
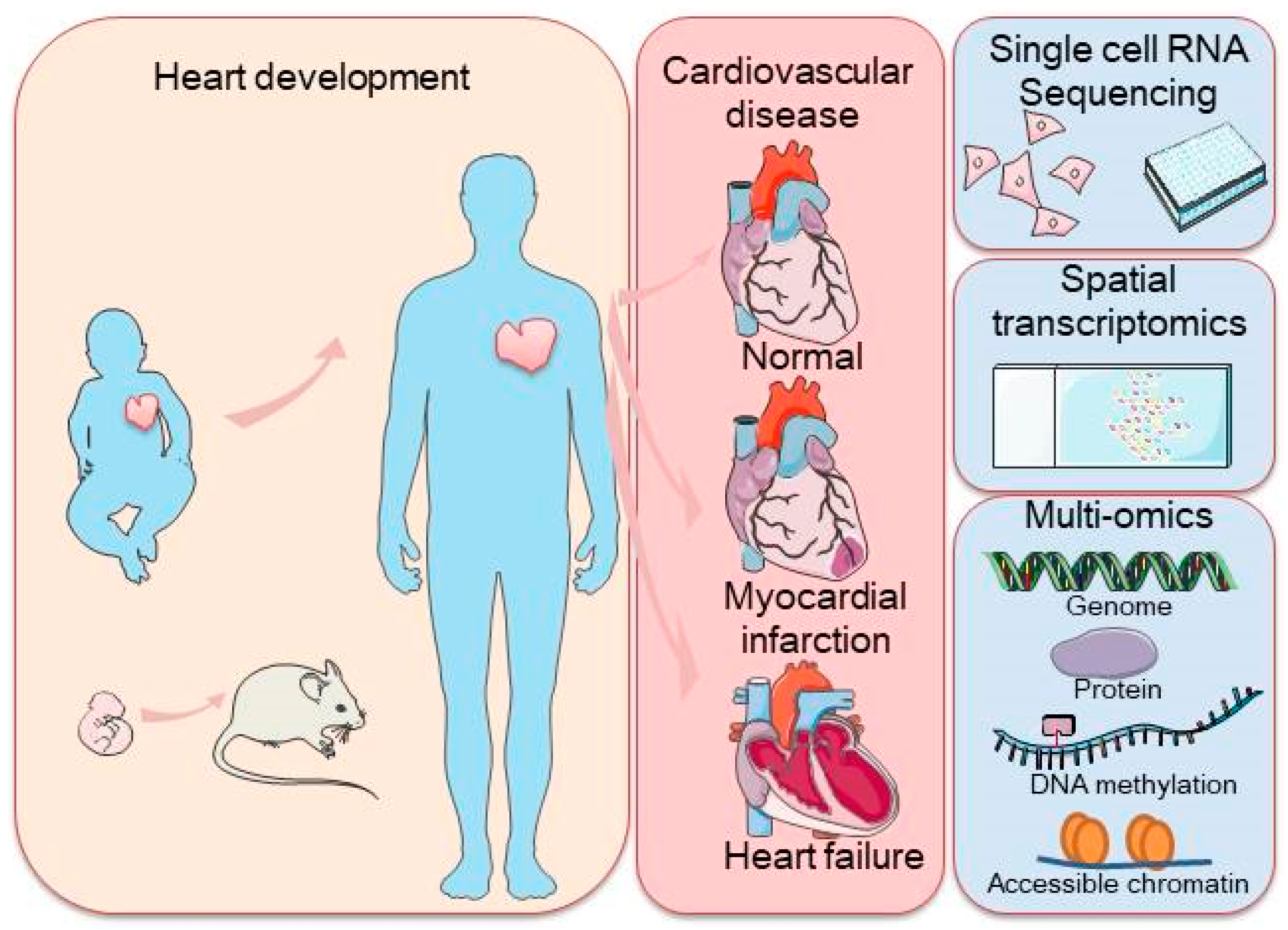
Single-Cell RNA Sequencing and Combinatorial Approaches for Understanding Heart Biology and Disease

Abstract
Simple Summary
Abstract
1. Introduction
2. The Application of scRNA-Seq in Understanding Cardiovascular Development and Disease
2.1. Pioneering scRNA-Seq Studies in Heart Development
2.2. Cardiovascular Progenitor Cells (CPCs) and Epicardium-Derived Progenitor Cells (EPDCs) in Heart Development
2.3. Cardiomyocyte Heterogeneity and Disease-Driven Changes
2.4. Charting the Cellular Landscape of the Adult Human Heart
2.5. Postnatal Heart Development and Maturation
2.6. Limitations and Future Directions for Single-Cell Techniques
3. Combination of scRNA-Seq with Other Single-Cell-Level Assessments
3.1. scRNA-Seq and Spatial Transcriptomics
3.2. scRNA-Seq and Epigenomics
3.3. scRNA-Seq and Proteomics
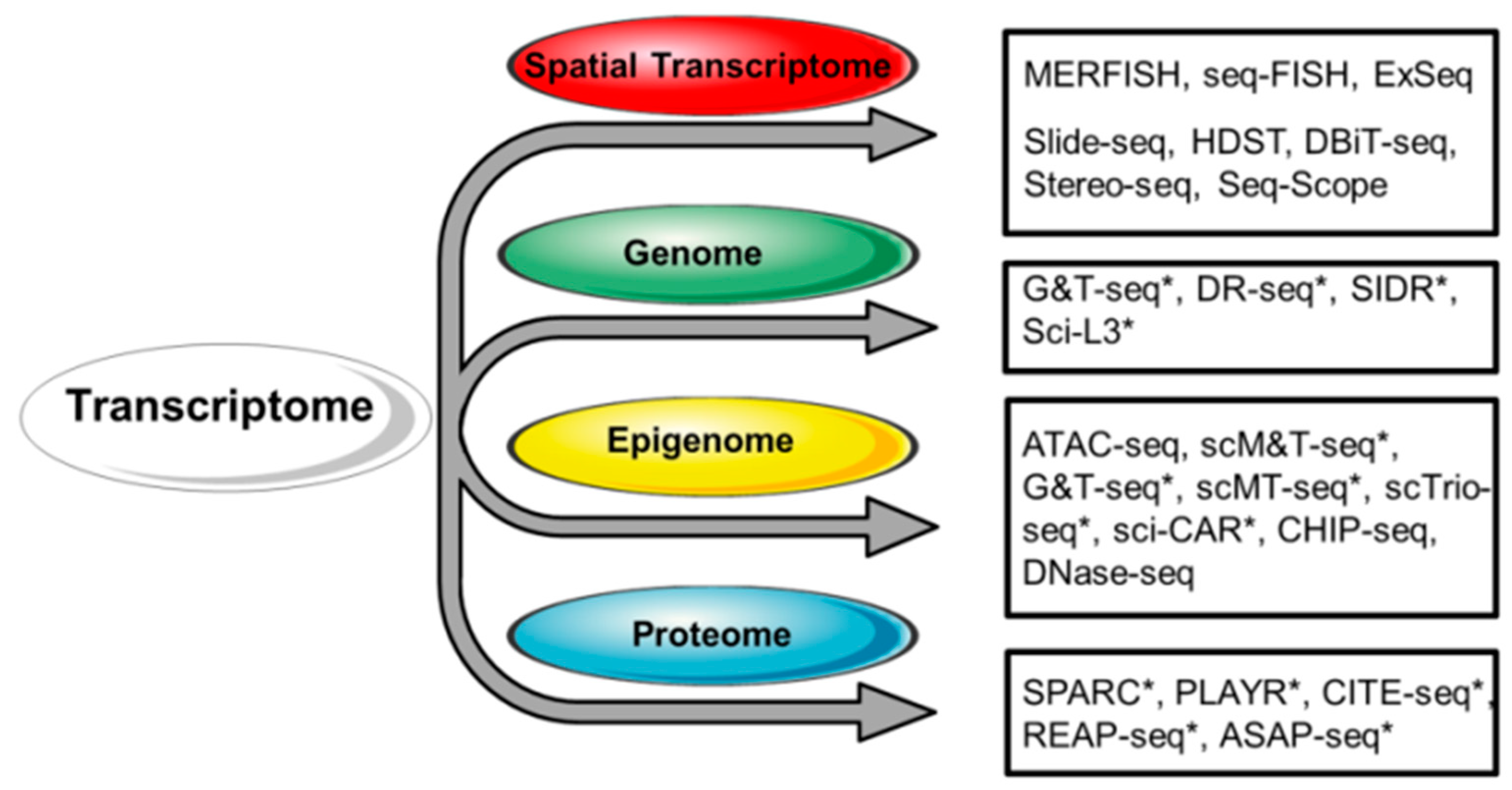
4. Multi-Omic Study of Single Cells
4.1. Single-Cell Genome and Transcriptome
4.2. Single-Cell Epigenome and Transcriptome
4.3. Single-Cell Protein Expression and Transcriptome
5. Perspectives and Significance
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, L.; Yu, P.; Zhou, B.; Song, J.; Li, Z.; Zhang, M.; Guo, G.; Wang, Y.; Chen, X.; Han, L.; et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat. Cell Biol. 2020, 22, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Tucker, N.R.; Chaffin, M.; Fleming, S.J.; Hall, A.W.; Parsons, V.A.; Bedi, K.C.; Akkad, A.-D.; Herndon, C.N.; Arduini, A.; Papangeli, I.; et al. Transcriptional and Cellular Diversity of the Human Heart. Circulation 2020, 142, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Paik, D.T.; Cho, S.; Tian, L.; Chang, H.Y.; Wu, J.C. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat. Rev. Cardiol. 2020, 17, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Bondue, A.; Blanpain, C. Mesp1: A key regulator of cardiovascular lineage commitment. Circ. Res. 2010, 107, 1414–1427. [Google Scholar] [CrossRef] [PubMed]
- Lyons, I.; Parsons, L.M.; Hartley, L.; Li, R.; Andrews, J.E.; Robb, L.; Harvey, R.P. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes. Dev. 1995, 9, 1654–1666. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.L.; Liang, X.; Shi, Y.; Chu, P.H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Brade, T.; Pane, L.S.; Moretti, A.; Chien, K.R.; Laugwitz, K.L. Embryonic heart progenitors and cardiogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a013847. [Google Scholar] [CrossRef] [PubMed]
- Cochain, C.; Vafadarnejad, E.; Arampatzi, P.; Pelisek, J.; Winkels, H.; Ley, K.; Wolf, D.; Saliba, A.E.; Zernecke, A. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ. Res. 2018, 122, 1661–1674. [Google Scholar] [CrossRef]
- Sun, K.; Li, Y.Y.; Jin, J. A double-edged sword of immuno-microenvironment in cardiac homeostasis and injury repair. Signal Transduct. Target. Ther. 2021, 6, 79. [Google Scholar] [CrossRef]
- Asp, M.; Salmén, F.; Ståhl, P.L.; Vickovic, S.; Felldin, U.; Löfling, M.; Fernandez Navarro, J.; Maaskola, J.; Eriksson, M.J.; Persson, B.; et al. Spatial detection of fetal marker genes expressed at low level in adult human heart tissue. Sci. Rep. 2017, 7, 12941. [Google Scholar] [CrossRef]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Budnik, B.; Levy, E.; Harmange, G.; Slavov, N. SCoPE-MS: Mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome. Biol. 2018, 19, 161. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. A dream of single-cell proteomics. Nat. Methods 2019, 16, 809–812. [Google Scholar] [CrossRef] [PubMed]
- Specht, H.; Slavov, N. Transformative Opportunities for Single-Cell Proteomics. J. Proteome Res. 2018, 17, 2565–2571. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Xu, A.; Sim, S.; Priest, J.R.; Tian, X.; Khan, T.; Quertermous, T.; Zhou, B.; Tsao, P.S.; Quake, S.R.; et al. Transcriptomic Profiling Maps Anatomically Patterned Subpopulations among Single Embryonic Cardiac Cells. Dev. Cell 2016, 39, 491–507. [Google Scholar] [CrossRef]
- DeLaughter, D.M.; Bick, A.G.; Wakimoto, H.; McKean, D.; Gorham, J.M.; Kathiriya, I.S.; Hinson, J.T.; Homsy, J.; Gray, J.; Pu, W.; et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell 2016, 39, 480–490. [Google Scholar] [CrossRef] [PubMed]
- de Soysa, T.Y.; Ranade, S.S.; Okawa, S.; Ravichandran, S.; Huang, Y.; Salunga, H.T.; Schricker, A.; Del Sol, A.; Gifford, C.A.; Srivastava, D. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature 2019, 572, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Sereti, K.-I.; Nguyen, N.B.; Kamran, P.; Zhao, P.; Ranjbarvaziri, S.; Park, S.; Sabri, S.; Engel, J.L.; Sung, K.; Kulkarni, R.P.; et al. Analysis of cardiomyocyte clonal expansion during mouse heart development and injury. Nat. Commun. 2018, 9, 754. [Google Scholar] [CrossRef]
- Xiong, H.; Luo, Y.; Yue, Y.; Zhang, J.; Ai, S.; Li, X.; Wang, X.; Zhang, Y.-L.; Wei, Y.; Li, H.-H.; et al. Single-Cell Transcriptomics Reveals Chemotaxis-Mediated Intraorgan Crosstalk During Cardiogenesis. Circ. Res. 2019, 125, 398–410. [Google Scholar] [CrossRef]
- Jia, G.; Preussner, J.; Chen, X.; Guenther, S.; Yuan, X.; Yekelchyk, M.; Kuenne, C.; Looso, M.; Zhou, Y.; Teichmann, S.; et al. Single cell RNA-seq and ATAC-seq analysis of cardiac progenitor cell transition states and lineage settlement. Nat. Commun. 2018, 9, 4877. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.; Giacomello, S.; Larsson, L.; Wu, C.; Fürth, D.; Qian, X.; Wärdell, E.; Custodio, J.; Reimegård, J.; Salmén, F.; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660.e19. [Google Scholar] [CrossRef] [PubMed]
- Lescroart, F.; Wang, X.; Lin, X.; Swedlund, B.; Gargouri, S.; Sànchez-Dànes, A.; Moignard, V.; Dubois, C.; Paulissen, C.; Kinston, S.; et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 2018, 359, 1177–1181. [Google Scholar] [CrossRef]
