
Comprehensive Review and Assessment of Computational Methods for Prediction of N6-Methyladenosine Sites


Abstract
Simple Summary
Abstract
1. Introduction
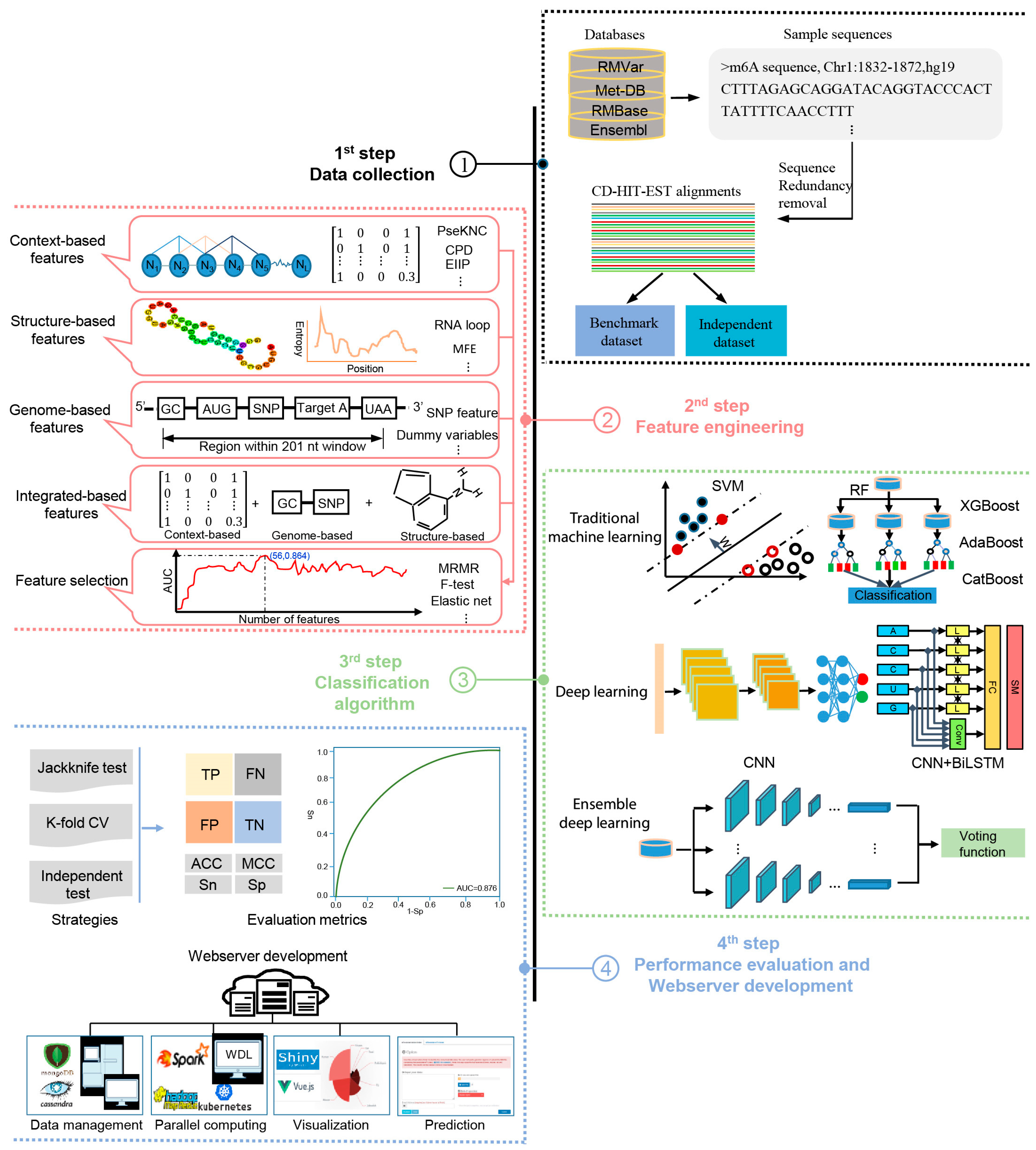
2. Systematic Comparison of Computational Approaches for m6A Site Prediction
2.1. Existing Methods for m6A Site Prediction
2.2. Construction of Training Dataset

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Species | Latest Version | Feature | Website (URL) |
|---|---|---|---|---|
| Met-DB [78,79] | H. sapiens, M. musculus | 1.0 (November 2014) | MeT-DB is the first comprehensive resource for m6A in the mammalian transcriptome and provides ∼300 k m6A methylation sites in 74 MeRIP-Seq samples from 22 different experimental conditions. | http://compgenomics.utsa.edu/methylation/ (accessed on 12 September 2022) |
| RMBase [76,77] | H. sapiens, M. musculus, Rhesus, Chimpanzee, Rat, Pig, Zebrafish, S. cerevisiae, Fly, A. thaliana, S. pombe, E. coli, P. aetuginosa | 2.0 (October 2017) | RMBase v2.0 was expanded with ∼600 datasets and ∼1,397,000 modification sites from 47 studies among 13 species, including ∼1,373,000 m6A sites at a single nucleotide or very high resolution. | http://rna.sysu.edu.cn/rmbase/ (accessed on 12 September 2022) |
| RMVar [75] | H. sapiens, M. musculus | 2.0 (October 2020) | RMVar is an updated version of m6Avar and contains 179,270 high-confidence m6A sites from H. sapiens and 10,760 from M. musculus in total. | http://rmvar.renlab.org (accessed on 12 September 2022) |
| m6A-Atlas [84] | H. sapiens, M. musculus, A. thaliana, Fly, Rat, Yeast, Zebrafish, virus | 1.0 (August 2020) | m6A-Atlas is a comprehensive knowledge base for the unraveling of the m6A epitranscriptome and provides 442,162 high-confidence m6A sites identified from seven base-resolution technologies. | www.xjtlu.edu.cn/biologicalsciences/atlas (accessed on 12 September 2022) |
| ConsRM [65] | H. sapiens | 1.0 (February 2021) | ConsRM is a database on the collection and large-scale prediction of evolutionarily conserved RNA methylation sites and includes 177,998 base-resolution human m6A RNA methylation sites with ConsRM scores. | https://www.xjtlu.edu.cn/biologicalsciences/con (accessed on 12 September 2022) |
| Ensembl | H. sapiens, M. musculus | 106 (April 2022) | Ensembl annotates genes, collects disease data, and provides m6A site information from mammalian species. | https://asia.ensembl.org/index.html (accessed on 12 September 2022) |
2.3. Construction of Independent Test Dataset
| Species | Dataset Name | Source | Positive-to-Negative Ratio |
|---|---|---|---|
| 1:1 | |||
| H. sapiens | Hg38_Human | scDART-seq data containing single-nucleotide m6A sites in single cells [22] | 22,248 |
| hg19_Human1 | Single-nucleotide m6A data [86] | 2064 | |
| hg19_Human2 | Single-nucleotide m6A data [85] | 37,372 | |
| hg19_Human3 | Single-nucleotide m6A data [88] | 930 | |
| hg19_Human4 | Data intersection between ConsRM, RMBase, and m6A-Atlas | 12,588 | |
| M. musculus | mm10_Mouse | Data intersection between RMVar, RMBase, and m6A-Atlas | 3330 |
| Rhesus | rheMac8_Rhesus | RMBase | 12,098 |
| Chimpanzee | panTro4_Chimpanzee | RMBase | 15,424 |
