
Cross-Tissue Regulatory Network Analyses Reveal Novel Susceptibility Genes and Potential Mechanisms for Endometriosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
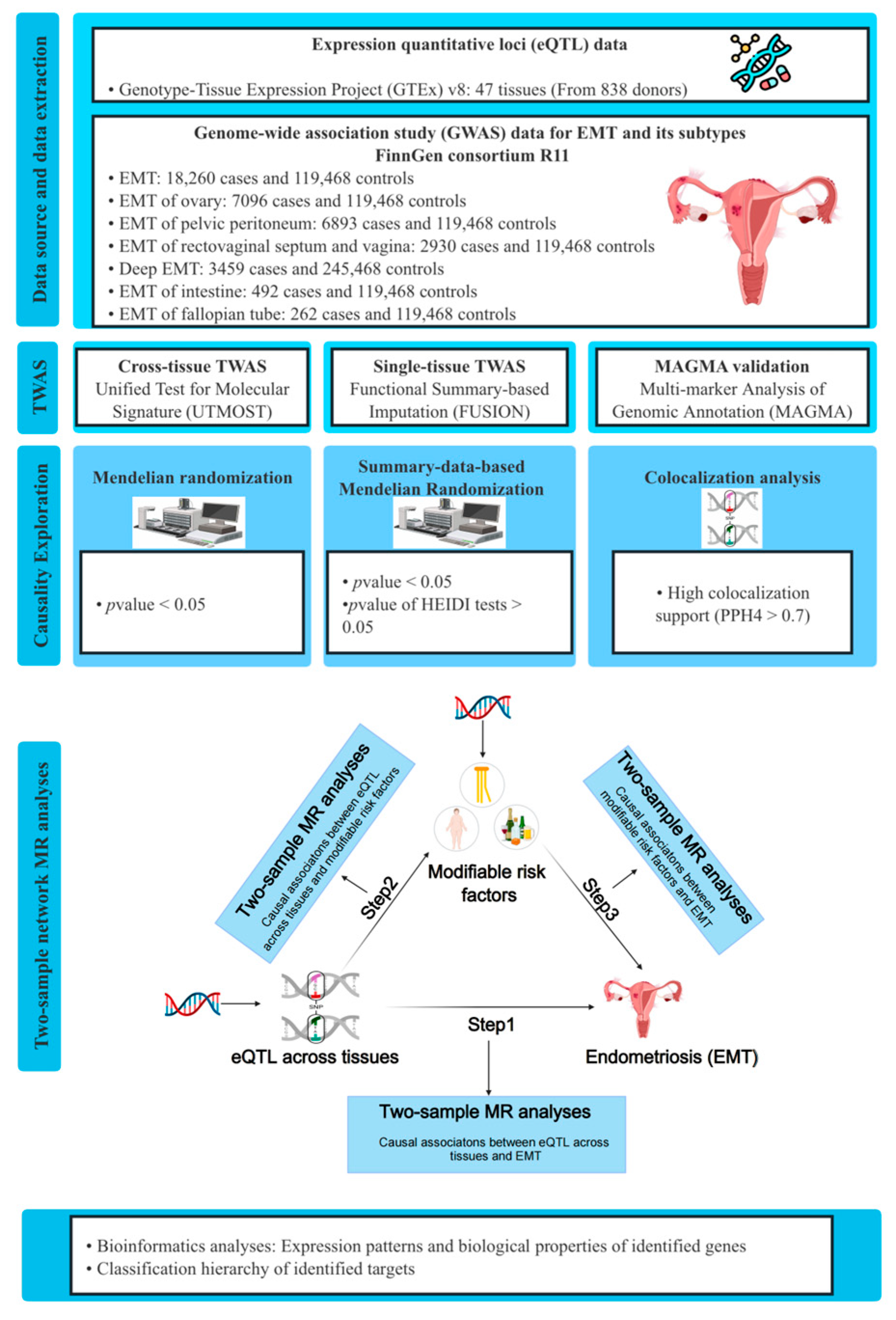
2. Materials and Methods
2.1. Data Source
2.2. Cross-Tissue TWAS Analyses
2.3. Single-Tissue TWAS Analyses
2.4. Gene-Based Analyses for Validation
2.5. Conditional and Joint Analyses
2.6. Mendelian Randomization and Colocalization Analyses Between Identified Genes and EMT
2.7. Two-Step Network MR Analyses
2.8. Transcriptome Differential Analysis and GeneMANIA Analysis
2.9. Classification Hierarchy of Identified Gene Targets
3. Results
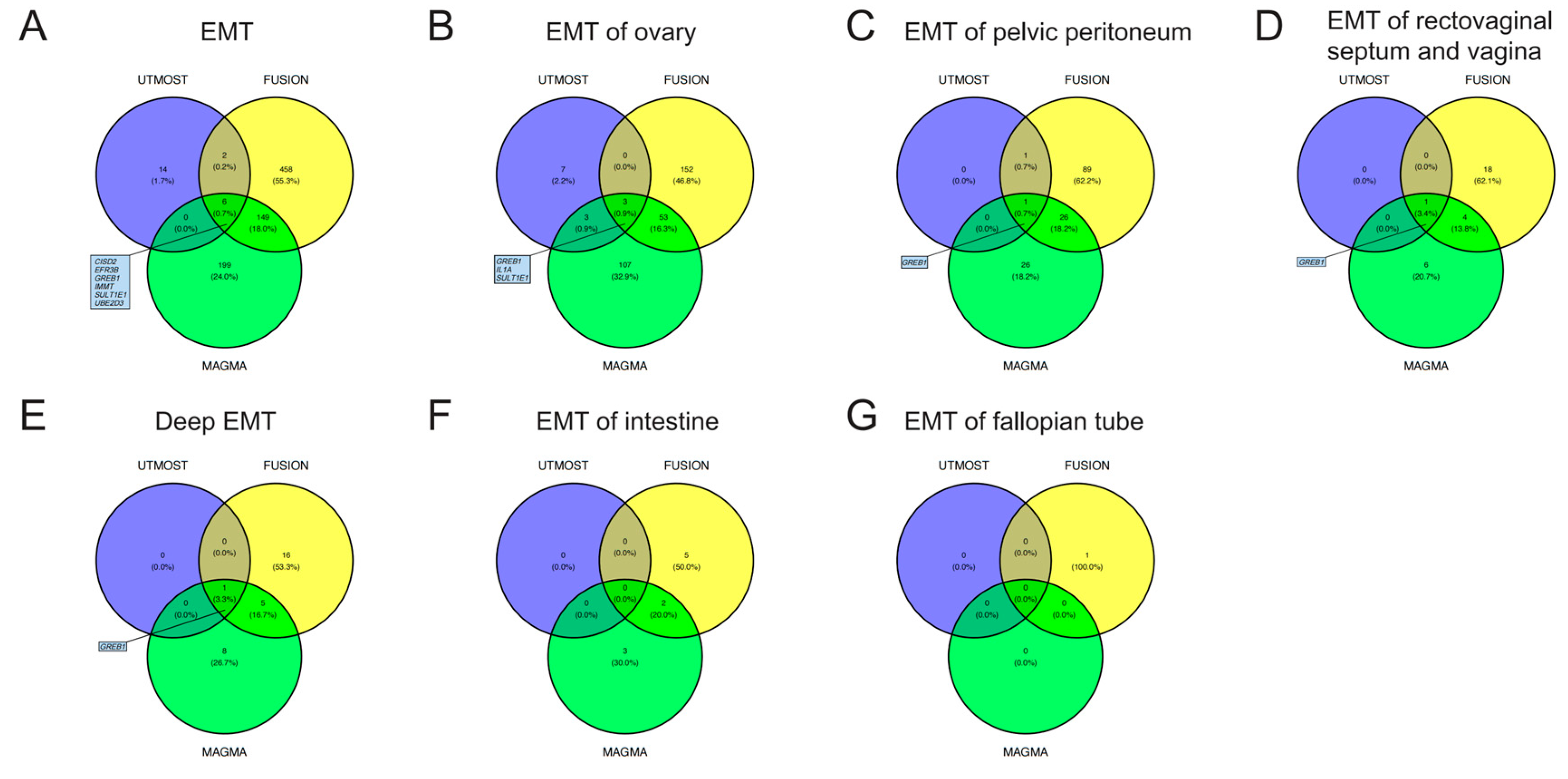
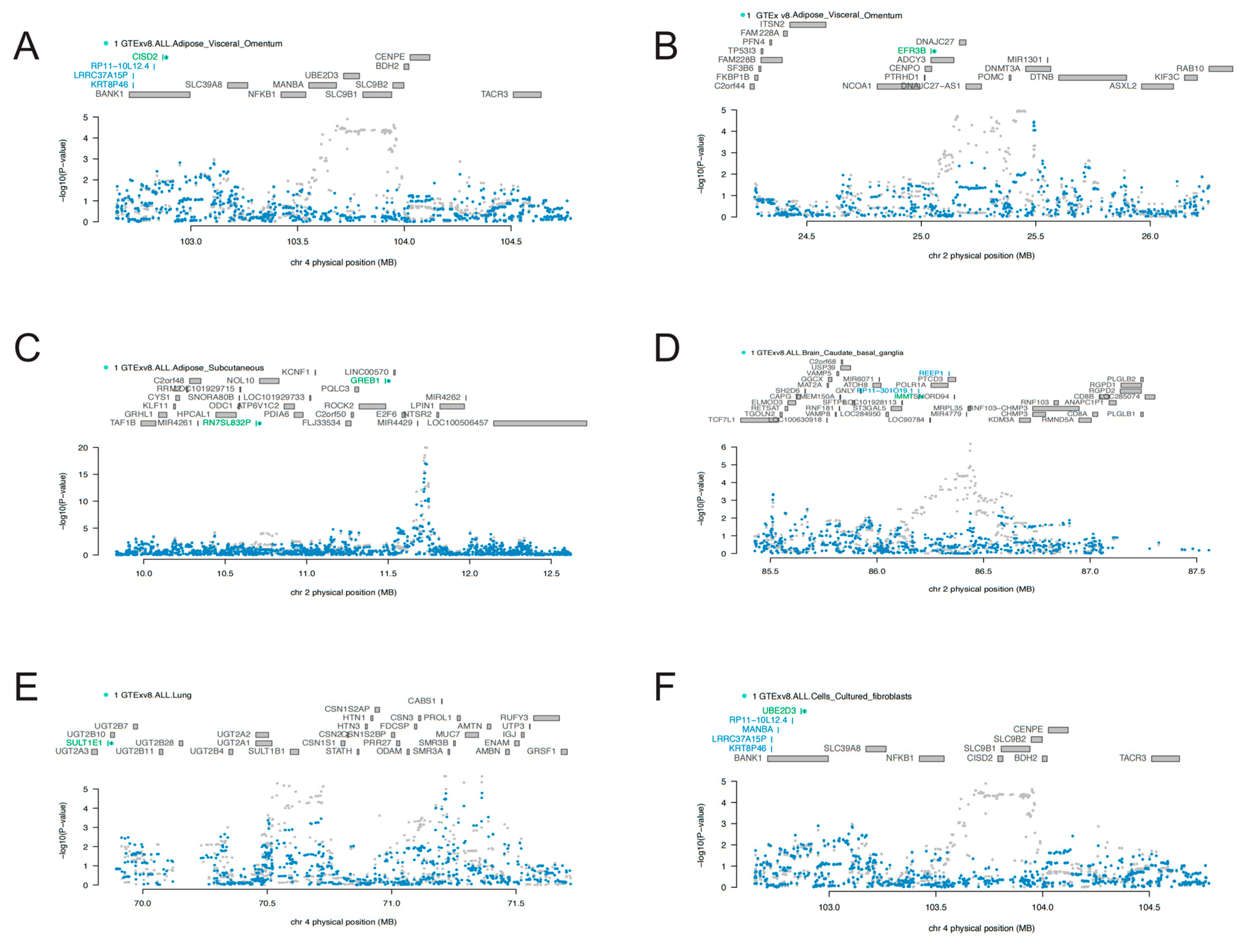
3.1. TWAS Analyses in Cross-Tissue and Single-Tissue
3.2. COJO Analyses
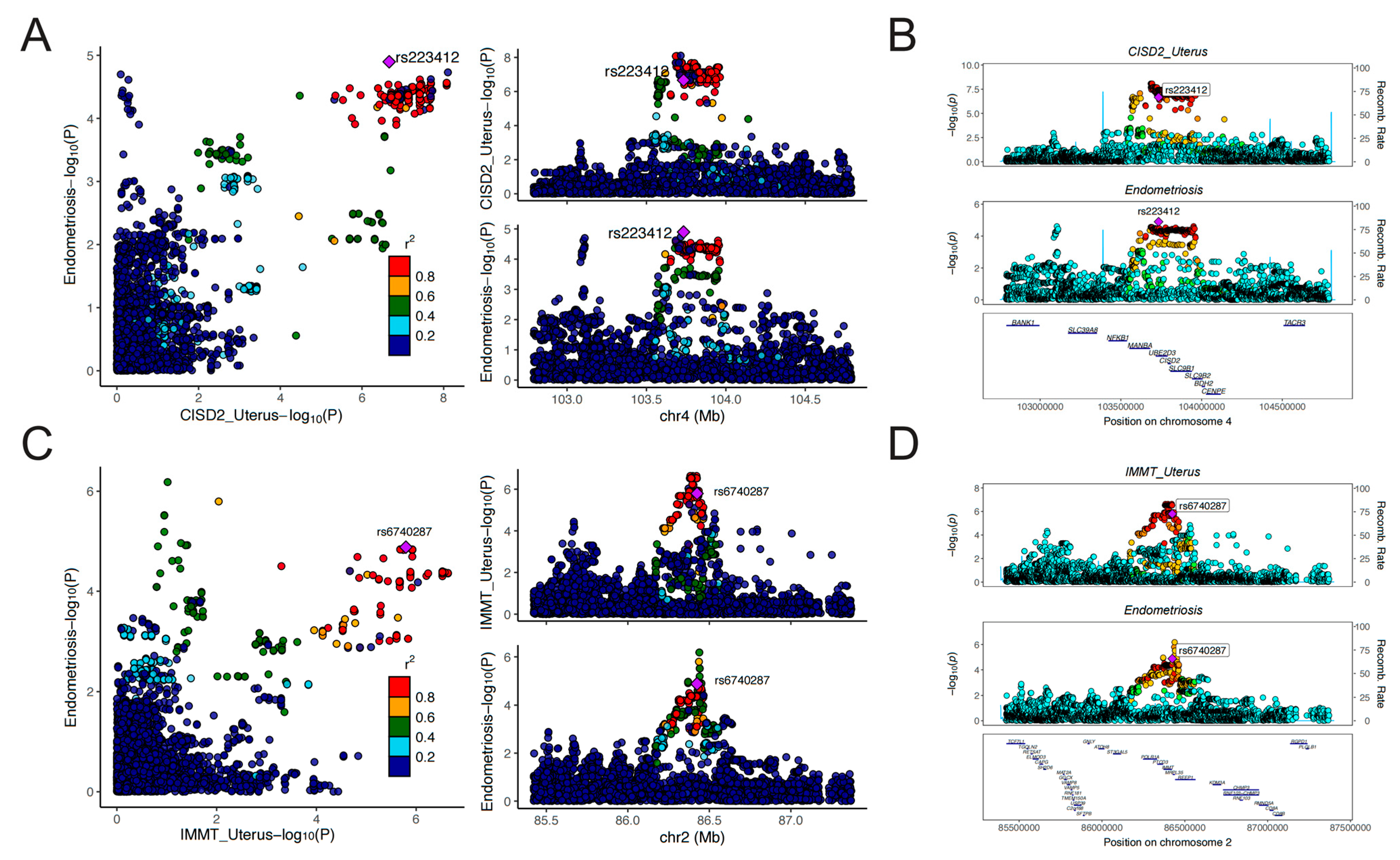
3.3. MR and Colocalization Analyses
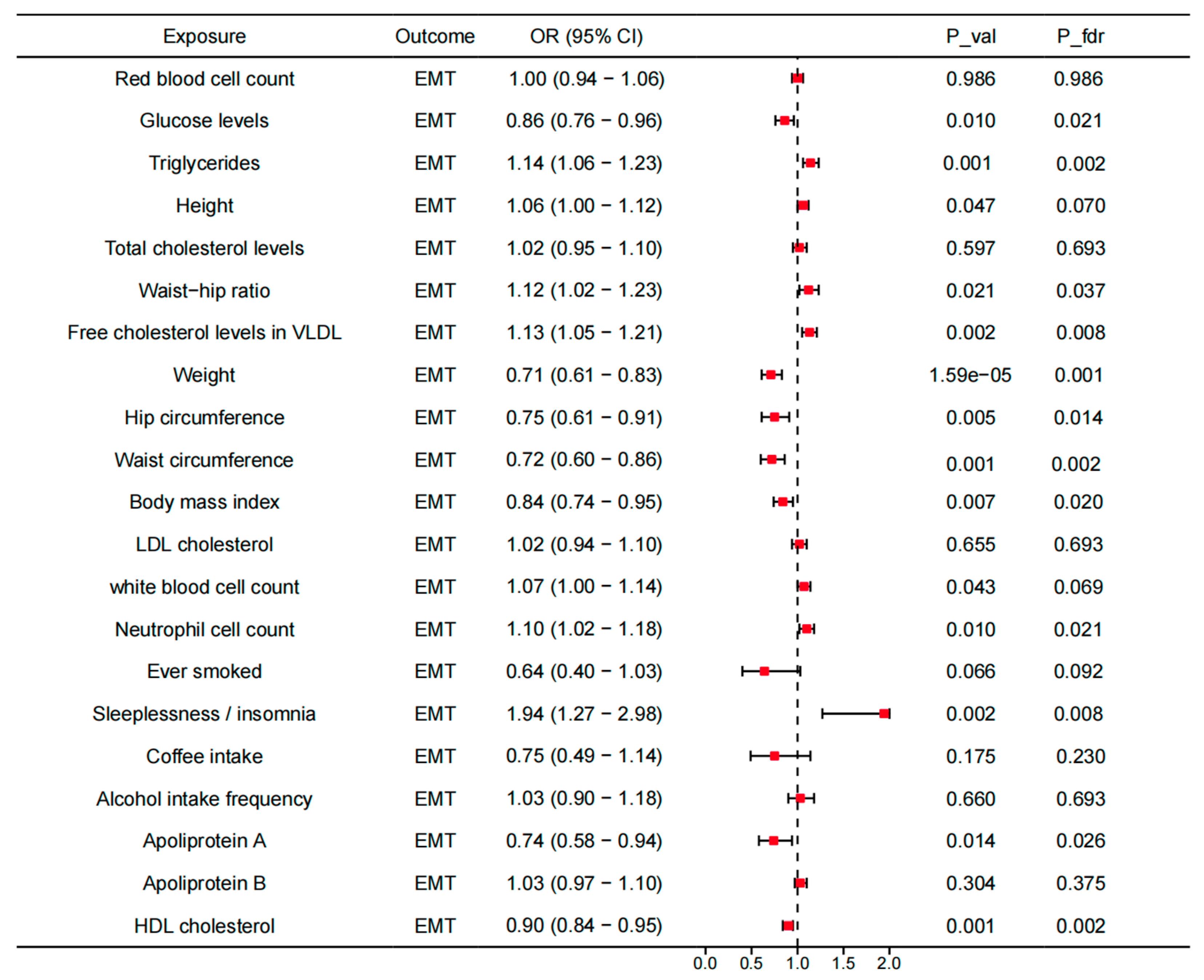
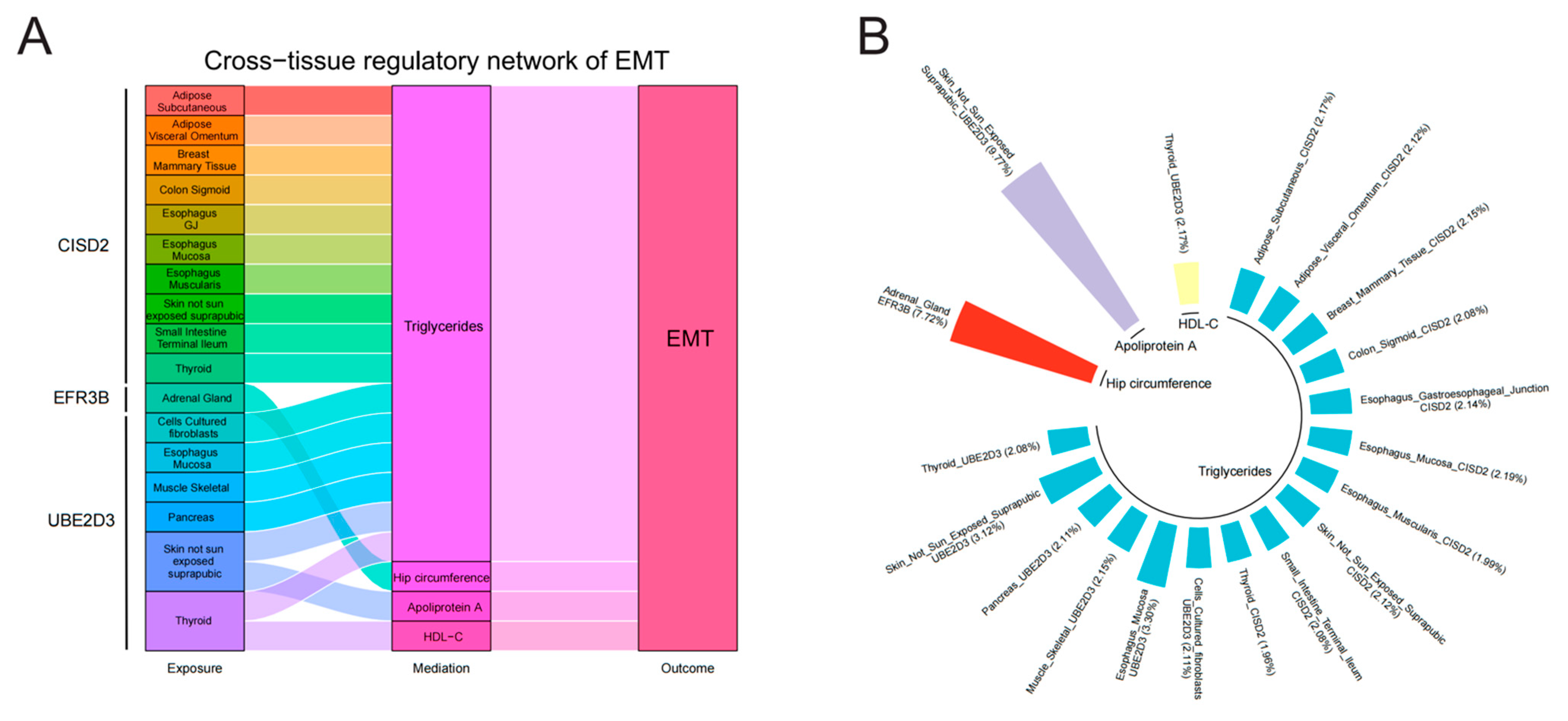
3.4. Mediating Roles of Modifiable Risk Factors in the Association Between Identified Genes Across Tissues and EMT
3.5. Further Bioinformatics Analyses
3.6. Classification Hierarchy of Identified Gene Targets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zondervan, K.T.; Becker, C.M.; Koga, K.; Missmer, S.A.; Taylor, R.N.; Viganò, P. Endometriosis. Nat. Rev. Dis. Primers 2018, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Simoens, S.; Dunselman, G.; Dirksen, C.; Hummelshoj, L.; Bokor, A.; Brandes, I.; Brodszky, V.; Canis, M.; Colombo, G.L.; DeLeire, T.; et al. The burden of endometriosis: Costs and quality of life of women with endometriosis and treated in referral centres. Hum. Reprod. 2012, 27, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Greene, R.; Stratton, P.; Cleary, S.D.; Ballweg, M.L.; Sinaii, N. Diagnostic experience among 4,334 women reporting surgically diagnosed endometriosis. Fertil. Steril. 2009, 91, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.S.; Kotlyar, A.M.; Flores, V.A. Endometriosis is a chronic systemic disease: Clinical challenges and novel innovations. Lancet 2021, 397, 839–852. [Google Scholar] [CrossRef]