- Smits, A.M.; Dronkers, E.; Goumans, M.-J. The epicardium as a source of multipotent adult cardiac progenitor cells: Their origin, role and fate. Pharmacol. Res. 2018, 127, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Hill, M.C.; Zhang, M.; Martin, T.J.; Morikawa, Y.; Wang, S.; Moise, A.R.; Wythe, J.D.; Martin, J.F. Hippo Signaling Plays an Essential Role in Cell State Transitions during Cardiac Fibroblast Development. Dev. Cell 2018, 45, 153–169.e6. [Google Scholar] [CrossRef]
- See, K.; Tan, W.L.W.; Lim, E.H.; Tiang, Z.; Lee, L.T.; Li, P.Y.Q.; Luu, T.D.A.; Ackers-Johnson, M.; Foo, R.S. Single cardiomyocyte nuclear transcriptomes reveal a lincRNA-regulated de-differentiation and cell cycle stress-response in vivo. Nat. Commun. 2017, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- King, K.R.; Aguirre, A.D.; Ye, Y.-X.; Sun, Y.; Roh, J.D.; Ng, R.P.; Kohler, R.H.; Arlauckas, S.P.; Iwamoto, Y.; Savol, A.; et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 2017, 23, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Skelly, D.A.; Squiers, G.T.; McLellan, M.A.; Bolisetty, M.T.; Robson, P.; Rosenthal, N.A.; Pinto, A.R. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 2018, 22, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Schafer, S.; Viswanathan, S.; Widjaja, A.A.; Lim, W.-W.; Moreno-Moral, A.; DeLaughter, D.M.; Ng, B.; Patone, G.; Chow, K.; Khin, E.; et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017, 552, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Alter, C.; Henseler, A.S.; Owenier, C.; Hesse, J.; Ding, Z.; Lautwein, T.; Bahr, J.; Hayat, S.; Kramann, R.; Kostenis, E.; et al. IL-6 in the infarcted heart is preferentially formed by fibroblasts and modulated by purinergic signaling. J. Clin. Investig. 2023, 133, e163799. [Google Scholar] [CrossRef]
- Nicin, L.; Schroeter, S.M.; Glaser, S.F.; Schulze-Brüning, R.; Pham, M.-D.; Hille, S.S.; Yekelchyk, M.; Kattih, B.; Abplanalp, W.T.; Tombor, L.; et al. A human cell atlas of the pressure-induced hypertrophic heart. Nat. Cardiovasc. Res. 2022, 1, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Gladka, M.M.; Molenaar, B.; de Ruiter, H.; van der Elst, S.; Tsui, H.; Versteeg, D.; Lacraz, G.P.A.; Huibers, M.M.H.; van Oudenaarden, A.; van Rooij, E. Single-Cell Sequencing of the Healthy and Diseased Heart Reveals Cytoskeleton-Associated Protein 4 as a New Modulator of Fibroblasts Activation. Circulation 2018, 138, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.; Satoh, M.; Fujita, T.; Higo, T.; Sumida, T.; Ko, T.; Yamaguchi, T.; Tobita, T.; Naito, A.T.; Ito, M.; et al. Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat. Commun. 2018, 9, 4435. [Google Scholar] [CrossRef] [PubMed]
- Yekelchyk, M.; Guenther, S.; Preussner, J.; Braun, T. Mono- and multi-nucleated ventricular cardiomyocytes constitute a transcriptionally homogenous cell population. Basic Res. Cardiol. 2019, 114, 36. [Google Scholar] [CrossRef]
- Ren, Z.; Yu, P.; Li, D.; Li, Z.; Liao, Y.; Wang, Y.; Zhou, B.; Wang, L. Single-Cell Reconstruction of Progression Trajectory Reveals Intervention Principles in Pathological Cardiac Hypertrophy. Circulation 2020, 141, 1704–1719. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Wehrens, M.; de Leeuw, A.E.; Wright-Clark, M.; Eding, J.E.C.; Boogerd, C.J.; Molenaar, B.; van der Kraak, P.H.; Kuster, D.W.D.; van der Velden, J.; Michels, M.; et al. Single-cell transcriptomics provides insights into hypertrophic cardiomyopathy. Cell Rep. 2022, 39, 110809. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yao, F.; Wang, L.; Li, Z.; Ren, Z.; Li, D.; Zhang, M.; Han, L.; Wang, S.-Q.; Zhou, B.; et al. Single-cell analysis of murine fibroblasts identifies neonatal to adult switching that regulates cardiomyocyte maturation. Nat. Commun. 2020, 11, 2585. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Fabyanic, E.; Kwon, D.Y.; Tang, S.; Zhou, Z.; Wu, H. Dissecting Cell-Type Composition and Activity-Dependent Transcriptional State in Mammalian Brains by Massively Parallel Single-Nucleus RNA-Seq. Mol. Cell 2017, 68, 1006–1015.e7. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Liu, J.; Zhao, J.; Wilkins, B.J.; Lupino, K.; Wu, H.; Pei, L. Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev. 2018, 32, 1344–1357. [Google Scholar] [CrossRef] [PubMed]
- Nawy, T. In situ sequencing. Nat. Methods 2014, 11, 29. [Google Scholar] [CrossRef]
- Femino, A.M.; Fay, F.S.; Fogarty, K.; Singer, R.H. Visualization of single RNA transcripts in situ. Science 1998, 280, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sun, Y.-C.; Church, G.M.; Lee, J.H.; Zador, A.M. Efficient in situ barcode sequencing using padlock probe-based BaristaSeq. Nucleic Acids Res. 2018, 46, e22. [Google Scholar] [CrossRef]
- Wang, X.; Allen, W.E.; Wright, M.A.; Sylwestrak, E.L.; Samusik, N.; Vesuna, S.; Evans, K.; Liu, C.; Ramakrishnan, C.; Liu, J.; et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 2018, 361, eaat5691. [Google Scholar] [CrossRef]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef]
- Lubeck, E.; Coskun, A.F.; Zhiyentayev, T.; Ahmad, M.; Cai, L. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 2014, 11, 360–361. [Google Scholar] [CrossRef]
- Lee, J.H.; Daugharthy, E.R.; Scheiman, J.; Kalhor, R.; Yang, J.L.; Ferrante, T.C.; Terry, R.; Jeanty, S.S.F.; Li, C.; Amamoto, R.; et al. Highly multiplexed subcellular RNA sequencing in situ. Science 2014, 343, 1360–1363. [Google Scholar] [CrossRef] [PubMed]
- Alon, S.; Goodwin, D.R.; Sinha, A.; Wassie, A.T.; Chen, F.; Daugharthy, E.R.; Bando, Y.; Kajita, A.; Xue, A.G.; Marrett, K.; et al. Expansion sequencing: Spatially precise in situ transcriptomics in intact biological systems. Science 2021, 371, eaax2656. [Google Scholar] [CrossRef]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, C.; Navarro, J.F.; Jurek, A.; Märtin, A.; Lundeberg, J.; Meletis, K. Molecular atlas of the adult mouse brain. Sci. Adv. 2020, 6, eabb3446. [Google Scholar] [CrossRef]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Vickovic, S.; Eraslan, G.; Salmén, F.; Klughammer, J.; Stenbeck, L.; Schapiro, D.; Äijö, T.; Bonneau, R.; Bergenstråhle, L.; Navarro, J.F.; et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 2019, 16, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, M.; Deng, Y.; Su, G.; Enninful, A.; Guo, C.C.; Tebaldi, T.; Zhang, D.; Kim, D.; Bai, Z.; et al. High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue. Cell 2020, 183, 1665–1681.e18. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Yang, J.; Li, W.; Xu, J.; Hao, S.; et al. Large field of view-spatially resolved transcriptomics at nanoscale resolution. BioRxiv 2021, 2021, 427004. [Google Scholar] [CrossRef]
