of nPVB, red arrow indicates the banding region. IVC: inferior vena cava; PA: pulmonary artery; DA: ductus arteriosus; Ao: aorta; RPA: right pulmonary artery; PV: pulmonary vein; SVC: superior vena cava; RA: right atrium; MPA: main pulmonary artery. (A) was adopted ref. [23] under a Creative Commons Attribution-NonCommercial-NoDerivs License; (B) was adopted from ref. [115] under a Creative Commons Attribution-NonCommercial-NoDerivs License; (C) was adopted from ref. [30] with the permission of the publisher; (D) was adopted from ref. [116] with the permission of the publisher; (H) was adopted from ref. [28] under a Creative Commons Attribution-NonCommercial-NoDerivs License).
4.2. Key Points of Constructing Neonatal Surgical Rodent Models of CHDs
4.2.1. nACF Surgery
nACF surgery [33] (Figure 3A,B) includes the following four steps: (1) anesthesia and fixation; (2) abdominal aorta (AA) and inferior vena cava (IVC) exposure; (3) fistula creation, and (4) abdominal closure. There are two key points of nACF surgery: (1) During fistula creation, a mixing of arteriovenous blood in IVC should be observed under a microscope to make sure a successful fistula was created. (2) When exposing the AA and IVC, the small intestine and large intestine should be slowly moved with a cotton swab to prevent bleeding.
4.2.2. nPAB Surgery
nPAB surgery [30] (Figure 3C,D) includes the following four steps: (1) anesthesia and fixation; (2) pulmonary artery (PA) exposure; (3) PA banding; and (4) closure of the thoracic cavity. There are two key points of nPAB surgery: (1) The PA should be exposed with an incision as small as possible to reduce the total time for the thoracotomy surgery, which is critical for the neonatal pups’ recovery from anesthesia. (2) The PA should be separated from its adjacent small vessels with a blunt needle to avoid bleeding.
4.2.3. nTAC Surgery
nTAC surgery [32] (Figure 3E,F) includes the following four steps: (1) anesthesia and fixation; (2) ascending aorta exposure; (3) aorta banding; and (4) closure of the thoracic cavity. There are two key points of nTAC surgery: (1) Blunt needles should be used to prevent intraoperative bleeding, which is a primary cause of postoperative mortality. (2) During the surgery, the pericardium must be carefully separated from the aortic surface to expose the aortic site for banding. The pericardium should be kept intact as much as possible to avoid postoperative adhesion.
4.2.4. nPVB Surgery
The nPVB surgery [28] (Figure 3G,H) includes the following four steps: (1) anesthesia and fixation; (2) pulmonary vein (PV) exposure; (3) PV banding; and (4) closure of the thoracic cavity. There are two key points of nPVB surgery: (1) The right upper and right middle PVs, which are thicker than the other PVs, are selected for banding to replicate PVS. (2) The chest is closed layer by layer to avoid postoperative pneumothorax.
In summary, nACF surgery, which does not require opening the thoracic cavity, is easier to perform than the other three neonatal surgeries. All neonatal surgeries require ice-cooling anesthesia to reduce the operation time as much as possible, which increases the rate of neonatal pup recovery from anesthesia. A blunt needle is required to avoid intraoperative bleeding. For thoracotomy surgery, preventing postoperative adhesions and pneumothorax is key for the neonatal pups’ long term survival. Surgical videos for each neonatal cardiac surgery can be found at public access websites, which are summarized in Table 2.
Table 2.
Neonatal cardiac surgery teaching videos.
5. Hemodynamic Melody of Pediatric Heart and Lung Development
5.1. Cardiomyocyte Proliferation
Promoting cardiomyocyte proliferation is a fundamental method for treating heart failure that not only improves heart failure caused by myocardial infarction [117,118], but also improves pediatric heart failure caused by CHDs [78,89]. A previous study has demonstrated that children with TOF have impaired RV cardiomyocyte proliferation due to a reduced expression of epithelial cell transforming 2 (ECT2) [119]. The study also found that beta blockers can promote cardiomyocyte proliferation by enhancing the expression of ECT2 [119]. Thus, beta blockers have been recommended to treat TOF [119]. The New England Journal of Medicine has highlighted the study, commenting that the treatment of CHDs may not rely on surgical correction; this study opened a new approach for treating CHDs [120].
However, this study lacked a nPAB model to match the most important TOF clinical feature—RVOT obstruction, which produces RV PO–and made its conclusion based on an increase in the percentage of polyploidic cardiomyocytes in children with TOF [119]. An increased percentage of polyploidic cardiomyocytes could be a consequence of cardiomyocyte proliferation (Figure 4). Increasingly, more studies have shown that PO promoted neonatal cardiomyocyte proliferation in both RV and LV [31,121,122,123,124,125]. We have also found that PO promoted RV cardiomyocyte proliferation in children with TOF [31]. Therefore, it is inappropriate and unwise to ignore the contribution of PO to cardiomyocyte proliferation when studying pediatric TOF. Moreover, CHD is a polygenic disease, and mutations of ECT2 have rarely been reported in TOF patients [119]. Thus, it is more reasonable to open a new approach for CHD treatment focused on abnormal hemodynamics than a single gene modification.
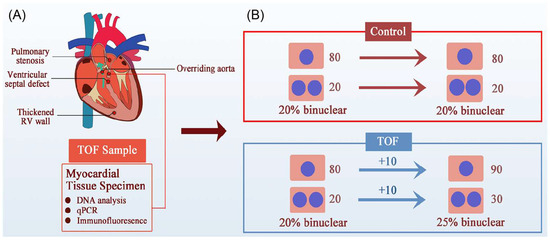
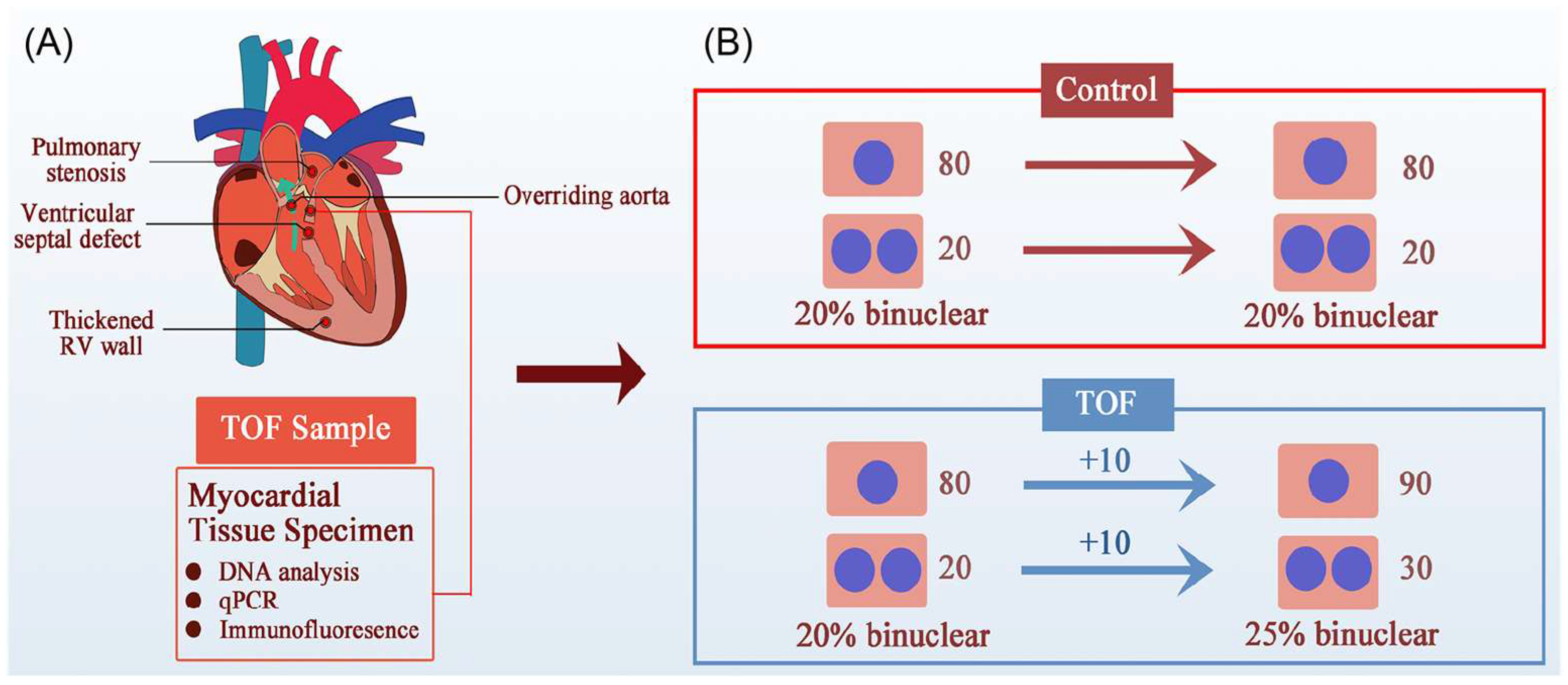
Figure 4.