- Guo, S.W.; Groothuis, P.G. Is it time for a paradigm shift in drug research and development in endometriosis/adenomyosis? Hum. Reprod. Update 2018, 24, 577–598. [Google Scholar] [CrossRef]
- Guo, S.W. Recurrence of endometriosis and its control. Hum. Reprod. Update 2009, 15, 441–461. [Google Scholar] [CrossRef] [PubMed]
- Borghese, B.; Zondervan, K.T.; Abrao, M.S.; Chapron, C.; Vaiman, D. Recent insights on the genetics and epigenetics of endometriosis. Clin. Genet. 2017, 91, 254–264. [Google Scholar] [CrossRef]
- Sapkota, Y.; Steinthorsdottir, V.; Morris, A.P.; Fassbender, A.; Rahmioglu, N.; De Vivo, I.; Buring, J.E.; Zhang, F.; Edwards, T.L.; Jones, S.; et al. Meta-analysis identifies five novel loci associated with endometriosis highlighting key genes involved in hormone metabolism. Nat. Commun. 2017, 8, 15539. [Google Scholar] [CrossRef]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar]
- Gusev, A.; Ko, A.; Shi, H.; Bhatia, G.; Chung, W.; Penninx, B.W.; Jansen, R.; de Geus, E.J.; Boomsma, D.I.; Wright, F.A.; et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016, 48, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, M.; Lu, Q.; Weng, H.; Wang, J.; Zekavat, S.M.; Yu, Z.; Li, B.; Gu, J.; Muchnik, S.; et al. A statistical framework for cross-tissue transcriptome-wide association analysis. Nat. Genet. 2019, 51, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wang, P.; Yin, K.J.; Yang, X.K.; Cen, H.; Sui, C.; Wu, G.C.; Pan, H.F. Novel insight into the aetiology of rheumatoid arthritis gained by a cross-tissue transcriptome-wide association study. RMD Open 2022, 8, e002529. [Google Scholar] [CrossRef]
- Gui, J.; Yang, X.; Tan, C.; Wang, L.; Meng, L.; Han, Z.; Liu, J.; Jiang, L. A cross-tissue transcriptome-wide association study reveals novel susceptibility genes for migraine. J. Headache Pain 2024, 25, 94. [Google Scholar] [CrossRef]