- Cho, C.-S.; Xi, J.; Si, Y.; Park, S.-R.; Hsu, J.-E.; Kim, M.; Jun, G.; Kang, H.M.; Lee, J.H. Microscopic examination of spatial transcriptome using Seq-Scope. Cell 2021, 184, 3559–3572.e22. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Sun, L.; Chen, J.Y.; Dong, R.; Lin, Y.; Palmiter, R.D.; Lin, S.; Gu, L. Continuous Polony Gels for Tissue Mapping with High Resolution and RNA Capture Efficiency. BioRxiv 2021, 2021, 435795. [Google Scholar] [CrossRef]
- Mantri, M.; Scuderi, G.J.; Abedini-Nassab, R.; Wang, M.F.Z.; McKellar, D.; Shi, H.; Grodner, B.; Butcher, J.T.; De Vlaminck, I. Spatiotemporal single-cell RNA sequencing of developing chicken hearts identifies interplay between cellular differentiation and morphogenesis. Nat. Commun. 2021, 12, 1771. [Google Scholar] [CrossRef]
- Chen, W.-T.; Lu, A.; Craessaerts, K.; Pavie, B.; Sala Frigerio, C.; Corthout, N.; Qian, X.; Laláková, J.; Kühnemund, M.; Voytyuk, I.; et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell 2020, 182, 976–991.e19. [Google Scholar] [CrossRef]
- Hoogaars, W.M.H.; Tessari, A.; Moorman, A.F.M.; de Boer, P.A.J.; Hagoort, J.; Soufan, A.T.; Campione, M.; Christoffels, V.M. The transcriptional repressor Tbx3 delineates the developing central conduction system of the heart. Cardiovasc. Res. 2004, 62, 489–499. [Google Scholar] [CrossRef]
- Papait, R.; Cattaneo, P.; Kunderfranco, P.; Greco, C.; Carullo, P.; Guffanti, A.; Viganò, V.; Stirparo, G.G.; Latronico, M.V.G.; Hasenfuss, G.; et al. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 20164–20169. [Google Scholar] [CrossRef]
- Huo, J.-L.; Jiao, L.; An, Q.; Chen, X.; Qi, Y.; Wei, B.; Zheng, Y.; Shi, X.; Gao, E.; Liu, H.-M.; et al. Myofibroblast Deficiency of LSD1 Alleviates TAC-Induced Heart Failure. Circ. Res. 2021, 129, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Goodyer, W.R.; Beyersdorf, B.M.; Paik, D.T.; Tian, L.; Li, G.; Buikema, J.W.; Chirikian, O.; Choi, S.; Venkatraman, S.; Adams, E.L.; et al. Transcriptomic Profiling of the Developing Cardiac Conduction System at Single-Cell Resolution. Circ. Res. 2019, 125, 379–397. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-M.; Lin, M.-S.; Chang, N.-C. Inhibition of histone deacetylase on ventricular remodeling in infarcted rats. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H968–H977. [Google Scholar] [CrossRef] [PubMed]
- Rotem, A.; Ram, O.; Shoresh, N.; Sperling, R.A.; Goren, A.; Weitz, D.A.; Bernstein, B.E. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 2015, 33, 1165–1172. [Google Scholar] [CrossRef]
- Nagano, T.; Lubling, Y.; Stevens, T.J.; Schoenfelder, S.; Yaffe, E.; Dean, W.; Laue, E.D.; Tanay, A.; Fraser, P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 2013, 502, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Tang, Q.; Wan, M.; Cui, K.; Zhang, Y.; Ren, G.; Ni, B.; Sklar, J.; Przytycka, T.M.; Childs, R.; et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 2015, 528, 142–146. [Google Scholar] [CrossRef]
- Adli, M.; Zhu, J.; Bernstein, B.E. Genome-wide chromatin maps derived from limited numbers of hematopoietic progenitors. Nat. Methods 2010, 7, 615–618. [Google Scholar] [CrossRef] [PubMed]
- Lara-Astiaso, D.; Weiner, A.; Lorenzo-Vivas, E.; Zaretsky, I.; Jaitin, D.A.; David, E.; Keren-Shaul, H.; Mildner, A.; Winter, D.; Jung, S.; et al. Immunogenetics. Chromatin state dynamics during blood formation. Science 2014, 345, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Chen, C.; He, B.; Tan, K.; Lu, C. A microfluidic device for epigenomic profiling using 100 cells. Nat. Methods 2015, 12, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Hsieh, Y.-P.; Ma, J.; Lu, C. Low-input and multiplexed microfluidic assay reveals epigenomic variation across cerebellum and prefrontal cortex. Sci. Adv. 2018, 4, eaar8187. [Google Scholar] [CrossRef] [PubMed]
- Grosselin, K.; Durand, A.; Marsolier, J.; Poitou, A.; Marangoni, E.; Nemati, F.; Dahmani, A.; Lameiras, S.; Reyal, F.; Frenoy, O.; et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet. 2019, 51, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, Y.; Qian, Y.; Liu, J.; Qian, L. Delineating chromatin accessibility re-patterning at single cell level during early stage of direct cardiac reprogramming. J. Mol. Cell. Cardiol. 2021, 162, 62–71. [Google Scholar] [CrossRef]
- Liu, J.; Liu, S.; Gao, H.; Han, L.; Chu, X.; Sheng, Y.; Shou, W.; Wang, Y.; Liu, Y.; Wan, J.; et al. Genome-wide studies reveal the essential and opposite roles of ARID1A in controlling human cardiogenesis and neurogenesis from pluripotent stem cells. Genome. Biol. 2020, 21, 169. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhang, S.; Zhao, Y.; Wang, W.; Zhang, H. A multi-omics approach to identify molecular alterations in a mouse model of heart failure. Theranostics 2022, 12, 1607–1620. [Google Scholar] [CrossRef]
- Wang, Z.; Cui, M.; Shah, A.M.; Tan, W.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Cell-Type-Specific Gene Regulatory Networks Underlying Murine Neonatal Heart Regeneration at Single-Cell Resolution. Cell Rep. 2020, 33, 108472. [Google Scholar] [CrossRef]
- Stege, N.M.; de Boer, R.A.; van den Berg, M.P.; Silljé, H.H.W. The Time Has Come to Explore Plasma Biomarkers in Genetic Cardiomyopathies. Int. J. Mol. Sci. 2021, 22, 2955. [Google Scholar] [CrossRef]
- Roncarati, R.; Viviani Anselmi, C.; Losi, M.A.; Papa, L.; Cavarretta, E.; Da Costa Martins, P.; Contaldi, C.; Saccani Jotti, G.; Franzone, A.; Galastri, L.; et al. Circulating miR-29a, among other up-regulated microRNAs, is the only biomarker for both hypertrophy and fibrosis in patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2014, 63, 920–927. [Google Scholar] [CrossRef]