| Rat | rn5_Rat | RMBase | 24,380 |
| Pig | susScr3_Pig | RMBase | 42,838 |
| Zebrafish | danRer10_Zebrafish | RMBase | 8946 |
| S. cerevisiae | sacCer3_S_cerevisiae | Data intersection between m6A-Atlas and RMBase | 14,876 |
| A. thaliana | TAIR10_A_thaliana | Data intersection between m6A-Atlas and RMBase | 4516 |
2.4. Feature Engineering and Representation
2.4.1. Context-Based Features
2.4.2. Structure-Based Features
2.4.3. Genome-Based Features
2.4.4. Integrated Features
2.4.5. Feature Selection
2.5. Predictive Algorithms Employed
2.5.1. Traditional Machine Learning-Based Methods
2.5.2. Deep Learning-Based Methods
2.5.3. Ensemble Learning-Based Methods
2.6. Strategies and Measures for Performance Assessment
2.7. Webserver/Software Availability and Usability
3. Experimental Results
3.1. Performance Comparison of Species-Specific Predictors
3.2. Cross-Species Validation of State-of-the-Art Predictors
3.3. Performance Comparison for Prediction of m6A Sites in Single Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, X.; Xiong, X.; Yi, C. Epitranscriptome sequencing technologies: Decoding RNA modifications. Nat. Methods 2016, 14, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Ren, J.; Li, L.; Li, S.; Xiang, K.; Shang, D. Development and validation of a novel N6-methyladenosine (m6A)-related multi- long non-coding RNA (lncRNA) prognostic signature in pancreatic adenocarcinoma. Bioengineered 2021, 12, 2432–2448. [Google Scholar] [CrossRef] [PubMed]
- Maden, B.E. Locations of methyl groups in 28 S rRNA of Xenopus laevis and man. Clustering in the conserved core of molecule. J. Mol. Biol. 1988, 201, 289–314. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Chen, P.J.; Miao, Z.; Liu, D.R. Programmable m(6)A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat. Biotechnol. 2020, 38, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef]
- Zhong, X.; Yu, J.; Frazier, K.; Weng, X.; Li, Y.; Cham, C.M.; Dolan, K.; Zhu, X.; Hubert, N.; Tao, Y.; et al. Circadian Clock Regulation of Hepatic Lipid Metabolism by Modulation of m(6)A mRNA Methylation. Cell Rep. 2018, 25, 1816–1828.e1814. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, M.; Xie, D.; Huang, Z.; Zhang, L.; Yang, Y.; Ma, D.; Li, W.; Zhou, Q.; Yang, Y.G.; et al. METTL3-mediated N(6)-methyladenosine mRNA modification enhances long-term memory consolidation. Cell Res. 2018, 28, 1050–1061. [Google Scholar] [CrossRef]
- Patil, D.P.; Chen, C.K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef]
- Zhou, J.; Wan, J.; Gao, X.; Zhang, X.; Jaffrey, S.R.; Qian, S.B. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature 2015, 526, 591–594. [Google Scholar] [CrossRef]
- Xu, K.; Yang, Y.; Feng, G.H.; Sun, B.F.; Chen, J.Q.; Li, Y.F.; Chen, Y.S.; Zhang, X.X.; Wang, C.X.; Jiang, L.Y.; et al. Mettl3-mediated m(6)A regulates spermatogonial differentiation and meiosis initiation. Cell Res. 2017, 27, 1100–1114. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Liu, J.; Chen, C.; Dong, L.; Liu, Y.; Chang, R.; Huang, X.; Liu, Y.; Wang, J.; Dougherty, U.; et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature 2019, 566, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Laurent, B.; Hsu, C.H.; Nachtergaele, S.; Lu, Z.; Sheng, W.; Xu, C.; Chen, H.; Ouyang, J.; Wang, S.; et al. RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature 2017, 543, 573–576. [Google Scholar] [CrossRef]