- Zhu, M.; Fan, J.; Zhang, C.; Xu, J.; Yin, R.; Zhang, E.; Wang, Y.; Ji, M.; Sun, Q.; Dai, J.; et al. A cross-tissue transcriptome-wide association study identifies novel susceptibility genes for lung cancer in Chinese populations. Hum. Mol. Genet. 2021, 30, 1666–1676. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef]
- Zheng, J.; Haberland, V.; Baird, D.; Walker, V.; Haycock, P.C.; Hurle, M.R.; Gutteridge, A.; Erola, P.; Liu, Y.; Luo, S.; et al. Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat. Genet. 2020, 52, 1122–1131. [Google Scholar] [CrossRef]
- The GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Sun, R.; Hui, S.; Bader, G.D.; Lin, X.; Kraft, P. Powerful gene set analysis in GWAS with the Generalized Berk-Jones statistic. PLoS Genet. 2019, 15, e1007530. [Google Scholar] [CrossRef]
- Glickman, M.E.; Rao, S.R.; Schultz, M.R. False discovery rate control is a recommended alternative to Bonferroni-type adjustments in health studies. J. Clin. Epidemiol. 2014, 67, 850–857. [Google Scholar] [CrossRef]
- Li, S.J.; Shi, J.J.; Mao, C.Y.; Zhang, C.; Xu, Y.F.; Fan, Y.; Hu, Z.W.; Yu, W.K.; Hao, X.Y.; Li, M.J.; et al. Identifying causal genes for migraine by integrating the proteome and transcriptome. J. Headache Pain 2023, 24, 111. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Laporte, A.D.; Spiegelman, D.; Akçimen, F.; Joober, R.; Dion, P.A.; Rouleau, G.A. Transcriptome-wide association study of attention deficit hyperactivity disorder identifies associated genes and phenotypes. Nat. Commun. 2019, 10, 4450. [Google Scholar] [CrossRef] [PubMed]
- Emdin, C.A.; Khera, A.V.; Kathiresan, S. Mendelian Randomization. Jama 2017, 318, 1925–1926. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Qin, F.; Li, X.; Du, X.; Li, T. Identification of novel proteins for lacunar stroke by integrating genome-wide association data and human brain proteomes. BMC Med. 2022, 20, 211. [Google Scholar] [CrossRef]