- Xu, K.; Zhang, W.; Wang, C.; Hu, L.; Wang, R.; Wang, C.; Tang, L.; Zhou, G.; Zou, B.; Xie, H.; et al. Integrative analyses of scRNA-seq and scATAC-seq reveal CXCL14 as a key regulator of lymph node metastasis in breast cancer. Hum. Mol. Genet. 2021, 30, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Dirks, R.A.M.; Stunnenberg, H.G.; Marks, H. Genome-wide epigenomic profiling for biomarker discovery. Clin. Epigenetics 2016, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Ægidius, H.M.; Veidal, S.S.; Feigh, M.; Hallenborg, P.; Puglia, M.; Pers, T.H.; Vrang, N.; Jelsing, J.; Kornum, B.R.; Blagoev, B.; et al. Multi-omics characterization of a diet-induced obese model of non-alcoholic steatohepatitis. Sci. Rep. 2020, 10, 1148. [Google Scholar] [CrossRef]
- Rindler, K.; Krausgruber, T.; Thaler, F.M.; Alkon, N.; Bangert, C.; Kurz, H.; Fortelny, N.; Rojahn, T.B.; Jonak, C.; Griss, J.; et al. Spontaneously Resolved Atopic Dermatitis Shows Melanocyte and Immune Cell Activation Distinct From Healthy Control Skin. Front. Immunol. 2021, 12, 630892. [Google Scholar] [CrossRef]
- Meng, Q.; Chen, L.; Xiong, B.; Kang, B.; Zhang, P.; Tang, S.; Han, H.; Shen, W.; Feng, X.; Feng, S.; et al. Single-Cell Transcriptome Sequencing and Proteomics Reveal Neonatal Ileum Dynamic Developmental Potentials. Msystems 2021, 6, e0072521. [Google Scholar] [CrossRef] [PubMed]
- Winkels, H.; Ehinger, E.; Vassallo, M.; Buscher, K.; Dinh, H.Q.; Kobiyama, K.; Hamers, A.A.J.; Cochain, C.; Vafadarnejad, E.; Saliba, A.-E.; et al. Atlas of the Immune Cell Repertoire in Mouse Atherosclerosis Defined by Single-Cell RNA-Sequencing and Mass Cytometry. Circ. Res. 2018, 122, 1675–1688. [Google Scholar] [CrossRef]
- Ma, L.; Zhou, N.; Zou, R.; Shi, W.; Luo, Y.; Du, N.; Zhong, J.; Zhao, X.; Chen, X.; Xia, H.; et al. Single-Cell RNA Sequencing and Quantitative Proteomics Analysis Elucidate Marker Genes and Molecular Mechanisms in Hypoplastic Left Heart Patients With Heart Failure. Front. Cell Dev. Biol. 2021, 9, 617853. [Google Scholar] [CrossRef]
- Shen, S.; Zhang, M.; Wang, X.; Liu, Q.; Su, H.; Sun, B.; Guo, Z.; Tian, B.; Gan, H.; Gong, C.; et al. Single-cell RNA sequencing reveals S100a9(hi) macrophages promote the transition from acute inflammation to fibrotic remodeling after myocardial ischemia–reperfusion. Theranostics 2024, 14, 1241–1259. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zi, X.; Garmire, L.X.; Wu, Y.; Weissman, S.M.; Pan, X.; Fan, R. Co-detection and sequencing of genes and transcripts from the same single cells facilitated by a microfluidics platform. Sci. Rep. 2014, 4, 6485. [Google Scholar] [CrossRef] [PubMed]
- Han, K.Y.; Kim, K.-T.; Joung, J.-G.; Son, D.-S.; Kim, Y.J.; Jo, A.; Jeon, H.-J.; Moon, H.-S.; Yoo, C.E.; Chung, W.; et al. SIDR: Simultaneous isolation and parallel sequencing of genomic DNA and total RNA from single cells. Genome. Res. 2018, 28, 75–87. [Google Scholar] [CrossRef]
- Macaulay, I.C.; Haerty, W.; Kumar, P.; Li, Y.I.; Hu, T.X.; Teng, M.J.; Goolam, M.; Saurat, N.; Coupland, P.; Shirley, L.M.; et al. G&T-seq: Parallel sequencing of single-cell genomes and transcriptomes. Nat. Methods 2015, 12, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.S.; Kester, L.; Spanjaard, B.; Bienko, M.; van Oudenaarden, A. Integrated genome and transcriptome sequencing of the same cell. Nat. Biotechnol. 2015, 33, 285–289. [Google Scholar] [CrossRef]
- Yin, Y.; Jiang, Y.; Lam, K.-W.G.; Berletch, J.B.; Disteche, C.M.; Noble, W.S.; Steemers, F.J.; Camerini-Otero, R.D.; Adey, A.C.; Shendure, J. High-Throughput Single-Cell Sequencing with Linear Amplification. Mol. Cell. 2019, 76, 676–690.e10. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Wang, T.; Sun, X.; Zhang, Y.; Qiu, Y.; Sun, W.; Zhang, Y.; Teng, P.; Hu, Y.; Hu, X.; et al. Fibroblasts facilitate lymphatic vessel formation in transplanted heart. Theranostics 2024, 14, 1886–1908. [Google Scholar] [CrossRef] [PubMed]
- Angermueller, C.; Clark, S.J.; Lee, H.J.; Macaulay, I.C.; Teng, M.J.; Hu, T.X.; Krueger, F.; Smallwood, S.; Ponting, C.P.; Voet, T.; et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods 2016, 13, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhu, P.; Wu, X.; Li, X.; Wen, L.; Tang, F. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome. Res. 2013, 23, 2126–2135. [Google Scholar] [CrossRef]
- Hu, Y.; Huang, K.; An, Q.; Du, G.; Hu, G.; Xue, J.; Zhu, X.; Wang, C.-Y.; Xue, Z.; Fan, G. Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome. Biol. 2016, 17, 88. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Guo, H.; Cao, C.; Li, X.; Hu, B.; Zhu, P.; Wu, X.; Wen, L.; Tang, F.; Huang, Y.; et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016, 26, 304–319. [Google Scholar] [CrossRef]
- Chaligne, R.; Gaiti, F.; Silverbush, D.; Schiffman, J.S.; Weisman, H.R.; Kluegel, L.; Gritsch, S.; Deochand, S.D.; Gonzalez Castro, L.N.; Richman, A.R.; et al. Epigenetic encoding, heritability and plasticity of glioma transcriptional cell states. Nat. Genet. 2021, 53, 1469–1479. [Google Scholar] [CrossRef]
- Cooper, J.; Ding, Y.; Song, J.; Zhao, K. Genome-wide mapping of DNase I hypersensitive sites in rare cell populations using single-cell DNase sequencing. Nat. Protoc. 2017, 12, 2342–2354. [Google Scholar] [CrossRef]
- Cao, J.; Cusanovich, D.A.; Ramani, V.; Aghamirzaie, D.; Pliner, H.A.; Hill, A.J.; Daza, R.M.; McFaline-Figueroa, J.L.; Packer, J.S.; Christiansen, L.; et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 2018, 361, 1380–1385. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, Y.; Ma, H.; Xie, Y.; Xu, J.; Near, D.; Wang, H.; Garbutt, T.; Li, Y.; Liu, J.; et al. Single-cell dual-omics reveals the transcriptomic and epigenomic diversity of cardiac non-myocytes. Cardiovasc. Res. 2022, 118, 1548–1563. [Google Scholar] [CrossRef] [PubMed]
- Virant-Klun, I.; Leicht, S.; Hughes, C.; Krijgsveld, J. Identification of Maturation-Specific Proteins by Single-Cell Proteomics of Human Oocytes. Mol. Cell. Proteomics 2016, 15, 2616–2627. [Google Scholar] [CrossRef] [PubMed]
