
Long-Read Sequencing and De Novo Genome Assembly Pipeline of Two Plasmodium falciparum Clones (Pf3D7, PfW2) Using Only the PromethION Sequencer from Oxford Nanopore Technologies without Whole-Genome Amplification

 , , , , and
, , , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmodium falciparum Laboratory Cultures
2.2. DNA Extraction
2.3. Whole-Genome Library
2.4. Data Analysis
2.4.1. Genome Consensus of the Plasmodium falciparum 3D7 Clone
2.4.2. Genome Consensus of the Plasmodium falciparum W2 Clone
2.4.3. Final Consensus of the Two Clones
2.4.4. Genome Annotation
2.4.5. Apicoplast and Mitochondria Annotation
2.5. Plasmodium falciparum 3D7 Reference Genome Variant Caller
3. Results
3.1. Long-Read Sequencing Results
3.2. Plasmodium falciparum 3D7 De Novo Assembly
3.3. Plasmodium falciparum W2 De Novo Assembly
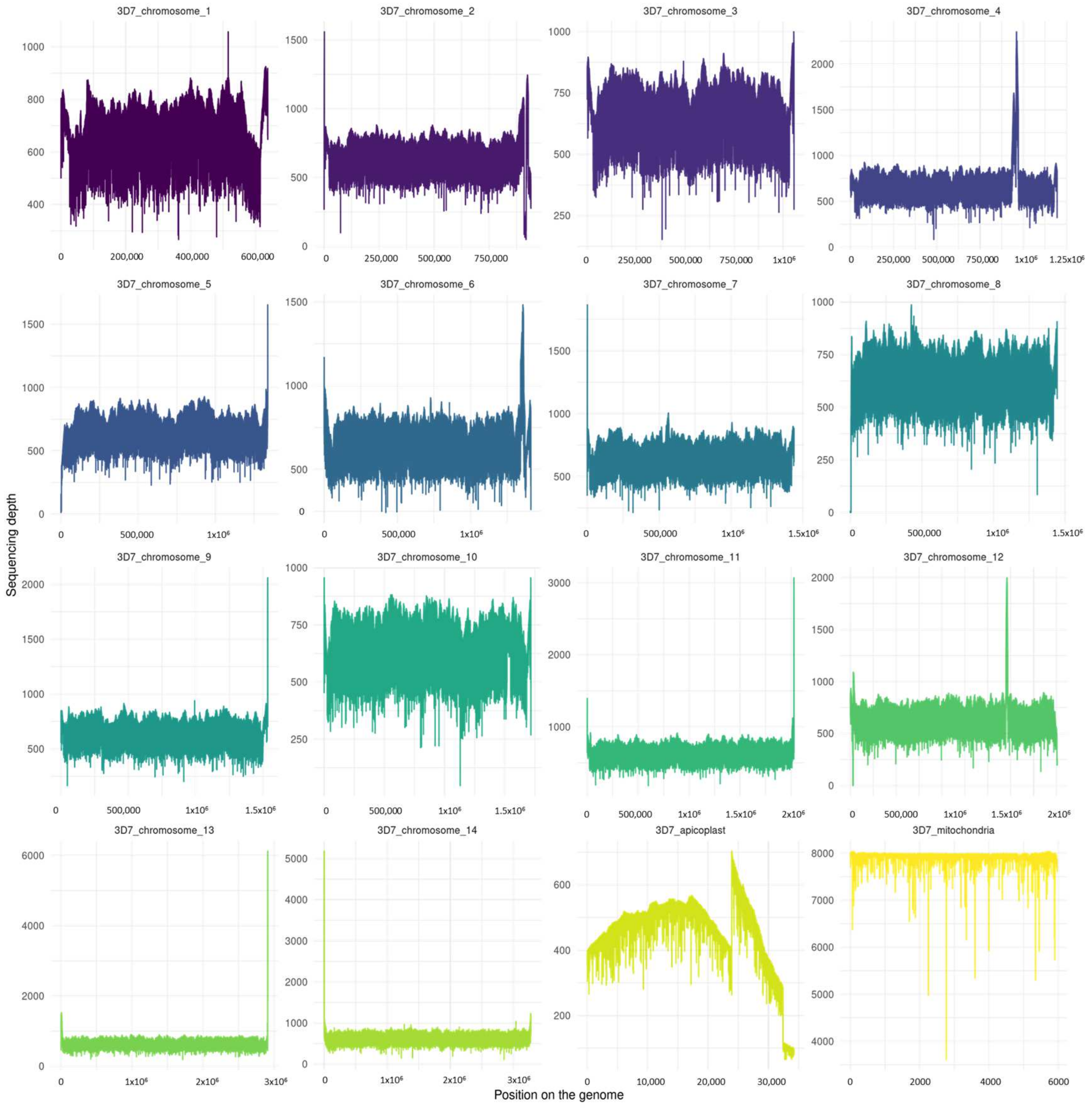
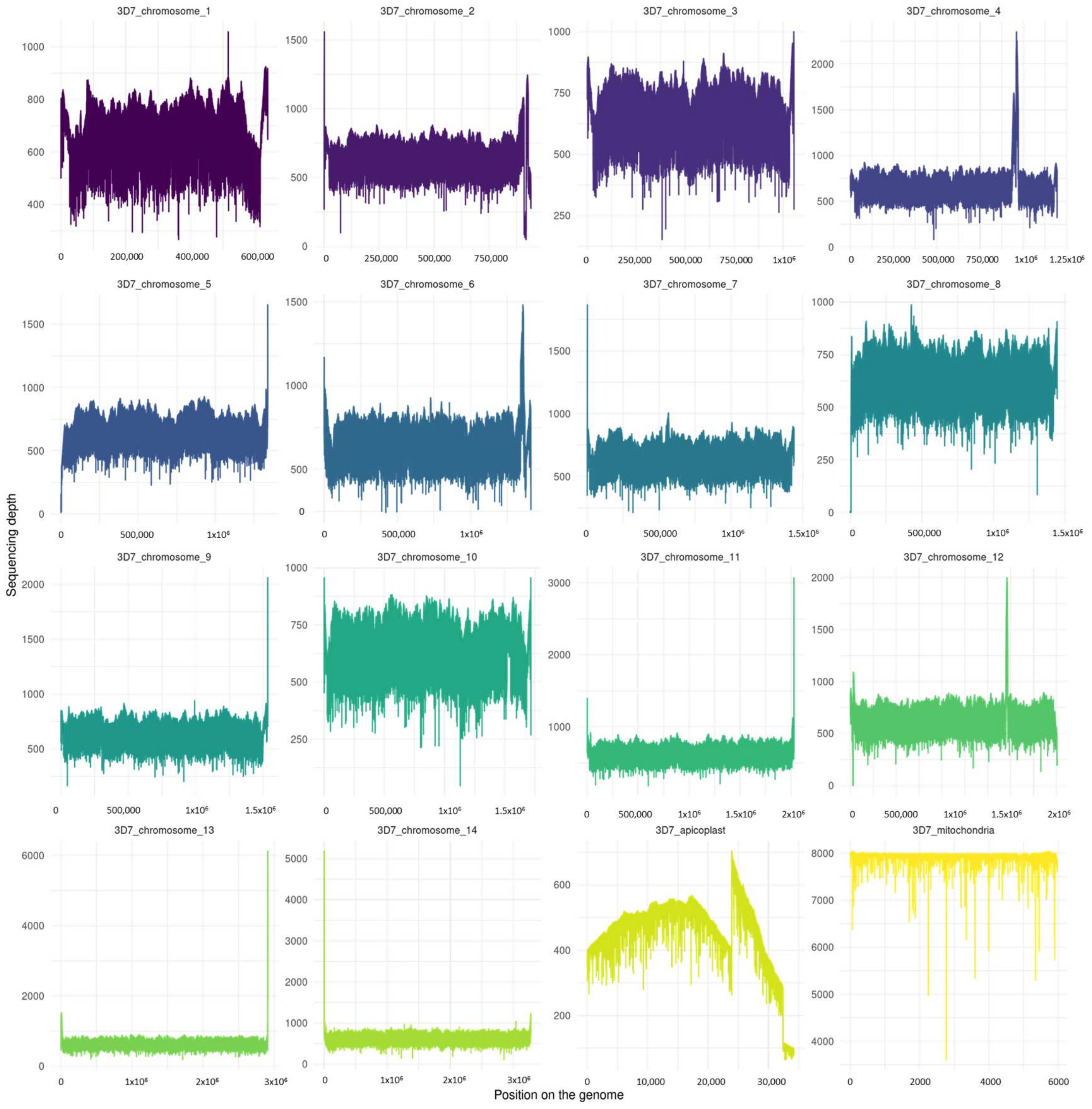
3.4. Genome Depth and Length
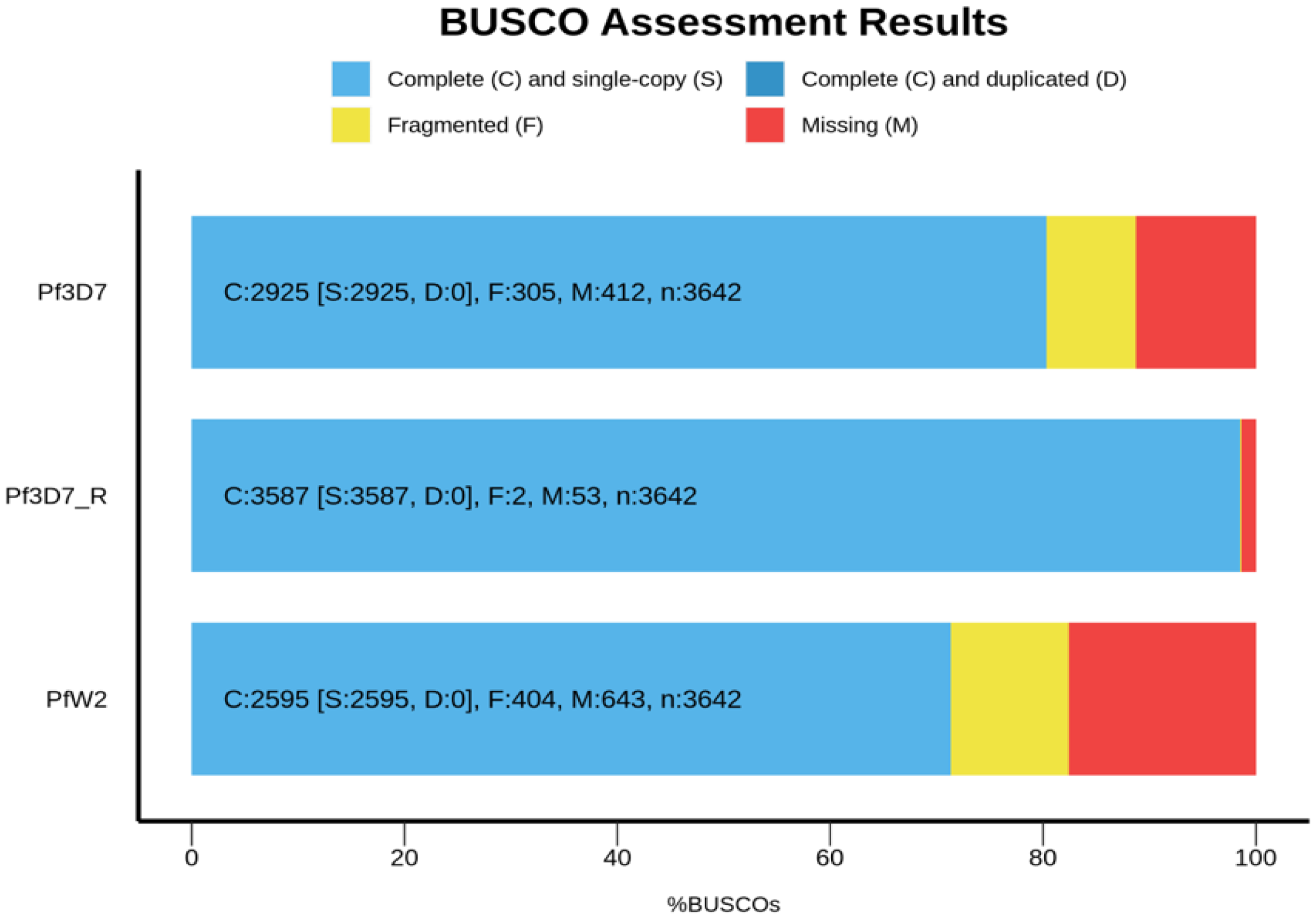
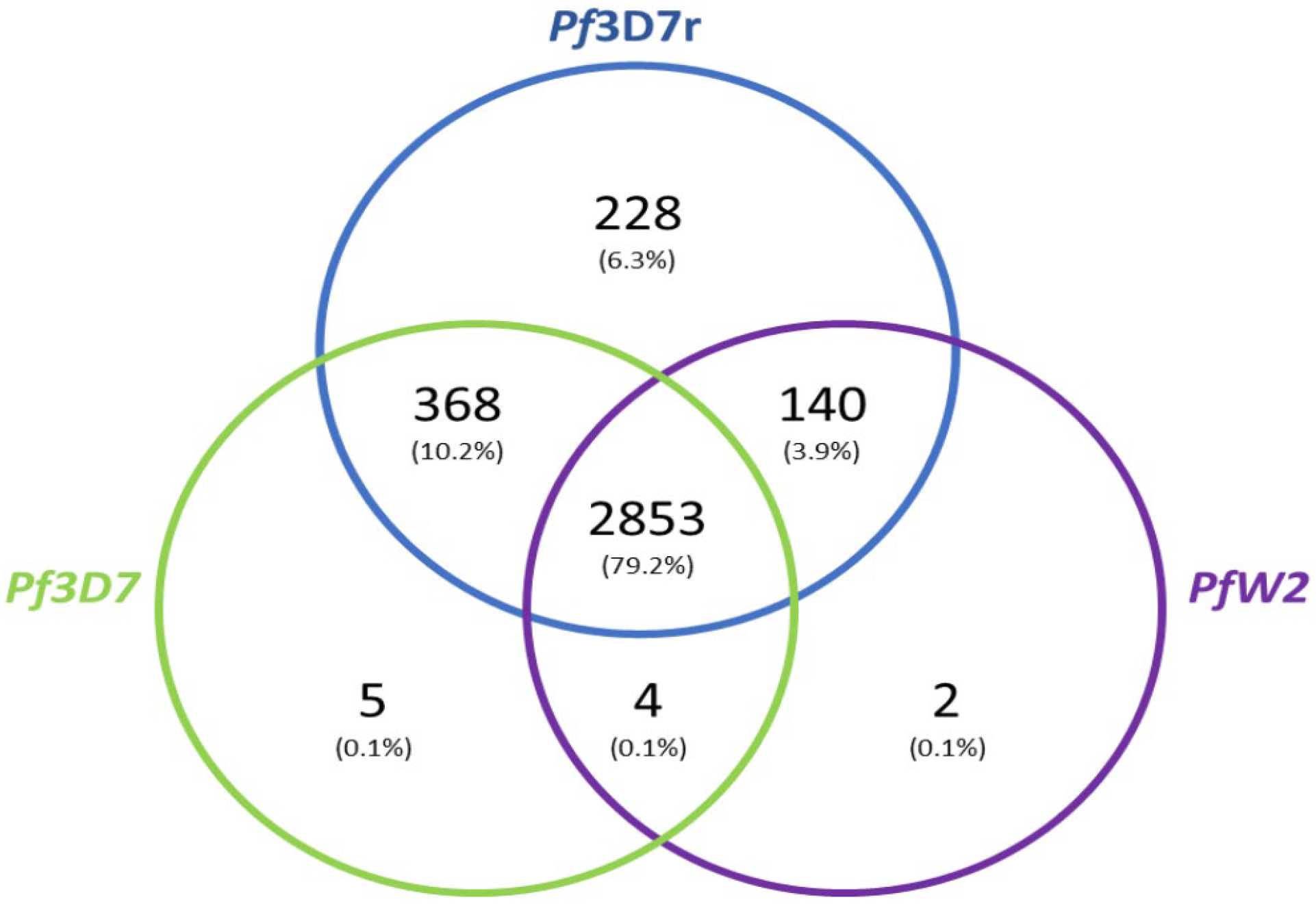
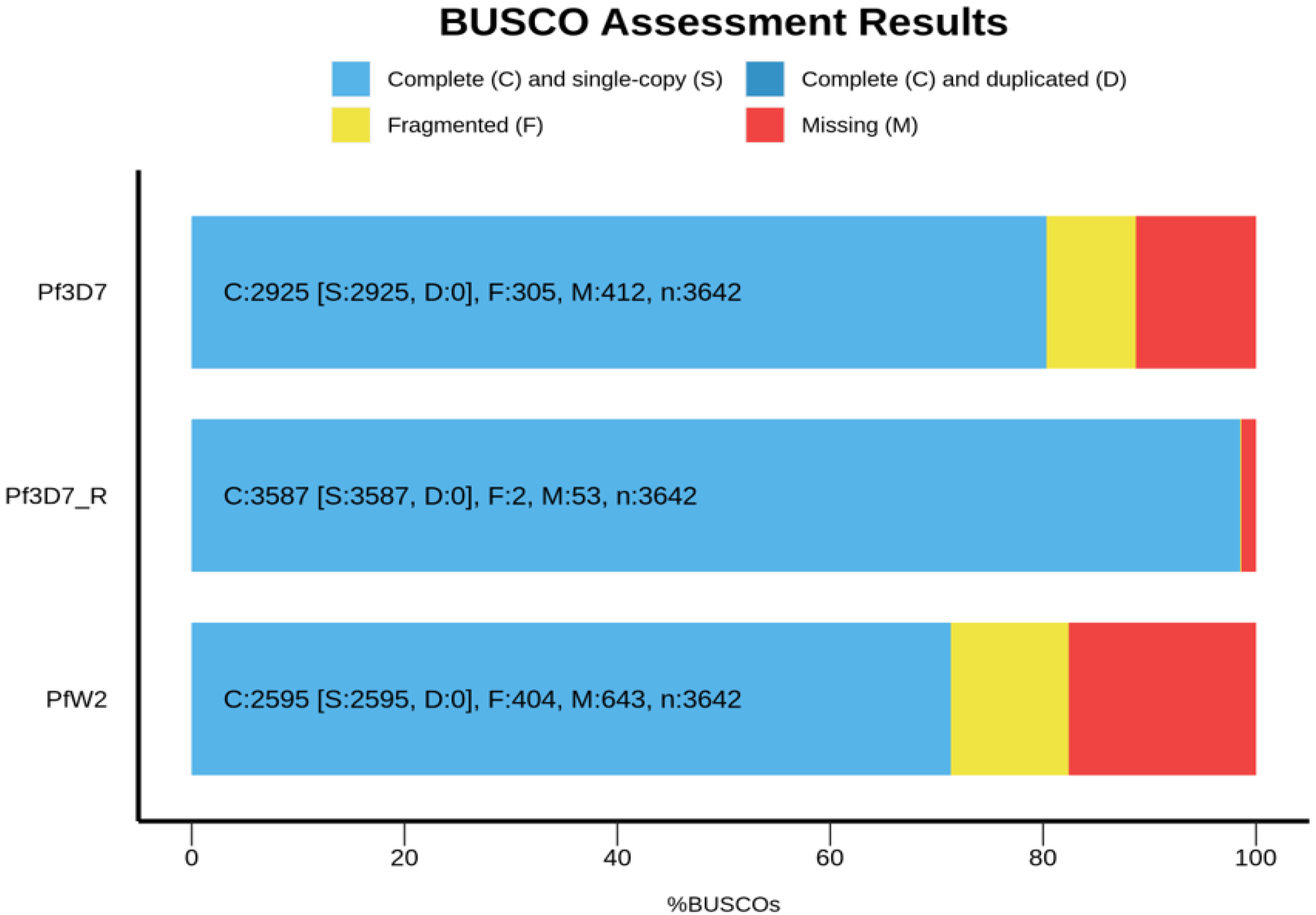