- Deng, Y.T.; Ou, Y.N.; Wu, B.S.; Yang, Y.X.; Jiang, Y.; Huang, Y.Y.; Liu, Y.; Tan, L.; Dong, Q.; Suckling, J.; et al. Identifying causal genes for depression via integration of the proteome and transcriptome from brain and blood. Mol. Psychiatry 2022, 27, 2849–2857. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef]
- Wallace, C. A more accurate method for colocalisation analysis allowing for multiple causal variants. PLoS Genet. 2021, 17, e1009440. [Google Scholar] [CrossRef]
- Burgess, S.; Scott, R.A.; Timpson, N.J.; Davey Smith, G.; Thompson, S.G. Using published data in Mendelian randomization: A blueprint for efficient identification of causal risk factors. Eur. J. Epidemiol. 2015, 30, 543–552. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef]
- Hartwig, F.P.; Davey Smith, G.; Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 2017, 46, 1985–1998. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef] [PubMed]
- VanderWeele, T.J. Mediation Analysis: A Practitioner’s Guide. Annu. Rev. Public Health 2016, 37, 17–32. [Google Scholar] [CrossRef]
- Carter, A.R.; Gill, D.; Davies, N.M.; Taylor, A.E.; Tillmann, T.; Vaucher, J.; Wootton, R.E.; Munafò, M.R.; Hemani, G.; Malik, R.; et al. Understanding the consequences of education inequality on cardiovascular disease: Mendelian randomisation study. BMJ 2019, 365, l1855. [Google Scholar] [CrossRef] [PubMed]
- Tamaresis, J.S.; Irwin, J.C.; Goldfien, G.A.; Rabban, J.T.; Burney, R.O.; Nezhat, C.; DePaolo, L.V.; Giudice, L.C. Molecular classification of endometriosis and disease stage using high-dimensional genomic data. Endocrinology 2014, 155, 4986–4999. [Google Scholar] [CrossRef] [PubMed]
- Mostafavi, S.; Ray, D.; Warde-Farley, D.; Grouios, C.; Morris, Q. GeneMANIA: A real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008, 9 (Suppl. S1), S4. [Google Scholar] [CrossRef]
- Tao, T.; Mo, X.; Zhao, L. Identifying novel potential drug targets for endometriosis via plasma proteome screening. Front. Endocrinol. 2024, 15, 1416978. [Google Scholar] [CrossRef]