- Nestorowa, S.; Hamey, F.K.; Pijuan Sala, B.; Diamanti, E.; Shepherd, M.; Laurenti, E.; Wilson, N.K.; Kent, D.G.; Göttgens, B. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 2016, 128, e20–e31. [Google Scholar] [CrossRef] [PubMed]
- Soh, K.T.; Tario, J.D.; Colligan, S.; Maguire, O.; Pan, D.; Minderman, H.; Wallace, P.K. Simultaneous, Single-Cell Measurement of Messenger RNA, Cell Surface Proteins, and Intracellular Proteins. Curr. Protoc. Cytom. 2016, 75, 7–45. [Google Scholar] [CrossRef] [PubMed]
- Kochan, J.; Wawro, M.; Kasza, A. Simultaneous detection of mRNA and protein in single cells using immunofluorescence-combined single-molecule RNA FISH. Biotechniques 2015, 59, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Assarsson, E.; Lundberg, M.; Holmquist, G.; Björkesten, J.; Thorsen, S.B.; Ekman, D.; Eriksson, A.; Rennel Dickens, E.; Ohlsson, S.; Edfeldt, G.; et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE 2014, 9, e95192. [Google Scholar] [CrossRef] [PubMed]
- Darmanis, S.; Gallant, C.J.; Marinescu, V.D.; Niklasson, M.; Segerman, A.; Flamourakis, G.; Fredriksson, S.; Assarsson, E.; Lundberg, M.; Nelander, S.; et al. Simultaneous Multiplexed Measurement of RNA and Proteins in Single Cells. Cell Rep. 2016, 14, 380–389. [Google Scholar] [CrossRef]
- Reimegård, J.; Tarbier, M.; Danielsson, M.; Schuster, J.; Baskaran, S.; Panagiotou, S.; Dahl, N.; Friedländer, M.R.; Gallant, C.J. A combined approach for single-cell mRNA and intracellular protein expression analysis. Commun. Biol. 2021, 4, 624. [Google Scholar] [CrossRef]
- Frei, A.P.; Bava, F.-A.; Zunder, E.R.; Hsieh, E.W.Y.; Chen, S.-Y.; Nolan, G.P.; Gherardini, P.F. Highly multiplexed simultaneous detection of RNAs and proteins in single cells. Nat. Methods 2016, 13, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Peterson, V.M.; Zhang, K.X.; Kumar, N.; Wong, J.; Li, L.; Wilson, D.C.; Moore, R.; McClanahan, T.K.; Sadekova, S.; Klappenbach, J.A. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 2017, 35, 936–939. [Google Scholar] [CrossRef]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Lareau, C.A.; Chen, K.Y.; Zorzetto-Fernandes, A.L.; Hao, Y.; Takeshima, Y.; Luo, W.; Huang, T.-S.; Yeung, B.Z.; Papalexi, E.; et al. Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nat. Biotechnol. 2021, 39, 1246–1258. [Google Scholar] [CrossRef] [PubMed]
- Trzupek, D.; Dunstan, M.; Cutler, A.J.; Lee, M.; Godfrey, L.; Jarvis, L.; Rainbow, D.B.; Aschenbrenner, D.; Jones, J.L.; Uhlig, H.H.; et al. Discovery of CD80 and CD86 as recent activation markers on regulatory T cells by protein-RNA single-cell analysis. Genome. Med. 2020, 12, 55. [Google Scholar] [CrossRef] [PubMed]
- Mahdessian, D.; Cesnik, A.J.; Gnann, C.; Danielsson, F.; Stenström, L.; Arif, M.; Zhang, C.; Le, T.; Johansson, F.; Shutten, R.; et al. Spatiotemporal dissection of the cell cycle with single-cell proteogenomics. Nature 2021, 590, 649–654. [Google Scholar] [CrossRef]
- Chelko, S.P.; Penna, V.R.; Engel, M.; Shiel, E.A.; Centner, A.M.; Farra, W.; Cannon, E.N.; Landim-Vieira, M.; Schaible, N.; Lavine, K.; et al. NFĸB signaling drives myocardial injury via CCR2+ macrophages in a preclinical model of arrhythmogenic cardiomyopathy. J. Clin. Investig. 2024, 134. [Google Scholar] [CrossRef]
- Vyas, V.; Sandhar, B.; Keane, J.M.; Wood, E.G.; Blythe, H.; Jones, A.; Shahaj, E.; Fanti, S.; Williams, J.; Metic, N.; et al. Tissue-resident memory T cells in epicardial adipose tissue comprise transcriptionally distinct subsets that are modulated in atrial fibrillation. Nat. Cardiovasc. Res. 2024, 3, 1067–1082. [Google Scholar] [CrossRef] [PubMed]
- Vafadarnejad, E.; Rizzo, G.; Krampert, L.; Arampatzi, P.; Arias-Loza, A.P.; Nazzal, Y.; Rizakou, A.; Knochenhauer, T.; Bandi, S.R.; Nugroho, V.A.; et al. Dynamics of Cardiac Neutrophil Diversity in Murine Myocardial Infarction. Circ. Res. 2020, 127, e232–e249. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, Z.; Zhang, Y.; Xu, Z.; Xin, H.; Huang, H.; Duerr, R.H.; Chen, K.; Ding, Y.; Chen, W. BREM-SC: A bayesian random effects mixture model for joint clustering single cell multi-omics data. Nucleic Acids Res. 2020, 48, 5814–5824. [Google Scholar] [CrossRef]
- Brunner, A.-D.; Thielert, M.; Vasilopoulou, C.G.; Ammar, C.; Coscia, F.; Mund, A.; Hoerning, O.B.; Bache, N.; Apalategui, A.; Lubeck, M.; et al. Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation. BioRxiv 2012, 18, e10789. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Scheibinger, M.; Ellwanger, D.C.; Krey, J.F.; Choi, D.; Kelly, R.T.; Heller, S.; Barr-Gillespie, P.G. Single-cell proteomics reveals changes in expression during hair-cell development. eLife 2019, 8, e50777. [Google Scholar] [CrossRef]
- Alfaro, J.A.; Bohländer, P.; Dai, M.; Filius, M.; Howard, C.J.; van Kooten, X.F.; Ohayon, S.; Pomorski, A.; Schmid, S.; Aksimentiev, A.; et al. The emerging landscape of single-molecule protein sequencing technologies. Nat. Methods 2021, 18, 604–617. [Google Scholar] [CrossRef]
- Paul, I.; White, C.; Turcinovic, I.; Emili, A. Imaging the future: The emerging era of single-cell spatial proteomics. FEBS J. 2020, 288, 6990–7001. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Krieg, C.; Nowicka, M.; Guglietta, S.; Schindler, S.; Hartmann, F.J.; Weber, L.M.; Dummer, R.; Robinson, M.D.; Levesque, M.P.; Becher, B. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med. 2018, 24, 144–153. [Google Scholar] [CrossRef]
- Hartmann, F.J.; Bernard-Valnet, R.; Quériault, C.; Mrdjen, D.; Weber, L.M.; Galli, E.; Krieg, C.; Robinson, M.D.; Nguyen, X.-H.; Dauvilliers, Y.; et al. High-dimensional single-cell analysis reveals the immune signature of narcolepsy. J. Exp. Med. 2016, 213, 2621–2633. [Google Scholar] [CrossRef]
- Newell, E.W.; Sigal, N.; Nair, N.; Kidd, B.A.; Greenberg, H.B.; Davis, M.M. Combinatorial tetramer staining and mass cytometry analysis facilitate T-cell epitope mapping and characterization. Nat. Biotechnol. 2013, 31, 623–629. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Study | Findings | Relevance to Heart Research |