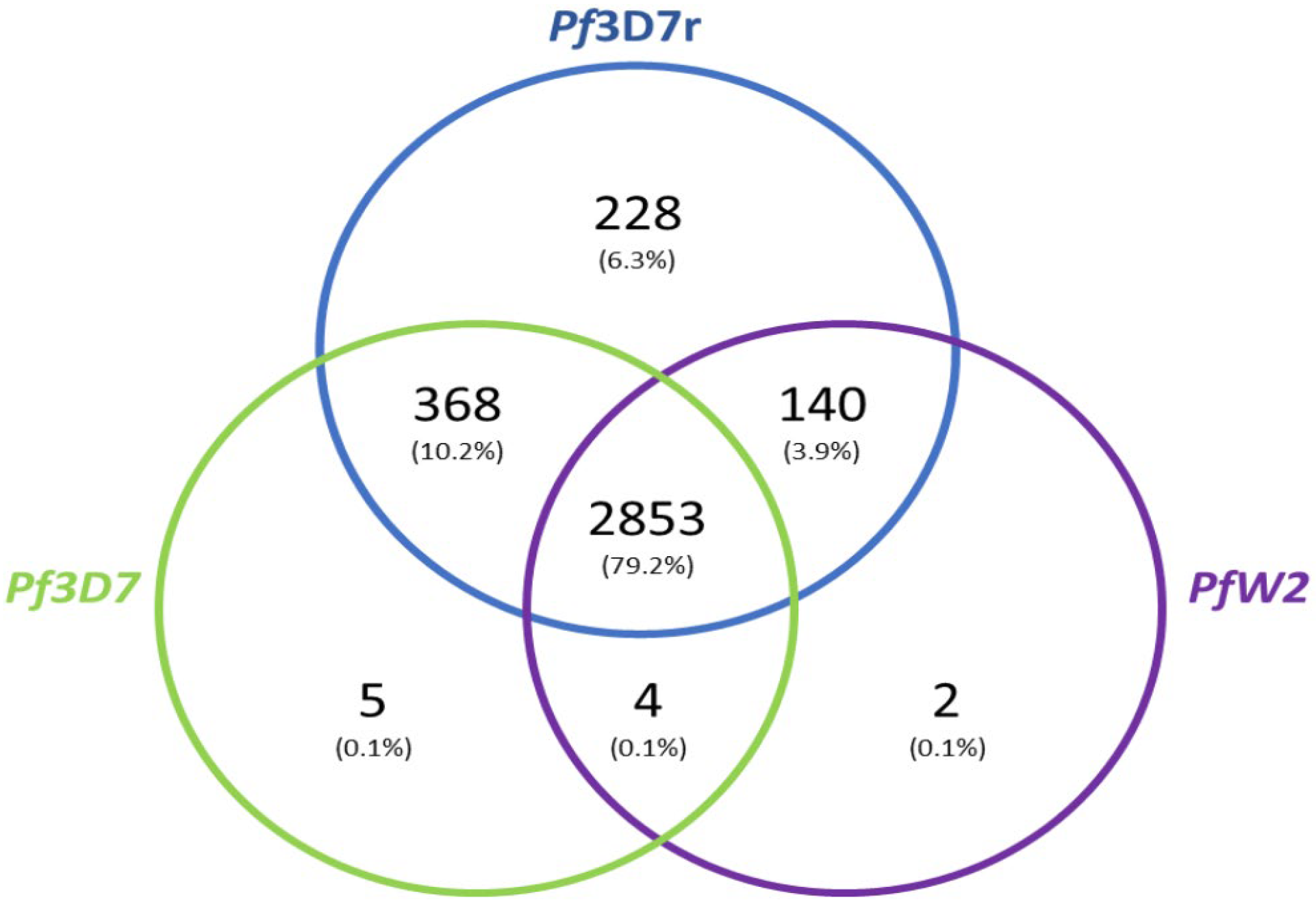
3.5. Genome Annotation
3.6. Apicoplast and Mitochondria Annotations
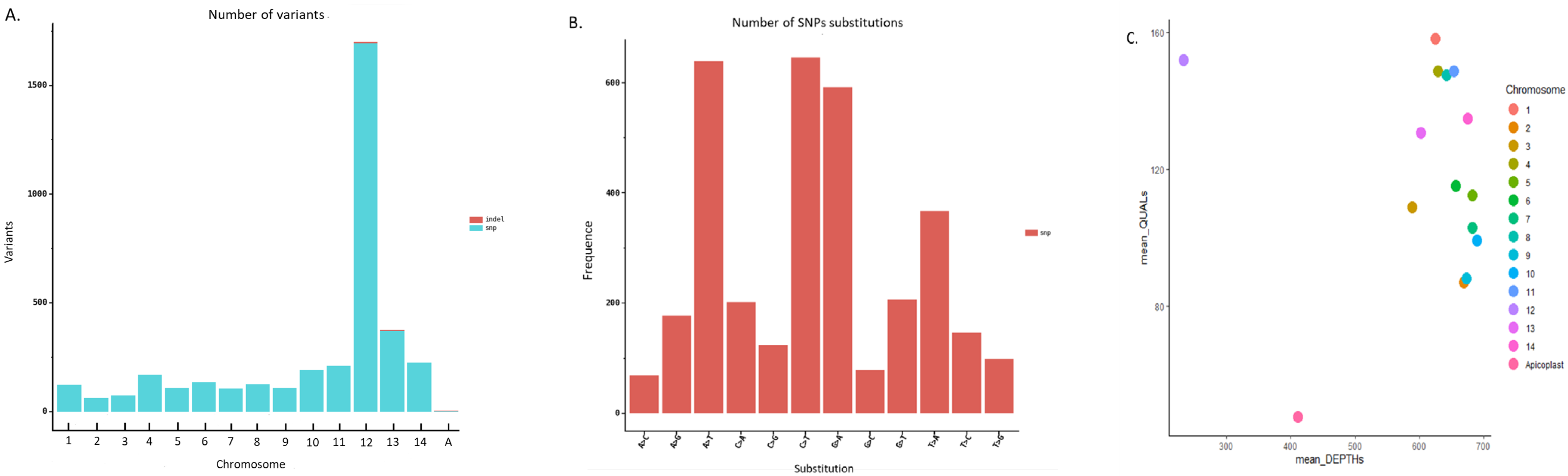
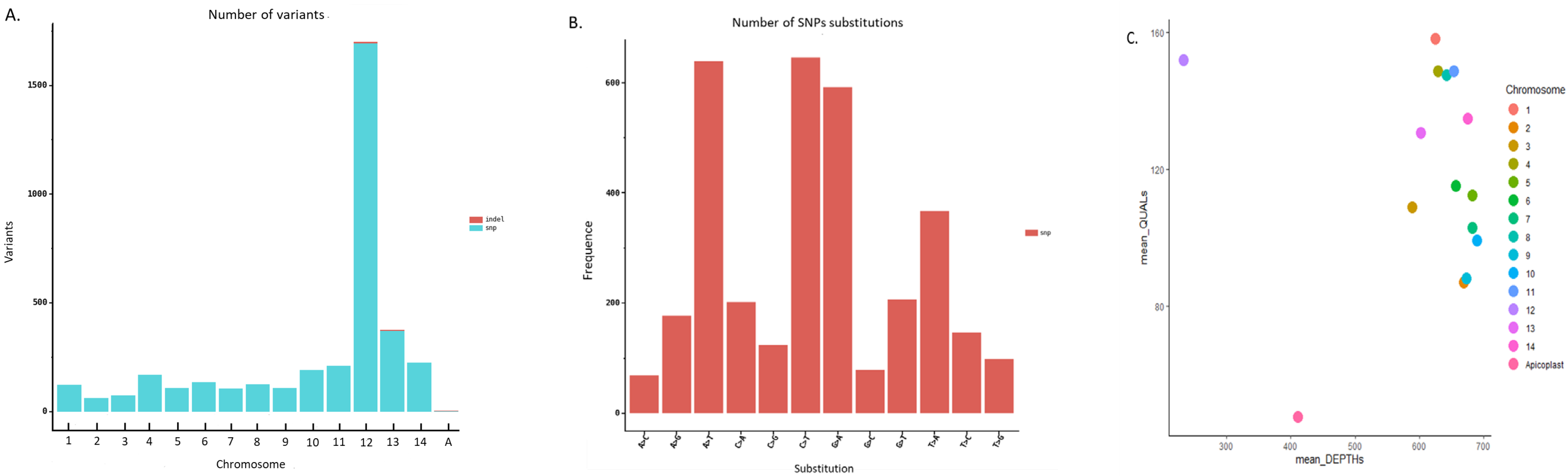
3.7. Genomic Variability of the Pf3D7 Clone against the Pf3D7 Reference Genome
3.8. Genomic Variability of the PfW2 Clone against the Pf3D7 Reference Genome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO World Malaria Report. 2023. Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2023 (accessed on 10 January 2024).