An alternative possibility for the increased percentage of polyploid cardiomyocytes in TOF children. (A) The four clinical features of TOF are RVOT obstruction, ventricular septal defect, thickened RV, and aortic riding, the most important of which is RVOT obstruction, which leads to RV PO and RV failure. (B) Illustration of the increased binuclear cardiomyocytes generated in TOF patients. Greater numbers of binuclear cardiomyocytes do not indicate a failure of cytokinesis. For example, in the beginning of a study, if both the control group and TOF group had 80 mononucleated and 20 binucleated cardiomyocytes (CMs), the proportion of binucleated CMs was 20%. Under PO conditions, both mononucleated and binucleated CMs in the TOF group increased by 10, and the proportion of binucleated CMs was 25%. Therefore, an increase in the proportion of binucleated CMs does not necessarily mean impaired cytokinesis and proliferation.
Nevertheless, the effect of PO on promoting proliferation of cardiomyocytes decreases with age, and PO cannot re-induce proliferation in differentiated and mature CMs at adult [31]. However, interestingly, VO re-induces proliferation in prepubertal cardiomyocytes in both the LV and RV [22,23], but does not promote proliferation in neonatal cardiomyocytes [126]. In contrast, with heart failure and at an adult stage, pressure unloading promotes cardiomyocyte proliferation [127]. These results show the complex roles of forces generated by abnormal hemodynamics on cardiomyocyte proliferation.
Force is one of the foundations of matter [128,129], and the maintenance of many life phenomena depend on a balance of forces [128,129]. How force regulates cardiac regeneration remains elusive, and perhaps changing a single mechanoreceptor can promote cardiac regeneration. In fact, a recent study has demonstrated that Plxnd1 is a necessary and sufficient condition for endothelial cells to sense force for regulating cardiovascular pathophysiology [130]. We have also found that Plxnd1 significantly increased in prepubertal cardiomyocytes under VO conditions (Figure 5). The introduction of neonatal ventricular PO and VO models provides a platform for researchers to explore the regulation of cardiac regeneration by forces, and the role of Plxnd1 may be one of the future directions. In addition to Plxnd1, other mechanoreceptors, such as YAP1, Sdc4, and Itgal1, have also been implicated in the PO and VO regulation of postnatal cardiac development (Figure 5). It should be noted that combinations of mechanical receptors undergo changes at different developmental stages and under different forces (VO or PO).
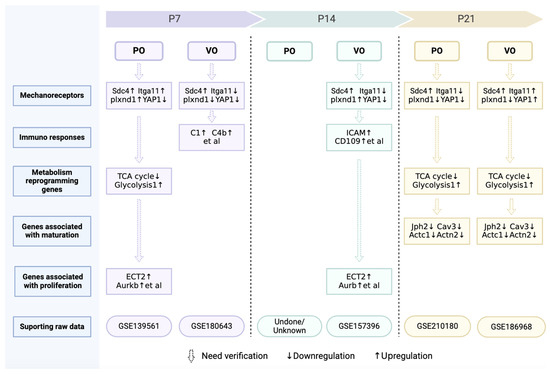
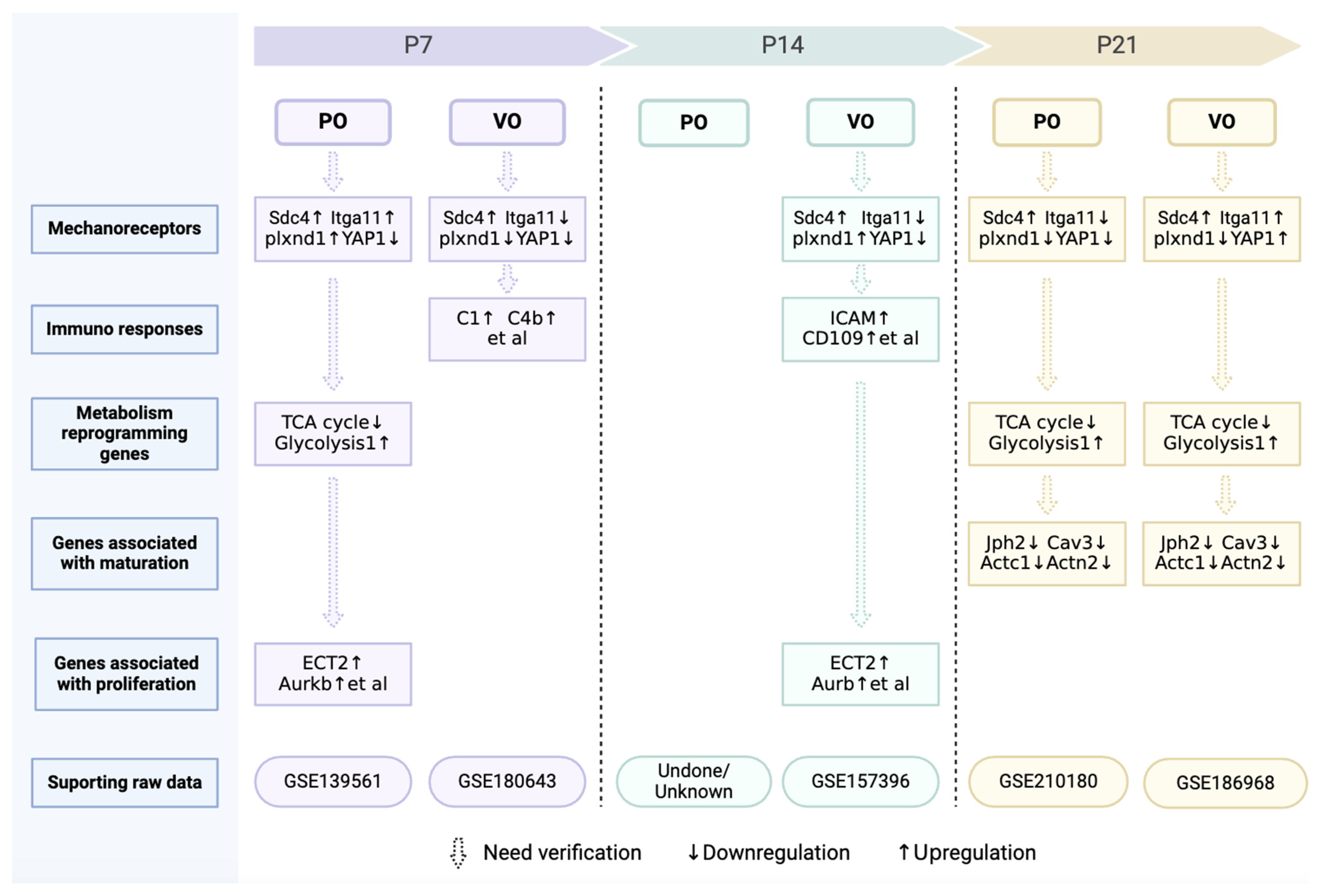
Figure 5.
Omics information on postnatal cardiac development under of VO or PO.
5.2. Cardiomyocyte Maturation
Maturation is the most important event of postnatal CM development in mammals [62,131]. Although the underlying mechanisms of cardiomyocyte maturation are largely unknown, the switch from pediatric to adult hemodynamics is undoubtedly an important contribution [132,133]. Due to the increase in body size, the preload and afterload faced by the adult heart are significantly greater than that of children [132,134]. In other words, it may be the force generated by the increasing preload and afterload that guide the maturation of cardiomyocytes.
The immaturity of CMs derives from induced pluripotent stem cells (iPSC-CMs) and their failure to mature in vivo limits their clinical use [131,132]. Therefore, in vitro experiments with various stimuli, including electrical rhythm, force, and 3D culture, were used to promote the maturation of iPSC-CMs [131,132]. Although their maturity has increased, there is still a large gap between stimuli-enhanced iPSC-CMs and mature adult CMs [131,132]. The differences between in vivo and in vitro conditions, as well as an insufficient understanding of force, may account for the limited effect of force on CM maturation in studies.