- Hong, K. Emerging function of N6-methyladenosine in cancer. Oncol. Lett. 2018, 16, 5519–5524. [Google Scholar] [CrossRef]
- Liu, J.; Li, K.; Cai, J.; Zhang, M.; Zhang, X.; Xiong, X.; Meng, H.; Xu, X.; Huang, Z.; Peng, J.; et al. Landscape and Regulation of m(6)A and m(6)Am Methylome across Human and Mouse Tissues. Mol. Cell 2020, 77, 426–440.e426. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Lu, Z.; Wang, X.; Fu, Y.; Luo, G.Z.; Liu, N.; Han, D.; Dominissini, D.; Dai, Q.; Pan, T.; et al. High-resolution N(6)-methyladenosine (m(6) A) map using photo-crosslinking-assisted m(6) A sequencing. Angew. Chem. Int. Ed. Engl. 2015, 54, 1587–1590. [Google Scholar] [CrossRef]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, L.Q.; Zhao, Y.L.; Yang, C.G.; Roundtree, I.A.; Zhang, Z.; Ren, J.; Xie, W.; He, C.; Luo, G.Z. Single-base mapping of m(6)A by an antibody-independent method. Sci. Adv. 2019, 5, eaax0250. [Google Scholar] [CrossRef]
- Meyer, K.D. DART-seq: An antibody-free method for global m(6)A detection. Nat. Methods 2019, 16, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Tegowski, M.; Flamand, M.N.; Meyer, K.D. scDART-seq reveals distinct m(6)A signatures and mRNA methylation heterogeneity in single cells. Mol. Cell 2022, 82, 868–878.e810. [Google Scholar] [CrossRef]
- Chen, X.; Sun, Y.Z.; Liu, H.; Zhang, L.; Li, J.Q.; Meng, J. RNA methylation and diseases: Experimental results, databases, Web servers and computational models. Brief Bioinform. 2019, 20, 896–917. [Google Scholar] [CrossRef]
- Zhu, X.; He, J.; Zhao, S.; Tao, W.; Xiong, Y.; Bi, S. A comprehensive comparison and analysis of computational predictors for RNA N6-methyladenosine sites of Saccharomyces cerevisiae. Brief Funct. Genom. 2019, 18, 367–376. [Google Scholar] [CrossRef]
- Zhang, S.Y.; Zhang, S.W.; Zhang, T.; Fan, X.N.; Meng, J. Recent advances in functional annotation and prediction of the epitranscriptome. Comput. Struct. Biotechnol. J. 2021, 19, 3015–3026. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.; Ding, H.; Lin, H.; Chou, K.C. iRNA-Methyl: Identifying N(6)-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem. 2015, 490, 26–33. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, X.; Yu, D.J.; Jia, J.; Qiu, W.R.; Chou, K.C. pRNAm-PC: Predicting N(6)-methyladenosine sites in RNA sequences via physical-chemical properties. Anal. Biochem. 2016, 497, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.Z.; Zhang, J.J.; Gu, W.Z. RNA-MethylPred: A high-accuracy predictor to identify N6-methyladenosine in RNA. Anal. Biochem. 2016, 510, 72–75. [Google Scholar] [CrossRef]
- Zhang, M.; Sun, J.W.; Liu, Z.; Ren, M.W.; Shen, H.B.; Yu, D.J. Improving N(6)-methyladenosine site prediction with heuristic selection of nucleotide physical-chemical properties. Anal. Biochem. 2016, 508, 104–113. [Google Scholar] [CrossRef]