- Zeng, P.; Lu, L.; Zhang, H.; Li, Y.; Tan, S.; Yu, T.; Zhou, H. Therapeutic targets for endometriosis: Genome-wide Mendelian randomization and colocalization analyses. Gene 2024, 893, 147970. [Google Scholar] [CrossRef]
- Mortlock, S.; Houshdaran, S.; Kosti, I.; Rahmioglu, N.; Nezhat, C.; Vitonis, A.F.; Andrews, S.V.; Grosjean, P.; Paranjpe, M.; Horne, A.W.; et al. Global endometrial DNA methylation analysis reveals insights into mQTL regulation and associated endometriosis disease risk and endometrial function. Commun. Biol. 2023, 6, 780. [Google Scholar] [CrossRef]
- Mortlock, S.; Kendarsari, R.I.; Fung, J.N.; Gibson, G.; Yang, F.; Restuadi, R.; Girling, J.E.; Holdsworth-Carson, S.J.; Teh, W.T.; Lukowski, S.W.; et al. Tissue specific regulation of transcription in endometrium and association with disease. Hum. Reprod. 2020, 35, 377–393. [Google Scholar] [CrossRef]
- Song, S.; Wang, L.; Hou, L.; Liu, J.S. Partitioning and aggregating cross-tissue and tissue-specific genetic effects to identify gene-trait associations. Nat. Commun. 2024, 15, 5769. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Kao, Y.C.; Lee, W.R.; Hsiao, Y.W.; Lu, T.P.; Chu, C.Y.; Lin, Y.J.; Huang, H.Y.; Hsieh, T.H.; Liu, Y.R.; et al. Clinicopathologic Characterization of GREB1-rearranged Uterine Sarcomas with Variable Sex-Cord Differentiation. Am. J. Surg. Pathol. 2019, 43, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Viana, P.C.S.; Mendes, A.; Delgado, L.F.; Tostes, G.; Gonçalves, L.; Gonçalves Júnior, H.; Raposo, N.R.B.; Vitral, G.S.F.; Gerheim, P. Association between Single Nucleotide Polymorphisms and Endometriosis in a Brazilian Population. Rev. Bras. Ginecol. Obstet. 2020, 42, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Chadchan, S.B.; Popli, P.; Liao, Z.; Andreas, E.; Dias, M.; Wang, T.; Gunderson, S.J.; Jimenez, P.T.; Lanza, D.G.; Lanz, R.B.; et al. A GREB1-steroid receptor feedforward mechanism governs differential GREB1 action in endometrial function and endometriosis. Nat. Commun. 2024, 15, 1947. [Google Scholar] [CrossRef]