Recent studies have indicated that CM proliferation and maturation are two opposite processes [134,135]. Because VO and PO promote CM proliferation at neonatal or prepubertal stages [23,31], it is not surprising to find that CM maturation was impaired because of VO [22,23]. We have also found that VO impeded CM maturation (Figure 5). The purpose of iPS cm transplantation is to treat patients with heart failure, the hemodynamic characteristic of which is VO. Thus, the finding that VO impedes CM maturation may further deter the transplantation of iPSC-CMs into patients with heart failure.
Another issue is whether VO or PO affect CM maturation temporarily or permanently. In other words, when VO or PO are released, can the CMs still mature? This is the situation that exists in children with CHD. When the structural defects of the hearts of children with CHDs are corrected, will the maturity of their adult CMs be affected? Clinical investigations have found that even with perfect anatomic correction in childhood, patients with TOF are still at a high risk of arrhythmia [136], a feature of cardiomyocyte immaturity, suggesting that CM maturation may be permanently impaired.
In summary, some force is helpful for CM maturation in vitro, and the force generated by age-increased preload and afterload may guide CM maturation in vivo. However, excessive force due to VO and PO impairs postnatal cardiomyocyte maturation. A key question is how force regulates CM maturation. Does maturation share the same mechanoreceptor(s) used in proliferation? After mechanoreceptor activation, different pathways involved in maturation and proliferation may be subsequently activated. In fact, we have found that VO initiated an immune response at neonatal and prepubertal stages in both the RV and LV, including macrophage activation [22,23,126]. Consistently, the activation of the mechanoreceptor Plxnd1 in endothelial cells has led to expression of macrophage chemokines, which recruited macrophages from the peripheral blood to the heart [130]. These interesting studies are a good foundation for us to understand how force, a basic component of the material world, affects cardiovascular pathophysiology. We summarize the genes and pathways identified from our omics studies in Figure 5, which shows that considerable further investigation and verification are still needed.
5.3. Lung Development
The interplay of the heart and lung profoundly, functionally, and anatomically determine a person’s quality of life [137,138,139]. About half a century ago, an autopsy study revealed a decrease in lung volume and pulmonary dysfunction in patients with RPF-associated CHDs [140]. However, the underlying mechanisms remained elusive until recent nPAB surgery was developed, which showed that RPF caused alveolar dysplasia, angiogenesis impairment, and inflammation [28,29]. RPF also impaired cell–cell communication and axon guidance, two critical events of late alveolar formation [29,103]. Axon guidance is required for the coronavirus infection, which may explain why children with RPF-associated CHDs are relatively insensitive to COVID-19 infection [29,141]. RPF reduces the intravascular pressure and gas exchange rate, which means a reduced force in the vessels of the lung. It is possible that force is the basic cause of RPF-induced pulmonary dysplasia. Interestingly, RPF also induces inflammation [25], similar to that of VO-induced cardiomyocyte proliferation, further suggesting that there may be a force-mediated regulation.
Current bronchopulmonary dysplasia (BPD) animal models for premature infants include models of hyperoxia, pulmonary ventilation, and lipopolysaccharide [96,142], all of which aim to induce inflammation, yet yield poor targets for improving lung development of premature infants [94,143]. This may be because inflammation is not the initiating factor. Complications of premature infants often include PH and PDA [94,144], both of which induce RPF. Thus, RPF may account for premature BPD, and the nPAB model may provide a new window into the study of BPD.
In contrast to RPF, IPF, which increases intravascular pressure, leads to pulmonary congestion and thickening of the pulmonary small blood vessels, a characteristic of PH, which in turn increases intravascular pressure, and ultimately a vicious circle forms. A nPVB model has shown a similar presentation as children with PVS, which included PV thickening, pulmonary small vessels thickening, pulmonary congestion, PH, and RV failure [28]. Consistently, the nACF model causing IPF also showed the thickening of pulmonary small blood vessels, but to a lesser extent [33]. Mechanoreceptors are less studied in the lungs than in the heart, including their regulation of force in lungs.
6. Summary and Prospects
Force is a basic component of our world and is generated by abnormal hemodynamics in CHDs, producing profound effects on postnatal heart and lung development, which we were previously unaware of due to the lack of neonatal rodent animal models. Nevertheless, we now know that both VO and PO promote prepubertal CM proliferation and impede CM maturation, and we also know that RPF leads to pulmonary dysplasia and IPF leads to the thickening of pulmonary blood vessels.
6.1. Clinical Decision Making
VO- and PO-impaired CM maturation are associated with arrhythmias and weakened cardiac systolic function [132,133], suggesting that childhood corrections of VO or PO or the promotion of CM maturation may improve adult cardiac performance. However, VO and PO promote prepubertal CM proliferation, which is fundamental to heart failure treatment [31,33], suggesting that VO and PO should be enhanced. Clearly, these two suggestions are contradictory. An in-depth understanding of how force regulates CM proliferation and maturation is a prerequisite for us to precisely regulate proliferation and maturation via VO and PO to improve the quality of life of children with CHD.
RPF and IPF are both detrimental to pulmonary performance, suggesting that they should be corrected as early as possible. If RPF or IPF cannot be corrected, mechanoreceptor blockers, axon guidance molecules, or vessel thickening inhibitors are suggested to be used as early as possible to improve pulmonary performance of children with CHD.
6.2. Limitations and Future Directions
Currently, the neonatal surgical rodent models only help us obtain a very primitive observation, leaving an abundance of unanswered questions: (1) Do mechanoreceptors for VO or PO promote cardiomyocyte proliferation or impede CM maturation? Apart from Plxnd1, other mechanoreceptors have also been revealed. Sdc4 is upregulated, while Itga11 is downregulated in the neonatal PO RV (Figure 5). The expression patterns of neonatal Sdc4 and Itga11 are different from that of adults, in whom both are upregulated [145,146]. How PO or VO regulate CM proliferation and maturation via Plxnd1, Sdc4, and Itga11 might be a future direction of research. Whether there are other mechanoreceptors that play a crucial role in regulating postnatal heart and lung development also needs to be explored. (2) Metabolic reprogramming has also occurred under the condition of neonatal PO and VO (Figure 5). Is metabolic reprogramming the cause or result of CM proliferation? How do PO and VO initiate metabolic reprogramming of CMs? (3) The chromatin openness of many genes associated with heart and lung development has been changed greatly by PO, VO, IPF, and RPF. The epigenetics study of hemodynamics should also be a future direction.
7. Conclusions
Abnormal hemodynamics and cardiac and pulmonary development intertwine to form the most important features of childhood CHDs. With the development of neonatal CHD rodent models, a window into the CHD-associated hemodynamic regulation of postnatal heart and lung development has been opened, allowing us a superficial but important view of postnatal heart and lung development (Figure 5). With continuous and in-depth research on neonatal CHD rodent models, the long-term quality of life of children with CHD may be improved, and the mechanisms underlying CM proliferation and maturation and lung development will be more comprehensively understood.
8. Searching Methods
We used “congenital heart disease AND neonatal OR animal models OR cardiomyocyte OR proliferation OR maturation OR alveoli” as the search method in PubMed.
Author Contributions
S.Z., writing—original draft preparation; L.Y., writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Shanghai Natural Science Foundation (22ZR1479900), the National Natural Science Foundation of China (NSFC) (82270314), and the Innovation Project of the Distinguished Medical Team in Ningbo (2022020405).
Institutional Review Board Statement
Not applicable to studies that do not involve humans or animals.
Informed Consent Statement
Not applicable to studies that do not involve humans.
Data Availability Statement
The raw data in Figure 5 were all generated by us and were deposited in NCBI’s Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo, accessed on 19 August 2020, 8 December 2021, 16 August 2021, 1 May 2024, and 7 April 2022) with accession number GSE139561, GSE180643, GSE157396, GSE210180, and GSE186968.