- Chen, W.; Ding, H.; Zhou, X.; Lin, H.; Chou, K.C. iRNA(m6A)-PseDNC: Identifying N(6)-methyladenosine sites using pseudo dinucleotide composition. Anal. Biochem. 2018, 561–562, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Su, R.; Wang, B.; Li, X.; Zou, Q.; Gao, X. Integration of deep feature representations and handcrafted features to improve the prediction of N6-methyladenosine sites. Neurocomputing 2019, 324, 3–9. [Google Scholar] [CrossRef]
- Khan, A.; Rehman, H.U.; Habib, U.; Ijaz, U. m6A-Finder: Detecting m6A methylation sites from RNA transcriptomes using physical and statistical properties based features. Comput. Biol. Chem. 2022, 97, 107640. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Rehman, H.U.; Habib, U.; Ijaz, U. Detecting N6-methyladenosine sites from RNA transcriptomes using random forest. J. Comput. Sci. 2020, 47, 101238. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Y.; Ning, Q.; Zhang, H.; Ji, J.; Yin, M. Identifying N(6)-methyladenosine sites using extreme gradient boosting system optimized by particle swarm optimizer. J. Theor. Biol. 2019, 467, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xing, P.; Zou, Q. Detecting N(6)-methyladenosine sites from RNA transcriptomes using ensemble Support Vector Machines. Sci. Rep. 2017, 7, 40242. [Google Scholar] [CrossRef]
- Wei, L.; Chen, H.; Su, R. M6APred-EL: A Sequence-Based Predictor for Identifying N6-methyladenosine Sites Using Ensemble Learning. Mol. Ther. Nucleic Acids 2018, 12, 635–644. [Google Scholar] [CrossRef]
- Chen, W.; Tran, H.; Liang, Z.; Lin, H.; Zhang, L. Identification and analysis of the N(6)-methyladenosine in the Saccharomyces cerevisiae transcriptome. Sci. Rep. 2015, 5, 13859. [Google Scholar] [CrossRef]
- Li, G.Q.; Liu, Z.; Shen, H.B.; Yu, D.J. TargetM6A: Identifying N(6)-Methyladenosine Sites From RNA Sequences via Position-Specific Nucleotide Propensities and a Support Vector Machine. IEEE Trans. Nanobioscience 2016, 15, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, O.; Wahab, A.; Chong, K.T. iMethyl-Deep: N6 Methyladenosine Identification of Yeast Genome with Automatic Feature Extraction Technique by Using Deep Learning Algorithm. Genes 2020, 11, 529. [Google Scholar] [CrossRef]
- Feng, P.; Ding, H.; Yang, H.; Chen, W.; Lin, H.; Chou, K.C. iRNA-PseColl: Identifying the Occurrence Sites of Different RNA Modifications by Incorporating Collective Effects of Nucleotides into PseKNC. Mol. Ther. Nucleic Acids 2017, 7, 155–163. [Google Scholar] [CrossRef]
- Tahir, M.; Hayat, M.; Chong, K.T. A convolution neural network-based computational model to identify the occurrence sites of various RNA modifications by fusing varied features. Chemom. Intell. Lab. Syst. 2021, 211, 104233. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.; Ding, H.; Lin, H. Identifying N (6)-methyladenosine sites in the Arabidopsis thaliana transcriptome. Mol. Genet. Genom. 2016, 291, 2225–2229. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Yan, Z.; Liu, K.; Zhang, Y.; Sun, Z. AthMethPre: A web server for the prediction and query of mRNA m(6)A sites in Arabidopsis thaliana. Mol. Biosyst. 2016, 12, 3333–3337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, J.; Li, X.; Liang, Y. M6A-GSMS: Computational identification of N(6)-methyladenosine sites with GBDT and stacking learning in multiple species. J. Biomol. Struct. Dyn. 2022, 40, 12380–12391. [Google Scholar] [CrossRef] [PubMed]