- Amaratunga, C.; Witkowski, B.; Khim, N.; Menard, D.; Fairhurst, R.M. Artemisinin resistance in Plasmodium falciparum. Lancet Infect. Dis. 2014, 14, 449–450. [Google Scholar] [CrossRef]
- Menard, D.; Dondorp, A. Antimalarial drug resistance: A threat to malaria elimination. Cold Spring Harb. Perspect. Med. 2017, 7, a025619. [Google Scholar] [CrossRef]
- Haldar, K.; Bhattacharjee, S.; Safeukui, I. Drug resistance in Plasmodium. Nat. Rev. Microbiol. 2018, 16, 156–170. [Google Scholar] [CrossRef]
- Böhme, U.; Otto, T.D.; Sanders, M.; Newbold, C.I.; Berriman, M. Progression of the canonical reference malaria parasite genome from 2002–2019. Wellcome Open Res. 2019, 4, 58. [Google Scholar] [CrossRef]
- Delemarre-van de Waal, H.A.; de Waal, F.C. A 2d patient with tropical malaria contracted in a natural way in the Netherlands. Ned. Tijdschr. Geneeskd. 1981, 125, 375–377. [Google Scholar]
- Gardner, M.J.; Hall, N.; Fung, E.; White, O.; Berriman, M.; Hyman, R.W.; Carlton, J.M.; Pain, A.; Nelson, K.E.; Bowman, S.; et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002, 419, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Bahl, A.; Brunk, B.; Crabtree, J.; Fraunholz, M.J.; Gajria, B.; Grant, G.R.; Ginsburg, H.; Gupta, D.; Kissinger, J.C.; Labo, P.; et al. PlasmoDB: The Plasmodium genome resource. A database integrating experimental and computational data. Nucleic Acids Res. 2003, 31, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Data Set Plasmodium falciparum 3D7 Genome Sequence and Annotation [Internet]. Available online: https://plasmodb.org/plasmo/app/record/dataset/DS_1d17c1883c (accessed on 21 July 2023).