Acknowledgments
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Aird, W.C. Discovery of the cardiovascular system: From Galen to William Harvey. J. Thromb. Haemost. 2011, 9 (Suppl. 1), 118–129. [Google Scholar] [CrossRef] [PubMed]
- Bynum, B.; Bynum, H. William Harvey’s demonstration rod. Lancet 2015, 386, 1933. [Google Scholar] [CrossRef]
- Dewan, S.; Krishnamurthy, A.; Kole, D.; Conca, G.; Kerckhoffs, R.; Puchalski, M.D.; Omens, J.H.; Sun, H.; Nigam, V.; McCulloch, A.D. Model of Human Fetal Growth in Hypoplastic Left Heart Syndrome: Reduced Ventricular Growth Due to Decreased Ventricular Filling and Altered Shape. Front. Pediatr. 2017, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Feit, L.R.; Copel, J.A.; Kleinman, C.S. Foramen ovale size in the normal and abnormal human fetal heart: An indicator of transatrial flow physiology. Ultrasound Obstet. Gynecol. 1991, 1, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Tanai, E.; Frantz, S. Pathophysiology of Heart Failure. Compr. Physiol. 2015, 6, 187–214. [Google Scholar] [CrossRef] [PubMed]
- Thandavarayan, R.A.; Chitturi, K.R.; Guha, A. Pathophysiology of Acute and Chronic Right Heart Failure. Cardiol. Clin. 2020, 38, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Nagalingam, R.S.; Chattopadhyaya, S.; Al-Hattab, D.S.; Cheung, D.Y.C.; Schwartz, L.Y.; Jana, S.; Aroutiounova, N.; Ledingham, D.A.; Moffatt, T.L.; Landry, N.M.; et al. Scleraxis and fibrosis in the pressure-overloaded heart. Eur. Heart J. 2022, 43, 4739–4750. [Google Scholar] [CrossRef] [PubMed]
- Burri, P.H. Fetal and postnatal development of the lung. Annu. Rev. Physiol. 1984, 46, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Whitsett, J.A.; Wert, S.E.; Trapnell, B.C. Genetic disorders influencing lung formation and function at birth. Hum. Mol. Genet. 2004, 13, R207–R215. [Google Scholar] [CrossRef]
- Pedra, C.A.; Haddad, J.; Pedra, S.F.; Peirone, A.; Pilla, C.B.; Marin-Neto, J.A. Paediatric and congenital heart disease in South America: An overview. Heart 2009, 95, 1385–1392. [Google Scholar] [CrossRef]
- Fernandes, P.S.; Magalhães, L.R.; Pezzini, T.R.; de Sousa Santos, E.F.; Calderon, M.G. Congenital heart diseases trends in São Paulo State, Brazil: A national live birth data bank analysis. World J. Pediatr. 2022, 18, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Galdos, F.X.; Guo, Y.; Paige, S.L.; VanDusen, N.J.; Wu, S.M.; Pu, W.T. Cardiac Regeneration: Lessons from Development. Circ. Res. 2017, 120, 941–959. [Google Scholar] [CrossRef] [PubMed]
- GBD 2017 Congenital Heart Disease Collaborators. Global, Regional, and National Burden of Congenital Heart Disease, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet Child Adolesc. Health 2020, 4, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Beaton, A.Z.; Boehme, A.K.; Buxton, A.E.; et al. Heart Disease and Stroke Statistics-2023 Update: A Report from the American Heart Association. Circulation 2023, 147, e93–e621. [Google Scholar] [CrossRef] [PubMed]
- Corporan, D.; Segura, A.; Padala, M. Ultrastructural Adaptation of the Cardiomyocyte to Chronic Mitral Regurgitation. Front. Cardiovasc. Med. 2021, 8, 714774. [Google Scholar] [CrossRef] [PubMed]
- Offen, S.M.; Baker, D.; Puranik, R.; Celermajer, D.S. Right ventricular volume and its relationship to functional tricuspid regurgitation. Int. J. Cardiol. Heart Vasc. 2021, 38, 100940. [Google Scholar] [CrossRef] [PubMed]
- Otani, H.; Kagaya, Y.; Yamane, Y.; Chida, M.; Ito, K.; Namiuchi, S.; Shiba, N.; Koseki, Y.; Ninomiya, M.; Ikeda, J.; et al. Long-term right ventricular volume overload increases myocardial fluorodeoxyglucose uptake in the interventricular septum in patients with atrial septal defect. Circulation 2000, 101, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.W.; Fisher, S.A.; Wehman, B.; Mehra, M.R. Adults with congenital heart disease and heart transplantation: Optimizing outcomes. J. Heart Lung Transplant. 2014, 33, 873–877. [Google Scholar] [CrossRef]
- Palomo-López, N.; Escalona-Rodríguez, S.; Martín-Villén, L.; Herruzo-Avilés, Á.; Hinojosa-Pérez, R.; Escoresca-Ortega, A.; Porras-López, M.; Corcia-Palomo, Y.; Adsuar-Gómez, A. Transplantation in Congenital Heart Disease: A Challenge. Transplant. Proc. 2020, 52, 577–579. [Google Scholar] [CrossRef]
- van der Bom, T.; Winter, M.M.; Bouma, B.J.; Groenink, M.; Vliegen, H.W.; Pieper, P.G.; van Dijk, A.P.; Sieswerda, G.T.; Roos-Hesselink, J.W.; Zwinderman, A.H.; et al. Effect of valsartan on systemic right ventricular function: A double-blind, randomized, placebo-controlled pilot trial. Circulation 2013, 127, 322–330. [Google Scholar] [CrossRef]
- Mathur, K.; Hsu, D.T.; Lamour, J.M.; Aydin, S.I. Safety of Enalapril in Infants: Data from the Pediatric Heart Network Infant Single Ventricle Trial. J. Pediatr. 2020, 227, 218–223. [Google Scholar] [CrossRef]
- Hu, Y.; Li, D.; Zhou, C.; Xiao, Y.; Sun, S.; Jiang, C.; Chen, L.; Liu, J.; Zhang, H.; Li, F.; et al. Molecular Changes in Prepubertal Left Ventricular Development under Experimental Volume Overload. Front. Cardiovasc. Med. 2022, 9, 850248. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Hu, Y.; Xiao, Y.; Wang, S.; Jiang, C.; Liu, J.; Zhang, H.; Hong, H.; Li, F.; Ye, L. Postnatal Right Ventricular Developmental Track Changed by Volume Overload. J. Am. Heart Assoc. 2021, 10, e020854. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Jiang, C.; Zhao, L.; Sun, S.; Xiao, Y.; Ye, L.; Sun, Q.; Li, J. Metabolic maturation during postnatal right ventricular development switches to heart-contraction regulation due to volume overload. J. Cardiol. 2022, 79, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Li, D.B.; Xu, X.X.; Hu, Y.Q.; Cui, Q.; Xiao, Y.Y.; Sun, S.J.; Chen, L.J.; Ye, L.C.; Sun, Q. Congenital heart disease-associated pulmonary dysplasia and its underlying mechanisms. Am. J. Physiol. Lung Cell Mol. Physiol. 2023, 324, L89–L101. [Google Scholar] [CrossRef]
- Zhou, C.; Li, D.; Cui, Q.; Sun, Q.; Hu, Y.; Xiao, Y.; Jiang, C.; Qiu, L.; Zhang, H.; Ye, L.; et al. Ability of the Right Ventricle to Serve as a Systemic Ventricle in Response to the Volume Overload at the Neonatal Stage. Biology 2022, 11, 1831. [Google Scholar] [CrossRef]
- Zhou, C.; Sun, S.; Hu, M.; Xiao, Y.; Yu, X.; Ye, L.; Qiu, L. Downregulated developmental processes in the postnatal right ventricle under the influence of a volume overload. Cell Death Discov. 2021, 7, 208. [Google Scholar] [CrossRef]
- Li, D.; Qiu, L.; Hong, H.; Chen, H.; Zhao, P.; Xiao, Y.; Zhang, H.; Sun, Q.; Ye, L. A neonatal rat model of pulmonary vein stenosis. Cell Biosci. 2023, 13, 112. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, J.; Fang, Y.; Hu, Y.; Xiao, Y.; Cui, Q.; Jiang, C.; Sun, S.; Chen, H.; Ye, L.; et al. Impaired cell-cell communication and axon guidance because of pulmonary hypoperfusion during postnatal alveolar development. Respir. Res. 2023, 24, 12. [Google Scholar] [CrossRef]