- Akbar, S.; Hayat, M. iMethyl-STTNC: Identification of N(6)-methyladenosine sites by extending the idea of SAAC into Chou’s PseAAC to formulate RNA sequences. J. Theor. Biol. 2018, 455, 205–211. [Google Scholar] [CrossRef]
- Chen, W.; Tang, H.; Lin, H. MethyRNA: A web server for identification of N(6)-methyladenosine sites. J. Biomol. Struct. Dyn. 2017, 35, 683–687. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chou, K.C. iRNA-3typeA: Identifying Three Types of Modification at RNA’s Adenosine Sites. Mol. Ther. Nucleic Acids 2018, 11, 468–474. [Google Scholar] [CrossRef]
- Xing, P.; Su, R.; Guo, F.; Wei, L. Identifying N(6)-methyladenosine sites using multi-interval nucleotide pair position specificity and support vector machine. Sci. Rep. 2017, 7, 46757. [Google Scholar] [CrossRef]
- Tahir, M.; Hayat, M.; Chong, K.T. Prediction of N6-methyladenosine sites using convolution neural network model based on distributed feature representations. Neural. Netw. 2020, 129, 385–391. [Google Scholar] [CrossRef]
- Nazari, I.; Tahir, M.; Tayara, H.; Chong, K.T. iN6-Methyl (5-step): Identifying RNA N6-methyladenosine sites using deep learning mode via Chou’s 5-step rules and Chou’s general PseKNC. Chemom. Intell. Lab. Syst. 2019, 193, 103811. [Google Scholar] [CrossRef]
- Pian, C.; Yang, Z.; Yang, Y.; Zhang, L.; Chen, Y. Identifying RNA N6-Methyladenine Sites in Three Species Based on a Markov Model. Front. Genet. 2021, 12, 650803. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.U.; Chong, K.T. A Neural Network Based Computational Model for Post-transcriptional Modification Site Identification. In Proceedings of the 2021 21st International Conference on Control, Automation and Systems (ICCAS), Jeju, Republic of Korea, 12–15 October 2021; pp. 1853–1857. [Google Scholar]
- Wang, Y.; Guo, R.; Huang, L.; Yang, S.; Hu, X.; He, K. m6AGE: A Predictor for N6-Methyladenosine Sites Identification Utilizing Sequence Characteristics and Graph Embedding-Based Geometrical Information. Front. Genet. 2021, 12, 670852. [Google Scholar] [CrossRef] [PubMed]
- Islam, N.; Park, J. bCNN-Methylpred: Feature-Based Prediction of RNA Sequence Modification Using Branch Convolutional Neural Network. Genes 2021, 12, 1155. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, L. Using Chou’s 5-steps rule to identify N(6)-methyladenine sites by ensemble learning combined with multiple feature extraction methods. J. Biomol. Struct. Dyn. 2022, 40, 796–806. [Google Scholar] [CrossRef]
- Rehman, M.U.; Hong, K.J.; Tayara, H.; to Chong, K. m6A-NeuralTool: Convolution neural tool for RNA N6-methyladenosine site identification in different species. IEEE Access 2021, 9, 17779–17786. [Google Scholar] [CrossRef]