- Mortlock, S.; Restuadi, R.; Levien, R.; Girling, J.E.; Holdsworth-Carson, S.J.; Healey, M.; Zhu, Z.; Qi, T.; Wu, Y.; Lukowski, S.W.; et al. Genetic regulation of methylation in human endometrium and blood and gene targets for reproductive diseases. Clin. Epigenetics 2019, 11, 49. [Google Scholar] [CrossRef]
- Sapkota, Y.; Vivo, I.; Steinthorsdottir, V.; Fassbender, A.; Bowdler, L.; Buring, J.E.; Edwards, T.L.; Jones, S.; O, D.; Peterse, D.; et al. Analysis of potential protein-modifying variants in 9000 endometriosis patients and 150000 controls of European ancestry. Sci. Rep. 2017, 7, 11380. [Google Scholar] [CrossRef]
- Osiński, M.; Mostowska, A.; Wirstlein, P.; Wender-Ożegowska, E.; Jagodziński, P.P.; Szczepańska, M. The assessment of GWAS-identified polymorphisms associated with infertility risk in Polish women with endometriosis. Ginekol. Pol. 2018, 89, 304–310. [Google Scholar] [CrossRef]
- Fung, J.N.; Holdsworth-Carson, S.J.; Sapkota, Y.; Zhao, Z.Z.; Jones, L.; Girling, J.E.; Paiva, P.; Healey, M.; Nyholt, D.R.; Rogers, P.A.; et al. Functional evaluation of genetic variants associated with endometriosis near GREB1. Hum. Reprod. 2015, 30, 1263–1275. [Google Scholar] [CrossRef]
- Rakers, C.; Schumacher, F.; Meinl, W.; Glatt, H.; Kleuser, B.; Wolber, G. In Silico Prediction of Human Sulfotransferase 1E1 Activity Guided by Pharmacophores from Molecular Dynamics Simulations. J. Biol. Chem. 2016, 291, 58–71. [Google Scholar] [CrossRef]
- Badie, A.; Saliminejad, K.; Salahshourifar, I.; Khorram Khorshid, H.R. Interleukin 1 alpha (IL1A) polymorphisms and risk of endometriosis in Iranian population: A case-control study. Gynecol. Endocrinol. 2020, 36, 135–138. [Google Scholar] [CrossRef]