- Wang, S.; Ye, L.; Hong, H.; Tang, C.; Li, M.; Zhang, Z.; Liu, J. A neonatal rat model of increased right ventricular afterload by pulmonary artery banding. J. Thorac. Cardiovasc. Surg. 2017, 154, 1734–1739. [Google Scholar] [CrossRef]
- Ye, L.; Wang, S.; Xiao, Y.; Jiang, C.; Huang, Y.; Chen, H.; Zhang, H.; Zhang, H.; Liu, J.; Xu, Z.; et al. Pressure Overload Greatly Promotes Neonatal Right Ventricular Cardiomyocyte Proliferation: A New Model for the Study of Heart Regeneration. J. Am. Heart Assoc. 2020, 9, e015574. [Google Scholar] [CrossRef] [PubMed]
- Malek Mohammadi, M.; Abouissa, A.; Heineke, J. A surgical mouse model of neonatal pressure overload by transverse aortic constriction. Nat. Protoc. 2021, 16, 775–790. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhu, H.; Wang, S.; Xu, X.; Ye, L. Establishment and Confirmation of a Postnatal Right Ventricular Volume Overload Mouse Model. J. Vis. Exp. 2023, 196, e65372. [Google Scholar] [CrossRef] [PubMed]
- McLennan, D.I.; Solano, E.C.R.; Handler, S.S.; Lincoln, J.; Mitchell, M.E.; Kirkpatrick, E.C. Pulmonary Vein Stenosis: Moving from Past Pessimism to Future Optimism. Front. Pediatr. 2021, 9, 747812. [Google Scholar] [CrossRef] [PubMed]
- Abu-Shaweesh, J.M.; Almidani, E. PDA: Does it matter? Int. J. Pediatr. Adolesc. Med. 2020, 7, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Manjunath, S.C.; Usha, M.K. Isolated Partial Anomalous Pulmonary Venous Connection: Development of Volume Overload and Elevated Estimated Pulmonary Pressure in Adults. J. Clin. Imaging Sci. 2019, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Calkoen, E.E.; Hazekamp, M.G.; Blom, N.A.; Elders, B.B.; Gittenberger-de Groot, A.C.; Haak, M.C.; Bartelings, M.M.; Roest, A.A.; Jongbloed, M.R. Atrioventricular septal defect: From embryonic development to long-term follow-up. Int. J. Cardiol. 2016, 202, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Grosse-Brockhoff, F.; Loogen, F. Ventricular septal defect. Circulation 1968, 38 (Suppl. 1), 13–20. [Google Scholar] [CrossRef] [PubMed]
- Hahn, R.T. Tricuspid Regurgitation. N. Engl. J. Med. 2023, 388, 1876–1891. [Google Scholar] [CrossRef]
- Holst, K.A.; Connolly, H.M.; Dearani, J.A. Ebstein’s Anomaly. Methodist. Debakey Cardiovasc. J. 2019, 15, 138–144. [Google Scholar] [CrossRef]
- Bouzas, B.; Kilner, P.J.; Gatzoulis, M.A. Pulmonary regurgitation: Not a benign lesion. Eur. Heart J. 2005, 26, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Tatewaki, H.; Shiose, A. Pulmonary valve replacement after repaired Tetralogy of Fallot. Gen. Thorac. Cardiovasc. Surg. 2018, 66, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Said, S.M.; Mainwaring, R.D.; Ma, M.; Tacy, T.A.; Hanley, F.L. Pulmonary Valve Repair for Patients with Acquired Pulmonary Valve Insufficiency. Ann. Thorac. Surg. 2016, 101, 2294–2301. [Google Scholar] [CrossRef] [PubMed]
- Sanz, J.; Sánchez-Quintana, D.; Bossone, E.; Bogaard, H.J.; Naeije, R. Anatomy, Function, and Dysfunction of the Right Ventricle: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 1463–1482. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Stan, M.N. Thyrotoxicosis: Diagnosis and Management. Mayo Clin. Proc. 2019, 94, 1048–1064. [Google Scholar] [CrossRef]
- Metcalf, M.K.; Rychik, J. Outcomes in Hypoplastic Left Heart Syndrome. Pediatr. Clin. N. Am. 2020, 67, 945–962. [Google Scholar] [CrossRef]
- Cohen, M.L.; Spray, T.; Gutierrez, F.; Barzilai, B.; Bauwens, D. Congenital tricuspid valve stenosis with atrial septal defect and left anterior fascicular block. Clin. Cardiol. 1990, 13, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Enriquez-Sarano, M.; Akins, C.W.; Vahanian, A. Mitral regurgitation. Lancet 2009, 373, 1382–1394. [Google Scholar] [CrossRef] [PubMed]
- Cramariuc, D.; Bahlmann, E.; Gerdts, E. Grading of Aortic Stenosis: Is it More Complicated in Women? Eur. Cardiol. 2022, 17, e21. [Google Scholar] [CrossRef]
- Bravo-Jaimes, K.; Prakash, S.K. Genetics in bicuspid aortic valve disease: Where are we? Prog. Cardiovasc. Dis. 2020, 63, 398–406. [Google Scholar] [CrossRef]
- Mandras, S.A.; Mehta, H.S.; Vaidya, A. Pulmonary Hypertension: A Brief Guide for Clinicians. Mayo Clin. Proc. 2020, 95, 1978–1988. [Google Scholar] [CrossRef]
- Chetan, D.; Mertens, L.L. Challenges in diagnosis and management of coarctation of the aorta. Curr. Opin. Cardiol. 2022, 37, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Dickey, J.; Phelan, C. Unrepaired Tetralogy of Fallot in Adulthood. N. Engl. J. Med. 2020, 382, e97. [Google Scholar] [CrossRef] [PubMed]
- Rali, P.M.; Criner, G.J. Submassive Pulmonary Embolism. Am. J. Respir. Crit. Care Med. 2018, 198, 588–598. [Google Scholar] [CrossRef]
- Patel, A.B.; Ratnayaka, K.; Bergersen, L. A review: Percutaneous pulmonary artery stenosis therapy: State-of-the-art and look to the future. Cardiol. Young 2019, 29, 93–99. [Google Scholar] [CrossRef]
- Marchini, F.; Meossi, S.; Passarini, G.; Campo, G.; Pavasini, R. Pulmonary Valve Stenosis: From Diagnosis to Current Management Techniques and Future Prospects. Vasc. Health Risk Manag. 2023, 19, 379–390. [Google Scholar] [CrossRef]
- Maron, B.J.; Maron, M.S. Hypertrophic cardiomyopathy. Lancet 2013, 381, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Abassi, H.; Gavotto, A.; Picot, M.C.; Bertet, H.; Matecki, S.; Guillaumont, S.; Moniotte, S.; Auquier, P.; Moreau, J.; Amedro, P. Impaired pulmonary function and its association with clinical outcomes, exercise capacity and quality of life in children with congenital heart disease. Int. J. Cardiol. 2019, 285, 86–92. [Google Scholar] [CrossRef]
- Diller, G.P.; Dimopoulos, K.; Okonko, D.; Li, W.; Babu-Narayan, S.V.; Broberg, C.S.; Johansson, B.; Bouzas, B.; Mullen, M.J.; Poole-Wilson, P.A.; et al. Exercise intolerance in adult congenital heart disease: Comparative severity, correlates, and prognostic implication. Circulation 2005, 112, 828–835. [Google Scholar] [CrossRef]
- Talman, V.; Teppo, J.; Pöhö, P.; Movahedi, P.; Vaikkinen, A.; Karhu, S.T.; Trošt, K.; Suvitaival, T.; Heikkonen, J.; Pahikkala, T.; et al. Molecular Atlas of Postnatal Mouse Heart Development. J. Am. Heart Assoc. 2018, 7, e010378. [Google Scholar] [CrossRef]
- Guo, Y.; Pu, W.T. Cardiomyocyte Maturation: New Phase in Development. Circ. Res. 2020, 126, 1086–1106. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, K.N.; Kikuchi, R.; Ngoh, G.A.; Coughlan, K.A.; Dominguez, I.; Stanley, W.C.; Walsh, K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ. Res. 2012, 111, 1012–1026. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Novotova, M.; Fortin, D.; Garnier, A.; Ventura-Clapier, R.; Veksler, V.; Joubert, F. Postnatal development of mouse heart: Formation of energetic microdomains. J. Physiol. 2010, 588 Pt 13, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.O.; Chiang, D.Y.; Wang, W.; Beavers, D.L.; Dixit, S.S.; Skapura, D.G.; Landstrom, A.P.; Song, L.S.; Ackerman, M.J.; Wehrens, X.H. Junctophilin-2 is necessary for T-tubule maturation during mouse heart development. Cardiovasc. Res. 2013, 100, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Yang, H.; Zhang, S.S.; Cho, H.C.; Kalashnikova, M.; Sun, B.; Zhang, H.; Bhargava, A.; Grabe, M.; Olgin, J.; et al. Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat. Med. 2014, 20, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Ong, L.P.; Bargehr, J.; Knight-Schrijver, V.R.; Lee, J.; Colzani, M.; Bayraktar, S.; Bernard, W.G.; Marchiano, S.; Bertero, A.; Murry, C.E.; et al. Epicardially secreted fibronectin drives cardiomyocyte maturation in 3D-engineered heart tissues. Stem Cell Rep. 2023, 18, 936–951. [Google Scholar] [CrossRef] [PubMed]