- Oduola, A.M.J.; Weatherly, N.F.; Bowdre, J.H.; Desjardins, R.E. Plasmodium falciparum: Cloning by single-erythrocyte micromanipulation and heterogeneity in vitro. Exp. Parasitol. 1988, 66, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Cardenas, J.A.; Garcia-Maroto, F.; Alvarez-Bermejo, J.A.; Manzano-Agugliaro, F. DNA sequencing sensors: An overview. Sensors 2017, 17, 588. [Google Scholar] [CrossRef] [PubMed]
- Le Roch, K.G.; Chung, D.-W.D.; Ponts, N. Genomics and integrated systems biology in Plasmodium falciparum: A path to malaria control and eradication. Parasite Immunol. 2012, 34, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Akoniyon, O.P.; Adewumi, T.S.; Maharaj, L.; Oyegoke, O.O.; Roux, A.; Adeleke, M.A.; Maharaj, R.; Okpeku, M. Whole genome sequencing contributions and challenges in disease reduction focused on malaria. Biology 2022, 11, 587. [Google Scholar] [CrossRef] [PubMed]
- De Cesare, M.; Mwenda, M.; Jeffreys, A.E.; Chirwa, J.; Drakeley, C.; Schneider, K.; Ghinai, I.; Busby, V.O.R.I.P.B.; Hamainza, B.; Hawela, M.; et al. Flexible and cost-effective genomic surveillance of P. falciparum malaria with targeted nanopore sequencing. bioRxiv 2023, arXiv:2023.02.06.527333. [Google Scholar]
- Girgis, S.T.; Adika, E.; Nenyewodey, F.E.; Jnr, D.K.S.; Ngoi, J.M.; Bandoh, K.; Lorenz, O.; van de Steeg, G.; Harrott, A.J.R.; Nsoh, S.; et al. Nanopore sequencing for real-time genomic surveillance of Plasmodium falciparum. bioRxiv 2022, arXiv:2022.12.20.521122. [Google Scholar]
- Runtuwene, L.R.; Tuda, J.S.B.; Mongan, A.E.; Makalowski, W.; Frith, M.C.; Imwong, M.; Srisutham, S.; Thi, L.A.N.; Tuan, N.N.; Eshita, Y.; et al. Nanopore sequencing of drug-resistance-associated genes in malaria parasites, Plasmodium falciparum. Sci. Rep. 2018, 8, 8286. [Google Scholar] [CrossRef] [PubMed]
- Sabin, S.; Jones, S.; Patel, D.; Subramaniam, G.; Kelley, J.; Aidoo, M.; Talundzic, E. Portable and cost-effective genetic detection and characterization of Plasmodium falciparum hrp2 using the MinION sequencer. Sci. Rep. 2023, 13, 2893. [Google Scholar] [CrossRef]
- Niaré, K.; Greenhouse, B.; Bailey, J.A. An optimized GATK4 pipeline for Plasmodium falciparum whole genome sequencing variant calling and analysis. Malar. J. 2023, 22, 207. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Wick, R. rrwick/Filtlong. 2023. Available online: https://github.com/rrwick/Filtlong (accessed on 9 February 2023).
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Lin, Y.; Yuan, J.; Kolmogorov, M.; Shen, M.W.; Chaisson, M.; Pevzner, P.A. Assembly of long error-prone reads using de Bruijn graphs. Proc. Natl. Acad. Sci. USA 2016, 113, E8396–E8405. [Google Scholar] [CrossRef] [PubMed]
- Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 2021, 37, 4572–4574. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2, pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, S.; Kajitani, R.; Itoh, T. GreenHill: A de novo chromosome-level scaffolding and phasing tool using Hi-C. Genome Biol. 2023, 24, 162. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Karin, E.L.; Mirdita, M.; Söding, J. MetaEuk—Sensitive, High-Throughput Gene Discovery, and Annotation for Large-Scale Eukaryotic Metagenomics. Microbiome 2020, 8, 48. Available online: https://link.springer.com/epdf/10.1186/s40168-020-00808-x (accessed on 25 July 2023).
- The Galaxy Community. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 2022, 50, W345–W351. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Steinbiss, S.; Silva-Franco, F.; Brunk, B.; Foth, B.; Hertz-Fowler, C.; Berriman, M.; Otto, T.D. Companion: A web server for annotation and analysis of parasite genomes. Nucleic Acids Res. 2016, 44, W29–W34. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Coppée, R.; Mama, A.; Sarrasin, V.; Kamaliddin, C.; Adoux, L.; Palazzo, L.; Ndam, N.T.; Letourneur, F.; Ariey, F.; Houzé, S.; et al. 5WBF: A low-cost and straightforward whole blood filtration method suitable for whole-genome sequencing of Plasmodium falciparum clinical isolates. Malar. J. 2022, 21, 51. [Google Scholar] [CrossRef]
- Oyola, S.O.; Ariani, C.V.; Hamilton, W.L.; Kekre, M.; Amenga-Etego, L.N.; Ghansah, A.; Rutledge, G.G.; Redmond, S.; Manske, M.; Jyothi, D.; et al. Whole genome sequencing of Plasmodium falciparum from dried blood spots using selective whole genome amplification. Malar. J. 2016, 15, 597. [Google Scholar] [CrossRef] [PubMed]