- Shen, Z.Q.; Huang, Y.L.; Teng, Y.C.; Wang, T.W.; Kao, C.H.; Yeh, C.H.; Tsai, T.F. CISD2 maintains cellular homeostasis. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118954. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.H.; Shen, Z.Q.; Hsiung, S.Y.; Wu, P.C.; Teng, Y.C.; Chou, Y.J.; Fang, S.W.; Chen, C.F.; Yan, Y.T.; Kao, L.S.; et al. Cisd2 is essential to delaying cardiac aging and to maintaining heart functions. PLoS Biol. 2019, 17, e3000508. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.Q.; Chen, Y.F.; Chen, J.R.; Jou, Y.S.; Wu, P.C.; Kao, C.H.; Wang, C.H.; Huang, Y.L.; Chen, C.F.; Huang, T.S.; et al. CISD2 Haploinsufficiency Disrupts Calcium Homeostasis, Causes Nonalcoholic Fatty Liver Disease, and Promotes Hepatocellular Carcinoma. Cell Rep. 2017, 21, 2198–2211. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.G.; Meng, F.G.; Wang, M.G. CISD2 promotes the proliferation of glioma cells via suppressing beclin-1-mediated autophagy and is targeted by microRNA-449a. Mol. Med. Rep. 2017, 16, 7939–7948. [Google Scholar] [PubMed]
- Zhou, Y.X.; Wei, J.; Deng, G.; Hu, A.; Sun, P.Y.; Zhao, X.; Song, B.L.; Luo, J. Delivery of low-density lipoprotein from endocytic carriers to mitochondria supports steroidogenesis. Nat. Cell Biol. 2023, 25, 937–949. [Google Scholar] [CrossRef]
- Yalçin, Z.; Koot, D.; Bezstarosti, K.; Salas-Lloret, D.; Bleijerveld, O.B.; Boersma, V.; Falcone, M.; González-Prieto, R.; Altelaar, M.; Demmers, J.A.A.; et al. Ubiquitinome Profiling Reveals in Vivo UBE2D3 Targets and Implicates UBE2D3 in Protein Quality Control. Mol. Cell. Proteom. 2023, 22, 100548. [Google Scholar] [CrossRef]
- Pan, Z.; Bao, J.; Zhang, L.; Wei, S. UBE2D3 Activates SHP-2 Ubiquitination to Promote Glycolysis and Proliferation of Glioma via Regulating STAT3 Signaling Pathway. Front. Oncol. 2021, 11, 674286. [Google Scholar] [CrossRef]
- Wang, X.; Yang, P.; Jiang, Y.; Xu, Y.; Wang, N.; Rao, P.; Yang, L.; Sun, L.; Lu, D. UBE2D3 contributes to myocardial ischemia-reperfusion injury by regulating autophagy in dependence of p62/SQSTM1. Cell Signal 2021, 87, 110118. [Google Scholar] [CrossRef]