- Reiser, P.J.; Portman, M.A.; Ning, X.H.; Schomisch Moravec, C. Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1814–H1820. [Google Scholar] [CrossRef]
- Cui, Y.; Zheng, Y.; Liu, X.; Yan, L.; Fan, X.; Yong, J.; Hu, Y.; Dong, J.; Li, Q.; Wu, X.; et al. Single-Cell Transcriptome Analysis Maps the Developmental Track of the Human Heart. Cell Rep. 2019, 26, 1934–1950.e5. [Google Scholar] [CrossRef] [PubMed]
- Kubalak, S.W.; Miller-Hance, W.C.; O’Brien, T.X.; Dyson, E.; Chien, K.R. Chamber specification of atrial myosin light chain-2 expression precedes septation during murine cardiogenesis. J. Biol. Chem. 1994, 269, 16961–16970. [Google Scholar] [CrossRef]
- O’Brien, T.X.; Lee, K.J.; Chien, K.R. Positional specification of ventricular myosin light chain 2 expression in the primitive murine heart tube. Proc. Natl. Acad. Sci. USA 1993, 90, 5157–5161. [Google Scholar] [CrossRef]
- Bedada, F.B.; Chan, S.S.; Metzger, S.K.; Zhang, L.; Zhang, J.; Garry, D.J.; Kamp, T.J.; Kyba, M.; Metzger, J.M. Acquisition of a quantitative, stoichiometrically conserved ratiometric marker of maturation status in stem cell-derived cardiac myocytes. Stem Cell Rep. 2014, 3, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., 2nd; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Uosaki, H.; Cahan, P.; Lee, D.I.; Wang, S.; Miyamoto, M.; Fernandez, L.; Kass, D.A.; Kwon, C. Transcriptional Landscape of Cardiomyocyte Maturation. Cell Rep. 2015, 13, 1705–1716. [Google Scholar] [CrossRef] [PubMed]
- Calmettes, G.; John, S.A.; Weiss, J.N.; Ribalet, B. Hexokinase-mitochondrial interactions regulate glucose metabolism differentially in adult and neonatal cardiac myocytes. J. Gen. Physiol. 2013, 142, 425–436. [Google Scholar] [CrossRef] [PubMed]
- DeLaughter, D.M.; Bick, A.G.; Wakimoto, H.; McKean, D.; Gorham, J.M.; Kathiriya, I.S.; Hinson, J.T.; Homsy, J.; Gray, J.; Pu, W.; et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell 2016, 39, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Goversen, B.; van der Heyden, M.A.G.; van Veen, T.A.B.; de Boer, T.P. The immature electrophysiological phenotype of iPSC-CMs still hampers in vitro drug screening: Special focus on IK1. Pharmacol. Ther. 2018, 183, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Haufe, V.; Camacho, J.A.; Dumaine, R.; Günther, B.; Bollensdorff, C.; von Banchet, G.S.; Benndorf, K.; Zimmer, T. Expression pattern of neuronal and skeletal muscle voltage-gated Na+ channels in the developing mouse heart. J. Physiol. 2005, 564 Pt 3, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Link, S.; Meissner, M.; Held, B.; Beck, A.; Weissgerber, P.; Freichel, M.; Flockerzi, V. Diversity and developmental expression of L-type calcium channel beta2 proteins and their influence on calcium current in murine heart. J. Biol. Chem. 2009, 284, 30129–30137. [Google Scholar] [CrossRef] [PubMed]
- Ellingsen, O.; Davidoff, A.J.; Prasad, S.K.; Berger, H.J.; Springhorn, J.P.; Marsh, J.D.; Kelly, R.A.; Smith, T.W. Adult rat ventricular myocytes cultured in defined medium: Phenotype and electromechanical function. Am. J. Physiol. 1993, 265 Pt 2, H747–H754. [Google Scholar] [CrossRef]
- Cho, G.S.; Lee, D.I.; Tampakakis, E.; Murphy, S.; Andersen, P.; Uosaki, H.; Chelko, S.; Chakir, K.; Hong, I.; Seo, K.; et al. Neonatal Transplantation Confers Maturation of PSC-Derived Cardiomyocytes Conducive to Modeling Cardiomyopathy. Cell Rep. 2017, 18, 571–582. [Google Scholar] [CrossRef]
- Heidi Au, H.T.; Cui, B.; Chu, Z.E.; Veres, T.; Radisic, M. Cell culture chips for simultaneous application of topographical and electrical cues enhance phenotype of cardiomyocytes. Lab Chip 2009, 9, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Jacot, J.G.; McCulloch, A.D.; Omens, J.H. Substrate stiffness affects the functional maturation of neonatal rat ventricular myocytes. Biophys. J. 2008, 95, 3479–3487. [Google Scholar] [CrossRef] [PubMed]
- Mihic, A.; Li, J.; Miyagi, Y.; Gagliardi, M.; Li, S.H.; Zu, J.; Weisel, R.D.; Keller, G.; Li, R.K. The effect of cyclic stretch on maturation and 3D tissue formation of human embryonic stem cell-derived cardiomyocytes. Biomaterials 2014, 35, 2798–2808. [Google Scholar] [CrossRef] [PubMed]
- Radisic, M.; Park, H.; Shing, H.; Consi, T.; Schoen, F.J.; Langer, R.; Freed, L.E.; Vunjak-Novakovic, G. Functional assembly of engineered myocardium by electrical stimulation of cardiac myocytes cultured on scaffolds. Proc. Natl. Acad. Sci. USA 2004, 101, 18129–18134. [Google Scholar] [CrossRef] [PubMed]
- Rog-Zielinska, E.A.; Richardson, R.V.; Denvir, M.A.; Chapman, K.E. Glucocorticoids and foetal heart maturation; implications for prematurity and foetal programming. J. Mol. Endocrinol. 2014, 52, R125–R135. [Google Scholar] [CrossRef]
- Krüger, M.; Sachse, C.; Zimmermann, W.H.; Eschenhagen, T.; Klede, S.; Linke, W.A. Thyroid hormone regulates developmental titin isoform transitions via the phosphatidylinositol-3-kinase/AKT pathway. Circ. Res. 2008, 102, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Butler, J.H. Parturition-related changes in insulin-like growth factors-I and -II in the perinatal lamb. J. Endocrinol. 1983, 99, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Rodriguez, M.L.; Leonard, A.; Sun, L.; Fischer, K.A.; Wang, Y.; Ritterhoff, J.; Zhao, L.; Kolwicz, S.C., Jr.; Pabon, L.; et al. Fatty Acids Enhance the Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2019, 13, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Jardin, B.D.; Zhou, P.; Sethi, I.; Akerberg, B.N.; Toepfer, C.N.; Ai, Y.; Li, Y.; Ma, Q.; Guatimosim, S.; et al. Hierarchical and stage-specific regulation of murine cardiomyocyte maturation by serum response factor. Nat. Commun. 2018, 9, 3837. [Google Scholar] [CrossRef]
- Friedman, C.E.; Nguyen, Q.; Lukowski, S.W.; Helfer, A.; Chiu, H.S.; Miklas, J.; Levy, S.; Suo, S.; Han, J.J.; Osteil, P.; et al. Single-Cell Transcriptomic Analysis of Cardiac Differentiation from Human PSCs Reveals HOPX-Dependent Cardiomyocyte Maturation. Cell Stem Cell 2018, 23, 586–598.e8. [Google Scholar] [CrossRef]