- Qiang, X.; Chen, H.; Ye, X.; Su, R.; Wei, L. M6AMRFS: Robust Prediction of N6-Methyladenosine Sites With Sequence-Based Features in Multiple Species. Front. Genet. 2018, 9, 495. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Huang, D.; Song, B.; Chen, K.; Song, Y.; Liu, G.; Su, J.; Magalhães, J.P.; Rigden, D.J.; Meng, J. Attention-based multi-label neural networks for integrated prediction and interpretation of twelve widely occurring RNA modifications. Nat. Commun. 2021, 12, 4011. [Google Scholar] [CrossRef]
- Chen, K.; Wei, Z.; Zhang, Q.; Wu, X.; Rong, R.; Lu, Z.; Su, J.; de Magalhães, J.P.; Rigden, D.J.; Meng, J. WHISTLE: A high-accuracy map of the human N6-methyladenosine (m6A) epitranscriptome predicted using a machine learning approach. Nucleic Acids Res. 2019, 47, e41. [Google Scholar] [CrossRef]
- Zhao, Z.; Peng, H.; Lan, C.; Zheng, Y.; Fang, L.; Li, J. Imbalance learning for the prediction of N(6)-Methylation sites in mRNAs. BMC Genom. 2018, 19, 574. [Google Scholar] [CrossRef]
- Körtel, N.; Rücklé, C.; Zhou, Y.; Busch, A.; Hoch-Kraft, P.; Sutandy, F.X.R.; Haase, J.; Pradhan, M.; Musheev, M.; Ostareck, D.; et al. Deep and accurate detection of m6A RNA modifications using miCLIP2 and m6Aboost machine learning. Nucleic Acids Res. 2021, 49, e92. [Google Scholar] [CrossRef]
- Xiang, S.; Liu, K.; Yan, Z.; Zhang, Y.; Sun, Z. RNAMethPre: A Web Server for the Prediction and Query of mRNA m6A Sites. PLoS ONE 2016, 11, e0162707. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zeng, P.; Li, Y.H.; Zhang, Z.; Cui, Q. SRAMP: Prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016, 44, e91. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xie, J.; Xu, S. M6A-BiNP: Predicting N(6)-methyladenosine sites based on bidirectional position-specific propensities of polynucleotides and pointwise joint mutual information. RNA Biol. 2021, 18, 2498–2512. [Google Scholar] [CrossRef]
- Song, B.; Chen, K.; Tang, Y.; Wei, Z.; Su, J.; de Magalhães, J.P.; Rigden, D.J.; Meng, J. ConsRM: Collection and large-scale prediction of the evolutionarily conserved RNA methylation sites, with implications for the functional epitranscriptome. Brief Bioinform. 2021, 22, bbab088. [Google Scholar] [CrossRef]
- Zhang, Y.; Hamada, M. DeepM6ASeq: Prediction and characterization of m6A-containing sequences using deep learning. BMC Bioinform. 2018, 19, 524. [Google Scholar] [CrossRef]
- Zou, Q.; Xing, P.; Wei, L.; Liu, B. Gene2vec: Gene subsequence embedding for prediction of mammalian N(6)-methyladenosine sites from mRNA. Rna 2019, 25, 205–218. [Google Scholar] [CrossRef]
- Xiong, Y.; He, X.; Zhao, D.; Tian, T.; Hong, L.; Jiang, T.; Zeng, J. Modeling multi-species RNA modification through multi-task curriculum learning. Nucleic Acids Res. 2021, 49, 3719–3734. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zou, Q.; Li, J. DeepM6ASeq-EL: Prediction of human N6-methyladenosine (m 6A) sites with LSTM and ensemble learning. Front. Comput. Sci. 2022, 16, 162302. [Google Scholar] [CrossRef]
- Dao, F.Y.; Lv, H.; Yang, Y.H.; Zulfiqar, H.; Gao, H.; Lin, H. Computational identification of N6-methyladenosine sites in multiple tissues of mammals. Comput. Struct. Biotechnol. J. 2020, 18, 1084–1091. [Google Scholar] [CrossRef]
- Liu, K.; Cao, L.; Du, P.; Chen, W. im6A-TS-CNN: Identifying the N(6)-Methyladenine Site in Multiple Tissues by Using the Convolutional Neural Network. Mol. Ther. Nucleic Acids 2020, 21, 1044–1049. [Google Scholar] [CrossRef]