- De Meulenaere, K.; Cuypers, W.L.; Gauglitz, J.M.; Guetens, P.; Rosanas-Urgell, A.; Laukens, K.; Cuypers, B. Selective whole-genome sequencing of Plasmodium parasites directly from blood samples by nanopore adaptive sampling. mBio 2023, 15, e0196723. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.L.; Reimering, S.; Escobar-Prieto, J.D.; Brancucci, N.M.B.; Echeverry, D.F.; I Abdi, A.; Marti, M.; Gómez-Díaz, E.; Otto, T.D. From contigs towards chromosomes: Automatic improvement of long read assemblies (ILRA). Brief. Bioinform. 2023, 24, bbad248. [Google Scholar] [CrossRef] [PubMed]
- Claessens, A.; Stewart, L.B.; Drury, E.; Ahouidi, A.D.; Amambua-Ngwa, A.; Diakite, M.; Kwiatkowski, D.P.; Awandare, G.A.; Conway, D.J. Genomic variation during culture adaptation of genetically complex Plasmodium falciparum clinical isolates. Microb. Genom. 2023, 9, 001009. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.; Kim, Y.; Humphreys, G.I.; Nardulli, A.M.; Kosari, F.; Vasmatzis, G.; Taylor, W.R.; Ahlquist, D.A.; Myong, S.; Bashir, R. Nanopore-based assay for detection of methylation in double-stranded DNA ragments. ACS Nano 2015, 9, 290–300. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanoplot Data | Pf3D7 Reads | Pf3D7 Filtered Reads | PfW2 Reads | PfW2 Filtered Reads |
|---|---|---|---|---|
| Mean read length * | 3440.2 | 15,843.6 | 7701.3 | 11,600.2 |
| Mean read quality | 14.4 | 17.0 | 13.9 | 14.5 |
| Median read length * | 1037.0 | 12,363.0 | 4068.0 | 7903.0 |
| Median read quality | 14.1 | 17.1 | 14.0 | 14.6 |
| Number of reads | 12,854,191.0 | 1,238,210.0 | 2,192,635.0 | 1,296,307.0 |
| Read length N50 * | 9731 | 18,448 | 16,305 | 17,502 |
| Total bases | 44,221,405,060.0 | 19,617,651,842.0 | 16,886,156,110.0 | 15,037,370,421.0 |
| Pf3D7 | PfW2 | |
|---|---|---|
| Total length | 23,477,924 | 23,302,768 |
| Fragments | 32 | 31 |
| Fragments N50 | 1,265,374 | 1,700,513 |
| Largest fragments | 3,284,512 | 3,249,547 |
| Scaffolds | 0 | 2 |
| Mean coverage | 787 | 653 |
| N50 (Kb) | 18,488 | 17,502 |
| N90 | 8461 | 5277 |
| Pf3D7 | PfW2 | |||
|---|---|---|---|---|
| Length | Average Depth | Length | Average Depth | |
| chromosome 1 | 638,193 | 754.4 ± 64.6 | 621,378 | 946 ± 1041.9 |
| chromosome 2 | 940,408 | 764 ± 95.5 | 942,789 | 731 ± 487.8 |
| chromosome 3 | 1,056,079 | 768.3 ± 56.3 | 955,041 | 669.7 ± 381 |
| chromosome 4 | 1,195,288 | 808.9 ± 161.5 | 969,939 | 749.4 ± 545.6 |
| chromosome 5 | 1,338,524 | 774 ± 81.4 | 1,353,934 | 705.9 ± 264.2 |
| chromosome 6 | 1,412,742 | 784 ± 81.7 | 1,294,864 | 669.7 ± 223.7 |
| chromosome 7 | 1,438,736 | 779.7 ± 59.8 | 1,272,765 | 685.7± 366.4 |
| chromosome 8 | 1,445,520 | 762 ± 68.1 | 1,320,440 | 660.6 ± 334.2 |
| chromosome 9 | 1,534,997 | 765 ± 59.8 | 1,420,162 | 632.8 ± 144.6 |
| chromosome 10 | 1,716,863 | 760.8 ± 56.2 | 1,528,816 | 627.5 ± 62.8 |
| chromosome 11 | 2,029,548 | 769.6 ± 73.7 | 1,894,263 | 620.9 ± 59.5 |
| chromosome 12 | 2,257,511 | 774.8 ± 109.7 | 2,097,388 | 635.9 ± 123.6 |
| chromosome 13 | 2,913,737 | 779.7 ± 126 | 2,786,778 | 624.8 ± 58.8 |
| chromosome 14 | 3,277,058 | 776.9 ± 91.4 | 3,213,288 | 630.5 ± 98.5 |
| apicoplast | 34,237 | 465.8 ± 113.4 | 34,226 | 348.2 ± 80.6 |
| mitochondria | 5966 | 7922.1 ± 151.9 | 5967 | 7823.6 ± 251.4 |
| total length | 23,235,407 | 21,712,038 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delandre, O.; Lamer, O.; Loreau, J.-M.; Papa Mze, N.; Fonta, I.; Mosnier, J.; Gomez, N.; Javelle, E.; Pradines, B. Long-Read Sequencing and De Novo Genome Assembly Pipeline of Two Plasmodium falciparum Clones (Pf3D7, PfW2) Using Only the PromethION Sequencer from Oxford Nanopore Technologies without Whole-Genome Amplification. Biology 2024, 13, 89. https://doi.org/10.3390/biology13020089
Delandre O, Lamer O, Loreau J-M, Papa Mze N, Fonta I, Mosnier J, Gomez N, Javelle E, Pradines B. Long-Read Sequencing and De Novo Genome Assembly Pipeline of Two Plasmodium falciparum Clones (Pf3D7, PfW2) Using Only the PromethION Sequencer from Oxford Nanopore Technologies without Whole-Genome Amplification. Biology. 2024; 13(2):89. https://doi.org/10.3390/biology13020089
Chicago/Turabian StyleDelandre, Océane, Ombeline Lamer, Jean-Marie Loreau, Nasserdine Papa Mze, Isabelle Fonta, Joel Mosnier, Nicolas Gomez, Emilie Javelle, and Bruno Pradines. 2024. "Long-Read Sequencing and De Novo Genome Assembly Pipeline of Two Plasmodium falciparum Clones (Pf3D7, PfW2) Using Only the PromethION Sequencer from Oxford Nanopore Technologies without Whole-Genome Amplification" Biology 13, no. 2: 89. https://doi.org/10.3390/biology13020089
APA StyleDelandre, O., Lamer, O., Loreau, J.-M., Papa Mze, N., Fonta, I., Mosnier, J., Gomez, N., Javelle, E., & Pradines, B. (2024). Long-Read Sequencing and De Novo Genome Assembly Pipeline of Two Plasmodium falciparum Clones (Pf3D7, PfW2) Using Only the PromethION Sequencer from Oxford Nanopore Technologies without Whole-Genome Amplification. Biology, 13(2), 89. https://doi.org/10.3390/biology13020089