- Lee, W.S.; Kim, J. Peroxisome Proliferator-Activated Receptors and the Heart: Lessons from the Past and Future Directions. PPAR Res. 2015, 2015, 271983. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; McDonald, C.; Petrenko, N.B.; Leblanc, M.; Wang, T.; Giguere, V.; Evans, R.M.; Patel, V.V.; Pei, L. Estrogen-related receptor α (ERRα) and ERRγ are essential coordinators of cardiac metabolism and function. Mol. Cell Biol. 2015, 35, 1281–1298. [Google Scholar] [CrossRef]
- Thébaud, B.; Goss, K.N.; Laughon, M.; Whitsett, J.A.; Abman, S.H.; Steinhorn, R.H.; Aschner, J.L.; Davis, P.G.; McGrath-Morrow, S.A.; Soll, R.F.; et al. Bronchopulmonary dysplasia. Nat. Rev. Dis. Primers 2019, 5, 78. [Google Scholar] [CrossRef] [PubMed]
- Salaets, T.; Aertgeerts, M.; Gie, A.; Vignero, J.; de Winter, D.; Regin, Y.; Jimenez, J.; Vande Velde, G.; Allegaert, K.; Deprest, J.; et al. Preterm birth impairs postnatal lung development in the neonatal rabbit model. Respir. Res. 2020, 21, 59. [Google Scholar] [CrossRef] [PubMed]
- Jobe, A.H. Animal Models, Learning Lessons to Prevent and Treat Neonatal Chronic Lung Disease. Front. Med. 2015, 2, 49. [Google Scholar] [CrossRef]
- Zepp, J.A.; Morley, M.P.; Loebel, C.; Kremp, M.M.; Chaudhry, F.N.; Basil, M.C.; Leach, J.P.; Liberti, D.C.; Niethamer, T.K.; Ying, Y.; et al. Genomic, epigenomic, and biophysical cues controlling the emergence of the lung alveolus. Science 2021, 371, eabc3172. [Google Scholar] [CrossRef]
- Frank, B.S.; Ivy, D.D. Pediatric Pulmonary Arterial Hypertension. Pediatr. Clin. N. Am. 2020, 67, 903–921. [Google Scholar] [CrossRef] [PubMed]
- Lvy, D.D.; Abman, S.H.; Barst, R.J.; Berger, R.M.; Bonnet, D.; Fleming, T.R.; Haworth, S.G.; Raj, J.U.; Rosenzweig, E.B.; Schulze Neick, I.; et al. Pediatric pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62 (Suppl. 25), D117–D126. [Google Scholar] [CrossRef]
- Herring, M.J.; Putney, L.F.; Wyatt, G.; Finkbeiner, W.E.; Hyde, D.M. Growth of alveoli during postnatal development in humans based on stereological estimation. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L338–L344. [Google Scholar] [CrossRef]
- Ochs, M.; Nyengaard, J.R.; Jung, A.; Knudsen, L.; Voigt, M.; Wahlers, T.; Richter, J.; Gundersen, H.J. The number of alveoli in the human lung. Am. J. Respir. Crit. Care Med. 2004, 169, 120–124. [Google Scholar] [CrossRef]
- Rippa, A.L.; Alpeeva, E.V.; Vasiliev, A.V.; Vorotelyak, E.A. Alveologenesis: What Governs Secondary Septa Formation. Int. J. Mol. Sci. 2021, 22, 12107. [Google Scholar] [CrossRef]
- Mund, S.I.; Stampanoni, M.; Schittny, J.C. Developmental alveolarization of the mouse lung. Dev. Dyn. 2008, 237, 2108–2116. [Google Scholar] [CrossRef]
- Beauchemin, K.J.; Wells, J.M.; Kho, A.T.; Philip, V.M.; Kamir, D.; Kohane, I.S.; Graber, J.H.; Bult, C.J. Temporal dynamics of the developing lung transcriptome in three common inbred strains of laboratory mice reveals multiple stages of postnatal alveolar development. PeerJ 2016, 4, e2318. [Google Scholar] [CrossRef]
- Frank, D.B.; Peng, T.; Zepp, J.A.; Snitow, M.; Vincent, T.L.; Penkala, I.J.; Cui, Z.; Herriges, M.J.; Morley, M.P.; Zhou, S.; et al. Emergence of a Wave of Wnt Signaling that Regulates Lung Alveologenesis by Controlling Epithelial Self-Renewal and Differentiation. Cell Rep. 2016, 17, 2312–2325. [Google Scholar] [CrossRef]
- Lee, J.H.; Tammela, T.; Hofree, M.; Choi, J.; Marjanovic, N.D.; Han, S.; Canner, D.; Wu, K.; Paschini, M.; Bhang, D.H.; et al. Anatomically and Functionally Distinct Lung Mesenchymal Populations Marked by Lgr5 and Lgr6. Cell 2017, 170, 1149–1163.e12. [Google Scholar] [CrossRef] [PubMed]
- Kugler, M.C.; Loomis, C.A.; Zhao, Z.; Cushman, J.C.; Liu, L.; Munger, J.S. Sonic Hedgehog Signaling Regulates Myofibroblast Function during Alveolar Septum Formation in Murine Postnatal Lung. Am. J. Respir Cell Mol. Biol. 2017, 57, 280–293. [Google Scholar] [CrossRef]
- He, H.; Snowball, J.; Sun, F.; Na, C.L.; Whitsett, J.A. IGF1R controls mechanosignaling in myofibroblasts required for pulmonary alveologenesis. JCI Insight 2021, 6, e144863. [Google Scholar] [CrossRef] [PubMed]
- Al Alam, D.; El Agha, E.; Sakurai, R.; Kheirollahi, V.; Moiseenko, A.; Danopoulos, S.; Shrestha, A.; Schmoldt, C.; Quantius, J.; Herold, S.; et al. Evidence for the involvement of fibroblast growth factor 10 in lipofibroblast formation during embryonic lung development. Development 2015, 142, 4139–4150. [Google Scholar] [CrossRef] [PubMed]
- Alanis, D.M.; Chang, D.R.; Akiyama, H.; Krasnow, M.A.; Chen, J. Two nested developmental waves demarcate a compartment boundary in the mouse lung. Nat. Commun. 2014, 5, 3923. [Google Scholar] [CrossRef]
- McGowan, S.; Jackson, S.K.; Jenkins-Moore, M.; Dai, H.H.; Chambon, P.; Snyder, J.M. Mice bearing deletions of retinoic acid receptors demonstrate reduced lung elastin and alveolar numbers. Am. J. Respir Cell Mol. Biol. 2000, 23, 162–167. [Google Scholar] [CrossRef]
- Zhang, K.; Yao, E.; Lin, C.; Chou, Y.T.; Wong, J.; Li, J.; Wolters, P.J.; Chuang, P.T. A mammalian Wnt5a-Ror2-Vangl2 axis controls the cytoskeleton and confers cellular properties required for alveologenesis. eLife 2020, 9, e53688. [Google Scholar] [CrossRef] [PubMed]
- Marino, B.S.; Cassedy, A.; Brown, K.L.; Franklin, R.; Gaynor, J.W.; Cvetkovic, M.; Laker, S.; Levinson, K.; MacGloin, H.; Mahony, L.; et al. Long-Term Quality of Life in Congenital Heart Disease Surgical Survivors: Multicenter Retrospective Study of Surgical and ICU Explanatory Factors. Pediatr. Crit. Care Med. 2023, 24, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, L.; Yang, T.; Huang, P.; Wang, L.; Zhao, L.; Zhang, S.; Ye, Z.; Chen, L.; Zheng, Z.; et al. Congenital Heart Disease and Risk of Cardiovascular Disease: A Meta-Analysis of Cohort Studies. J. Am. Heart Assoc. 2019, 8, e012030. [Google Scholar] [CrossRef]
- Shi, B.; Zhang, X.; Song, Z.; Dai, Z.; Luo, K.; Chen, B.; Zhou, Z.; Cui, Y.; Feng, B.; Zhu, Z.; et al. Targeting gut microbiota-derived kynurenine to predict and protect the remodeling of the pressure-overloaded young heart. Sci. Adv. 2023, 9, eadg7417. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Li, D.; Xiao, Y.; Sun, S.; Ye, L.; Hu, Y.; Hong, H. Volume overload impedes the maturation of sarcomeres and T-tubules in the right atria: A potential cause of atrial arrhythmia following delayed atrial septal defect closure. Front. Physiol. 2023, 14, 1237187. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Hong, H.; Li, M.; Xu, X.; Wang, S.; Xiao, Y.; Zheng, S.; Wang, Z.; Yan, Y.; Chen, H.; et al. A surgical mouse model of neonatal right ventricular outflow tract obstruction by pulmonary artery banding. J. Heart Lung Transplant. 2024, 43, 496–507. [Google Scholar] [CrossRef]
- Du, J.; Zheng, L.; Gao, P.; Yang, H.; Yang, W.J.; Guo, F.; Liang, R.; Feng, M.; Wang, Z.; Zhang, Z.; et al. A small-molecule cocktail promotes mammalian cardiomyocyte proliferation and heart regeneration. Cell Stem Cell 2022, 29, 545–558.e13. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, L.; Liu, H.; Duan, R.; Zhou, H.; Zhang, F.; He, X.; Lu, D.; Xiong, K.; Xiong, M.; et al. LRP6 downregulation promotes cardiomyocyte proliferation and heart regeneration. Cell Res. 2021, 31, 450–462. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, C.H.; Ammanamanchi, N.; Suresh, S.; Lewarchik, C.; Rao, K.; Uys, G.M.; Han, L.; Abrial, M.; Yimlamai, D.; et al. Control of cytokinesis by β-adrenergic receptors indicates an approach for regulating cardiomyocyte endowment. Sci. Transl. Med. 2019, 11, eaaw6419. [Google Scholar] [CrossRef]