|---|---|---|
| Li et al. [15] (2016) | Generated transcriptomic profiles of 2233 embryonic mouse cardiac cells using ATLAS-seq, accurately predicting the anatomical locations of individual cardiomyocytes during development. | Helps map the spatial and temporal dynamics of cardiomyocytes, aiding in understanding heart development at cellular resolution, which is crucial for dissecting lineage-traced cells like ISL1-marked cells. |
| DeLaughter et al. [16] (2016) | Profiled embryonic and postnatal cardiomyocytes, identifying stage-specific transcriptional programs and lineage-specific gene expression during early cardiac development. | Provides insights into the transcriptional mechanisms regulating heart development and maturation, facilitating the discovery of key genes involved in heart formation and function. |
| De Soysa et al. [17] (2017) | Identified Hand2 as a critical regulator of outflow tract (OFT) specification and right ventricle differentiation, using scRNA-seq to analyze cardiovascular progenitor cells (CPCs). | Offers new insights into how molecular regulators like Hand2 direct specific cardiac lineages, which is essential for understanding congenital heart defects and improving regenerative therapies. |
| Sereti et al. [18] (2018) | Identified Mesp1- and Nkx2-5-expressing CPCs as major sources of new cardiomyocytes, showing that α-MHC-expressing cells lose their proliferative potential after E12.5. | Contributes to understanding how cardiac progenitors contribute to heart cell populations and the timing of cardiomyocyte proliferation, which is key in cardiac regeneration research. |
| Xiong et al. (2019) [19] | Discovered distinct transcriptional programs for CPCs from the first and second heart fields (FHF and SHF), proposing a model of intraorgan crosstalk regulated by Nkx2-5. | Reveals the importance of cell communication in heart development and how CPCs migrate and differentiate into cardiomyocytes, providing potential therapeutic targets for heart repair. |
| Jia et al. [20] (2019) | Characterized CPCs marked by Nkx2-5 and Isl1, showing that Isl1 is crucial for endothelial and cardiomyocyte fate, while Nkx2-5 directs cardiomyocyte lineage. | Provides a detailed understanding of early cardiac lineage bifurcation, which is crucial for developing targeted therapies for heart diseases involving CPCs and early heart defects. |
| Asp et al. [21] (2020) | Created the first single-cell atlas of the human heart during early development, revealing the spatial distribution and heterogeneity of cell types at different developmental stages. | Offers a comprehensive reference for heart development, improving our ability to study congenital heart defects and develop 3D models for heart regeneration therapies. |
| Study | Findings | Relevance to Heart Research |
|---|---|---|
| Skelly et al. [27] (2018) | Mapped intercellular communication networks in mouse hearts, identifying fibroblasts as the most communicative cells. | Highlights the critical role of fibroblasts in heart function and their response to injury, offering new therapeutic approaches for cardiac fibrosis and heart repair post-injury. |
| Schafer et al. [28] (2018) | Identified IL-11-positive fibroblasts as key players in fibroblast differentiation into myofibroblasts during cardiovascular disease progression. | Provides potential therapeutic targets for controlling fibrosis in cardiovascular diseases, particularly by targeting IL-11-related pathways. |
| Christina et al. [29] (2020) | scRNA-seq revealed that IL-6 is primarily expressed by fibroblasts and regulated by T cell-derived adenosine, indicating a purinergic metabolic interaction between fibroblasts and T cells. | Highlights the role of immune–metabolic interactions in regulating IL-6 production, a key factor in inflammation, offering potential therapeutic targets for cardiac inflammation. |
| See et al. [25] (2020) | Single-nucleus RNA-seq on cardiomyocytes from healthy and failing hearts uncovered heterogeneity in the transcriptomic stress response, suggesting lincRNAs as targets for cardiac repair. | Identifies long intergenic non-coding RNAs (lincRNAs) as potential therapeutic targets for promoting cardiac repair, particularly in heart failure. |
| Nicin et al. [30] (2020) | snRNA-seq of the cardiac septum in hypertrophic patients identified 19 distinct cell clusters, emphasizing reduced Eph receptor signaling and intercellular crosstalk in disease. | Suggests that impaired Eph receptor signaling contributes to cardiac hypertrophy, offering insights into new therapeutic strategies targeting cell communication. |
| Nomura et al. [32] (2019) | Unveiled the heterogeneous gene expression of hypertrophying cardiomyocytes in a mouse model of pressure overload-induced cardiac hypertrophy, identifying the role of p53 in pathogenic gene programs. | Offers insights into the molecular mechanisms driving cardiac hypertrophy, which is crucial for developing treatments to prevent or reverse heart failure. |
| Dick et al. [39] (2020) | Showed the heterogeneity and plasticity of cardiac macrophages, revealing that resident macrophages in the infarct area provide cardioprotective effects after myocardial infarction. | Identifies cardiac macrophages as key players in heart injury and repair, providing insights into immune-based therapies for myocardial infarction recovery. |
| Wehrens et al. [40] (2020) | Identified subpopulations of cardiomyocytes in hypertrophic cardiomyopathy, with one population showing greater expression of NPPA. | Suggests that targeting specific cardiomyocyte subpopulations could offer novel therapeutic approaches for treating hypertrophic cardiomyopathy and improving heart function. |
| Wang et al. [1] (2020) | Created the first cellular map of the adult human heart, analyzing 11,492 single cells, showing the impact of endothelial cells on cardiomyocyte function. | Highlights endothelial cells’ role in heart function and potential as therapeutic targets for heart failure. |