- Hiyoshi, Y.; Sato, Y.; Ichinoe, M.; Nagashio, R.; Hagiuda, D.; Kobayashi, M.; Kusuhara, S.; Igawa, S.; Shiomi, K.; Goshima, N.; et al. Prognostic significance of IMMT expression in surgically-resected lung adenocarcinoma. Thorac. Cancer 2019, 10, 2142–2151. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, Q.; Xiong, D.; Li, D.; Du, J.; Huang, Y.; Yang, Y.; Chen, R. Suppressing mitochondrial inner membrane protein (IMMT) inhibits the proliferation of breast cancer cells through mitochondrial remodeling and metabolic regulation. Sci. Rep. 2024, 14, 12766. [Google Scholar] [CrossRef]
- Bojjireddy, N.; Guzman-Hernandez, M.L.; Reinhard, N.R.; Jovic, M.; Balla, T. EFR3s are palmitoylated plasma membrane proteins that control responsiveness to G-protein-coupled receptors. J. Cell Sci. 2015, 128, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Wang, J.; Yang, E.; Zhang, Y.; Qian, Q.; Li, X.; Huang, F.; Sun, B. Efr3b is essential for social recognition by modulating the excitability of CA2 pyramidal neurons. Proc. Natl. Acad. Sci. USA 2024, 121, e2314557121. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, M.; Lin, M.; Hu, K.-L.; Li, R. Cross-Tissue Regulatory Network Analyses Reveal Novel Susceptibility Genes and Potential Mechanisms for Endometriosis. Biology 2024, 13, 871. https://doi.org/10.3390/biology13110871
Zou M, Lin M, Hu K-L, Li R. Cross-Tissue Regulatory Network Analyses Reveal Novel Susceptibility Genes and Potential Mechanisms for Endometriosis. Biology. 2024; 13(11):871. https://doi.org/10.3390/biology13110871
Chicago/Turabian StyleZou, Mingrui, Mingmei Lin, Kai-Lun Hu, and Rong Li. 2024. "Cross-Tissue Regulatory Network Analyses Reveal Novel Susceptibility Genes and Potential Mechanisms for Endometriosis" Biology 13, no. 11: 871. https://doi.org/10.3390/biology13110871
APA StyleZou, M., Lin, M., Hu, K.-L., & Li, R. (2024). Cross-Tissue Regulatory Network Analyses Reveal Novel Susceptibility Genes and Potential Mechanisms for Endometriosis. Biology, 13(11), 871. https://doi.org/10.3390/biology13110871