- Yutzey, K.E. Cytokinesis, Beta-Blockers, and Congenital Heart Disease. N. Engl. J. Med. 2020, 382, 291–293. [Google Scholar] [CrossRef]
- Gu, J.; Chen, X.; Jin, Y.; Liu, M.; Xu, Q.; Liu, X.; Luo, Z.; Ling, S.; Liu, N.; Liu, S. A Neonatal Mouse Model for Pressure Overload: Myocardial Response Corresponds to Severity. Front. Cardiovasc. Med. 2021, 8, 660246. [Google Scholar] [CrossRef] [PubMed]
- Malek Mohammadi, M.; Abouissa, A.; Azizah, I.; Xie, Y.; Cordero, J.; Shirvani, A.; Gigina, A.; Engelhardt, M.; Trogisch, F.A.; Geffers, R.; et al. Induction of cardiomyocyte proliferation and angiogenesis protects neonatal mice from pressure overload-associated maladaptation. JCI Insight 2019, 5, e128336. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Wang, S.; Wang, Y.; Yang, J.; Bao, N.; Liu, J.; Zhang, Z. Neonatal Heart Responds to Pressure Overload with Differential Alterations in Various Cardiomyocyte Maturation Programs That Accommodate Simultaneous Hypertrophy and Hyperplasia. Front. Cell Dev. Biol. 2020, 8, 596960. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pu, W.; He, L.; Li, Y.; Zhao, H.; Li, Y.; Liu, K.; Huang, X.; Weng, W.; Wang, Q.D.; et al. Cell proliferation fate mapping reveals regional cardiomyocyte cell-cycle activity in subendocardial muscle of left ventricle. Nat. Commun. 2021, 12, 5784. [Google Scholar] [CrossRef] [PubMed]
- Bossers, G.P.L.; Günthel, M.; van der Feen, D.E.; Hagdorn, Q.A.J.; Koop, A.C.; van Duijvenboden, K.; Barnett, P.; Borgdorff, M.A.J.; Christoffels, V.M.; Silljé, H.H.W.; et al. Neuregulin-1 enhances cell-cycle activity, delays cardiac fibrosis, and improves cardiac performance in rat pups with right ventricular pressure load. J. Thorac. Cardiovasc. Surg. 2022, 164, e493–e510. [Google Scholar] [CrossRef]
- Cui, Q.; Sun, S.; Zhu, H.; Xiao, Y.; Jiang, C.; Zhang, H.; Liu, J.; Ye, L.; Shen, J. Volume Overload Initiates an Immune Response in the Right Ventricle at the Neonatal Stage. Front. Cardiovasc. Med. 2021, 8, 772336. [Google Scholar] [CrossRef] [PubMed]
- Canseco, D.C.; Kimura, W.; Garg, S.; Mukherjee, S.; Bhattacharya, S.; Abdisalaam, S.; Das, S.; Asaithamby, A.; Mammen, P.P.; Sadek, H.A. Human ventricular unloading induces cardiomyocyte proliferation. J. Am. Coll. Cardiol. 2015, 65, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Davoodianidalik, M.; Punzmann, H.; Kellay, H.; Xia, H.; Shats, M.; Francois, N. Fluctuation-Induced Interaction in Turbulent Flows. Phys. Rev. Lett. 2022, 128, 024503. [Google Scholar] [CrossRef] [PubMed]
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A tense situation: Forcing tumour progression. Nat. Rev. Cancer 2009, 9, 108–122. [Google Scholar] [CrossRef]
- Mehta, V.; Pang, K.L.; Rozbesky, D.; Nather, K.; Keen, A.; Lachowski, D.; Kong, Y.; Karia, D.; Ameismeier, M.; Huang, J.; et al. The guidance receptor plexin D1 is a mechanosensor in endothelial cells. Nature 2020, 578, 290–295. [Google Scholar] [CrossRef]
- Karbassi, E.; Fenix, A.; Marchiano, S.; Muraoka, N.; Nakamura, K.; Yang, X.; Murry, C.E. Cardiomyocyte maturation: Advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020, 17, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, M.; Hao, K.; Lei, W.; Tang, M.; Hu, S. Cardiomyocyte Maturation-the Road is not Obstructed. Stem Cell Rev. Rep. 2022, 18, 2966–2981. [Google Scholar] [CrossRef]
- Kannan, S.; Kwon, C. Regulation of cardiomyocyte maturation during critical perinatal window. J. Physiol. 2020, 598, 2941–2956. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.T.; Ye, S.; Su, J.; Garg, V. Cardiomyocyte Proliferation and Maturation: Two Sides of the Same Coin for Heart Regeneration. Front. Cell Dev. Biol. 2020, 8, 594226. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.E.; Li, L.; Xia, X.; Fu, W.; Liao, Q.; Lan, C.; Yang, D.; Chen, H.; Yue, R.; Zeng, C.; et al. Dedifferentiation, Proliferation, and Redifferentiation of Adult Mammalian Cardiomyocytes After Ischemic Injury. Circulation 2017, 136, 834–848. [Google Scholar] [CrossRef]
- Krieger, E.V.; Zeppenfeld, K.; DeWitt, E.S.; Duarte, V.E.; Egbe, A.C.; Haeffele, C.; Lin, K.Y.; Robinson, M.R.; Sillman, C.; Upadhyay, S.; et al. Arrhythmias in Repaired Tetralogy of Fallot: A Scientific Statement from the American Heart Association. Circ. Arrhythm. Electrophysiol. 2022, 15, e000084. [Google Scholar] [CrossRef]
- Badagliacca, R.; Papa, S.; Valli, G.; Pezzuto, B.; Poscia, R.; Reali, M.; Manzi, G.; Giannetta, E.; Berardi, D.; Sciomer, S.; et al. Right ventricular dyssynchrony and exercise capacity in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2017, 49, 1601419. [Google Scholar] [CrossRef]
- Van Aerde, N.; Meersseman, P.; Debaveye, Y.; Wilmer, A.; Casaer, M.P.; Gunst, J.; Wauters, J.; Wouters, P.J.; Goetschalckx, K.; Gosselink, R.; et al. Aerobic exercise capacity in long-term survivors of critical illness: Secondary analysis of the post-EPaNIC follow-up study. Intensive Care Med. 2021, 47, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, D.C. Respiratory physiology: Adaptations to high-level exercise. Br. J. Sports Med. 2012, 46, 381–384. [Google Scholar] [CrossRef]
- De Troyer, A.; Yernault, J.C.; Englert, M. Lung hypoplasia in congenital pulmonary valve stenosis. Circulation 1977, 56 Pt 1, 647–651. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Namba, F. An experimental animal model of bronchopulmonary dysplasia: Secondary publication. Pediatr. Int. 2021, 63, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Davis, J.M. Future applications of antioxidants in premature infants. Curr. Opin. Pediatr. 2011, 23, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Mourani, P.M.; Sontag, M.K.; Younoszai, A.; Miller, J.I.; Kinsella, J.P.; Baker, C.D.; Poindexter, B.B.; Ingram, D.A.; Abman, S.H. Early pulmonary vascular disease in preterm infants at risk for bronchopulmonary dysplasia. Am. J. Respir Crit. Care Med. 2015, 191, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Herum, K.M.; Lunde, I.G.; Skrbic, B.; Louch, W.E.; Hasic, A.; Boye, S.; Unger, A.; Brorson, S.H.; Sjaastad, I.; Tønnessen, T.; et al. Syndecan-4 is a key determinant of collagen cross-linking and passive myocardial stiffness in the pressure-overloaded heart. Cardiovasc. Res. 2015, 106, 217–226. [Google Scholar] [CrossRef]
- Yu, C.; Qiu, M.; Zhang, Z.; Song, X.; Du, H.; Peng, H.; Li, Q.; Yang, L.; Xiong, X.; Xia, B.; et al. Transcriptome sequencing reveals genes involved in cadmium-triggered oxidative stress in the chicken heart. Poult. Sci. 2021, 100, 100932. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).