| Tucker et al. [2] (2020) | Sequenced 287,269 nuclei, identifying nine major cell types and linking them to cardiovascular diseases through genome-wide association studies. | Expands understanding of heart disease and identifies adipocytes, cardiomyocytes, and fibroblasts as therapeutic targets. |
| Litviňuková et al. [11] (2020) | Provided transcriptomic data on six heart regions, identifying chamber and sex-specific differences and macrophage heterogeneity. | Offers insights into cellular diversity, heart homeostasis, and new therapeutic targets based on chamber-specific cell types. |
| Wang et al. [41] (2021) | Revealed cardiac fibroblast subtype switching is crucial for postnatal cardiomyocyte maturation. | Identifies fibroblasts’ role in heart maturation, offering targets for regenerative therapies. |
| Hu et al. [42] (2017) | Found a decrease in proliferating cardiomyocytes and an increase in mature cardiomyocytes between postnatal days 6 and 10. | Key to understanding postnatal heart development, with implications for regenerative medicine. |
| Technique | Summary | Advantages | Limitations |
|---|---|---|---|
| In situ sequencing (ISS) | Directly sequences RNA within fixed tissues, preserving spatial context. | High spatial resolution; preserves tissue architecture. | Limited to fixed samples; lower throughput. |
| MERFISH | Uses multiplexed error-robust fluorescence in situ hybridization for RNA detection. | High sensitivity; can detect thousands of RNA species. | Complex experimental setup; data analysis can be challenging. |
| seqFISH | Similar to MERFISH but employs sequential hybridization of fluorescent probes. | High multiplexing capability; quantitative measurements. | Time-consuming; requires extensive optimization. |
| ExSeq | Extracts mRNA from single cells and sequences it with spatial information. | Maintains spatial context; suitable for low-abundance transcripts. | Lower resolution compared to other methods. |
| Slide-seq | Transfers RNA from tissue sections to a DNA-barcoded slide for sequencing. | High spatial resolution; can map cellular neighborhoods. | Requires specialized equipment; potential for cross-contamination. |
| High-definition spatial transcriptomics (HDST) | Combines tissue sectioning with sequencing to capture spatial gene expression. | High resolution; robust quantitative data. | Technical complexity; expensive. |
| DBiT-seq | Utilizes DNA-barcoded probes for spatial transcriptomics via digital spatial profiling. | High spatial resolution; simultaneous protein/RNA detection. | Limited number of detectable transcripts in a single run. |
| Stereo-seq | Combines in situ transcriptome profiling with high spatial resolution imaging. | High throughput; accurate spatial mapping of transcripts. | Requires specialized equipment; analysis can be complex. |
| Seq-scope | Achieves high-resolution spatial transcriptomics using a miniaturized imaging system. | High throughput; real-time imaging capabilities. | Limited detection range; requires advanced imaging systems. |
| PIXEL-seq | Combines high-resolution imaging with RNA sequencing, providing spatial context. | High sensitivity; allows for multiplexing. | Potential resolution constraints and the necessity for advanced imaging systems. |
| Technique | Summary | Advantages | Limitations |
|---|---|---|---|
| ATAC-seq | Identifies regions of open chromatin, which are accessible to transposase and involved in gene regulation. | High sensitivity; low cell input; rapid protocol. | Limited to accessible chromatin regions; cannot distinguish between different types of open chromatin. |
| ChIP-seq | Analyzes protein–DNA interactions by combining chromatin immunoprecipitation with sequencing. | Precise localization of DNA-binding proteins; works for histone modifications. | Requires high-quality antibodies; resolution depends on fragment size. |
| Hi-C | Captures genome-wide chromatin interactions, identifying 3D genome structure. | Provides insight into 3D genome organization; genome-wide approach. | Low resolution; difficult to resolve small chromatin interactions. |
| DNase-seq | Maps DNase I hypersensitive sites to locate regions of open chromatin. | Effective in detecting regulatory elements; genome-wide mapping of accessible chromatin. | Requires high-quality enzymes; may produce biased results due to DNase sensitivity. |
| nano-ChIP | A miniaturized version of ChIP-seq, designed for very low input or single cells. | Suitable for low cell input; maintains ChIP-seq sensitivity in small samples. | Technically challenging; requires precise handling of small samples. |
| iChIP | Improved ChIP-seq with higher specificity, optimized for low-input or single-cell experiments. | Higher specificity and optimized for single-cell input; reduces noise. | Complex protocol; requires optimization for low-input samples. |
| MOWChIP | Microfluidic ChIP for small samples with high efficiency and reduced sample loss. | Efficient for small cell populations; reduces sample loss. | Expensive; requires specialized equipment. |
| SurfaceChIP | Surface-enhanced ChIP using specialized surfaces for improved chromatin capture. | Improved chromatin capture; suitable for small sample sizes. | Requires specialized surfaces and techniques; high complexity. |
| Drop-ChIP | Single-cell ChIP sequencing technology allowing for high-throughput profiling of chromatin states. | High-throughput profiling; compatible with single-cell experiments. | Complex data analysis; requires specialized tools for single-cell data. |
| Technique | Summary | Advantages | Limitations |
|---|---|---|---|
| SIDR (simultaneous isolation of gDNA and RNA) | A method for simultaneous isolation of genomic DNA (gDNA) and RNA from single cells for parallel sequencing. | Captures both genetic and transcriptomic information; preserves single-cell integrity. | Technically challenging; requires careful handling to prevent sample loss or degradation. |
| G&T-seq | Genome and transcriptome sequencing method for simultaneous separation and sequencing of gDNA and full-length mRNA from single cells. | Enables parallel analysis of genome and transcriptome; provides insights into gene expression and genetic variants. | Complex protocol; requires advanced sequencing tools and expertise in data integration. |
| DR-Seq (gDNA-mRNA sequencing) | A method that sequences both genomic DNA and mRNA from the same single cell, capturing the relationship between the genome and transcriptome. | Simultaneously provides genome and transcriptome data; useful for studying genotype–phenotype relationships. | Complex data analysis; requires optimized pipelines for both RNA and DNA sequencing data. |
| sci-L3-RNA/DNA | Single-cell combinatorial indexing method that allows for parallel sequencing of both RNA and DNA at the single-cell level. | High throughput; scalable to a large number of single cells; captures both RNA and DNA information. | Requires advanced bioinformatics analysis; expensive and technically complex. |
| Technique | Summary | Advantages | Limitations |
|---|---|---|---|
| scM&T-seq | Single-cell Multiome and Transcriptome Sequencing that simultaneously profiles both genome and transcriptome from the same cell. | Captures both genetic and epigenetic information; provides multi-omic data from a single cell. | Technically challenging; requires advanced bioinformatics for data integration. |
| G&T-seq | Genome and transcriptome sequencing method that separates and sequences gDNA and full-length mRNA from single cells. | Enables parallel analysis of genome and transcriptome; provides insights into gene expression and genetic variants. | Complex protocol; requires advanced sequencing tools and expertise in data integration. |
| ScTrioseq | A method that profiles the genome, transcriptome, and DNA methylation from a single cell, capturing multi-layered molecular information. | Captures genomic, transcriptomic, and epigenomic data in a single experiment; valuable for comprehensive analysis. | Complex workflow; requires advanced bioinformatics for multi-omic data analysis. |
| scATAC-seq | Single-cell ATAC-seq captures open chromatin regions at single-cell resolution. | High sensitivity; low-input requirements; captures open chromatin regions at single-cell resolution. | Requires specialized equipment; complex data analysis for single-cell resolution. |
| scDNase-seq | Single-cell DNase I hypersensitivity sequencing maps regions of open chromatin at the single-cell level. | Effective in detecting regulatory elements at single-cell level; captures dynamic chromatin accessibility. | Technically complex; requires high-quality enzymes and optimized analysis pipelines. |
| scChIP-seq | Single-cell ChIP-seq identifies protein–DNA interactions or histone modifications at single-cell resolution. | Provides high-resolution insight into chromatin states and protein–DNA interactions; works for histone modifications. | Requires high-quality antibodies and optimized protocols; complex data interpretation. |
| Technique | Summary | Advantages | Limitations |
|---|---|---|---|
| SPARC | SPARC (Single-cell Profiling using RNA and PEA) combines single-cell RNA sequencing with proximity extension assay (PEA) to detect proteins and transcripts simultaneously. | Simultaneously detects RNA and protein levels; provides complementary information about gene expression and protein activity. | Technically complex; requires optimization for PEA and RNA-seq data integration. |
| CITE-seq | CITE-seq (Cellular Indexing of Transcriptomes and Epitopes by sequencing) combines single-cell RNA-seq with surface protein detection using DNA-barcoded antibodies. | High throughput; simultaneous measurement of mRNA and surface proteins; scalable and flexible. | Requires high-quality antibodies; limited to surface protein detection; antibody availability can be a constraint. |
| REAP-seq | REAP-seq (RNA Expression and Protein Sequencing) profiles both RNA and proteins at the single-cell level using antibodies conjugated with oligonucleotides. | Profiles both RNA and protein from the same cell; provides multi-modal insights into cell function. | Requires high-quality antibodies; may face challenges in data normalization between RNA and protein readouts. |
| ASAP-seq | ASAP-seq (Antibody Sequencing for Assessing Protein modifications) integrates single-cell transcriptomics with detection of histone modifications and surface proteins. | Integrates transcriptomics with epigenomic modifications and protein detection; provides deeper insight into chromatin states and gene expression. | Technically demanding; requires expertise in both transcriptomics and epigenomics; data integration can be challenging. |
| AbSeq | AbSeq (Antibody Sequencing) combines single-cell sequencing with antibody-based protein detection, allowing for simultaneous measurement of RNA and protein levels. | Simultaneous profiling of RNA and protein at single-cell resolution; helps uncover relationships between gene expression and protein function. | Dependent on antibody quality and availability; complex data analysis required to integrate protein and RNA data. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Jin, B. Single-Cell RNA Sequencing and Combinatorial Approaches for Understanding Heart Biology and Disease. Biology 2024, 13, 783. https://doi.org/10.3390/biology13100783
Wang L, Jin B. Single-Cell RNA Sequencing and Combinatorial Approaches for Understanding Heart Biology and Disease. Biology. 2024; 13(10):783. https://doi.org/10.3390/biology13100783
Chicago/Turabian StyleWang, Le, and Bo Jin. 2024. "Single-Cell RNA Sequencing and Combinatorial Approaches for Understanding Heart Biology and Disease" Biology 13, no. 10: 783. https://doi.org/10.3390/biology13100783
APA StyleWang, L., & Jin, B. (2024). Single-Cell RNA Sequencing and Combinatorial Approaches for Understanding Heart Biology and Disease. Biology, 13(10), 783. https://doi.org/10.3390/biology13100783