- Jia, C.; Dong, J.; Wang, X.; Zhao, Q. Tissue specific prediction of N-methyladenine sites based on an ensemble of multi-input hybrid neural network. Biocell 2022, 46, 1105. [Google Scholar] [CrossRef]
- Zhang, L.; Qin, X.; Liu, M.; Xu, Z.; Liu, G. DNN-m6A: A Cross-Species Method for Identifying RNA N6-Methyladenosine Sites Based on Deep Neural Network with Multi-Information Fusion. Genes 2021, 12, 354. [Google Scholar] [CrossRef]
- Abbas, Z.; Tayara, H.; Zou, Q.; Chong, K.T. TS-m6A-DL: Tissue-specific identification of N6-methyladenosine sites using a universal deep learning model. Comput. Struct. Biotechnol. J. 2021, 19, 4619–4625. [Google Scholar] [CrossRef]
- Luo, X.; Li, H.; Liang, J.; Zhao, Q.; Xie, Y.; Ren, J.; Zuo, Z. RMVar: An updated database of functional variants involved in RNA modifications. Nucleic Acids Res. 2021, 49, D1405–D1412. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.J.; Li, J.H.; Liu, S.; Wu, J.; Zhou, H.; Qu, L.H.; Yang, J.H. RMBase: A resource for decoding the landscape of RNA modifications from high-throughput sequencing data. Nucleic Acids Res. 2016, 44, D259–D265. [Google Scholar] [CrossRef]
- Xuan, J.J.; Sun, W.J.; Lin, P.H.; Zhou, K.R.; Liu, S.; Zheng, L.L.; Qu, L.H.; Yang, J.H. RMBase v2.0: Deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018, 46, D327–D334. [Google Scholar] [CrossRef]
- Liu, H.; Flores, M.A.; Meng, J.; Zhang, L.; Zhao, X.; Rao, M.K.; Chen, Y.; Huang, Y. MeT-DB: A database of transcriptome methylation in mammalian cells. Nucleic Acids Res. 2015, 43, D197–D203. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, H.; Wei, Z.; Zhang, S.; Hua, G.; Zhang, S.-W.; Zhang, L.; Gao, S.-J.; Meng, J.; Chen, X. MeT-DB V2. 0: Elucidating context-specific functions of N 6-methyl-adenosine methyltranscriptome. Nucleic Acids Res. 2018, 46, D281–D287. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Huang, X.; Jiang, J. Deepm6A-MT: A deep learning-based method for identifying RNA N6-methyladenosine sites in multiple tissues. Methods 2024, 226, 1–8. [Google Scholar] [CrossRef]
- Wang, H.; Zeng, W.; Huang, X.; Liu, Z.; Sun, Y.; Zhang, L. MTTLm(6)A: A multi-task transfer learning approach for base-resolution mRNA m(6)A site prediction based on an improved transformer. Math. Biosci. Eng. 2024, 21, 272–299. [Google Scholar] [CrossRef]
- Tu, G.; Wang, X.; Xia, R.; Song, B. m6A-TCPred: A web server to predict tissue-conserved human m(6)A sites using machine learning approach. BMC Bioinform. 2024, 25, 127. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Chen, K.; Song, B.; Ma, J.; Wu, X.; Xu, Q.; Wei, Z.; Su, J.; Liu, G.; Rong, R.; et al. m6A-Atlas: A comprehensive knowledgebase for unraveling the N6-methyladenosine (m6A) epitranscriptome. Nucleic Acids Res. 2021, 49, D134–D143. [Google Scholar] [CrossRef]
- Dierks, D.; Garcia-Campos, M.A.; Uzonyi, A.; Safra, M.; Edelheit, S.; Rossi, A.; Sideri, T.; Varier, R.A.; Brandis, A.; Stelzer, Y.; et al. Multiplexed profiling facilitates robust m6A quantification at site, gene and sample resolution. Nat. Methods 2021, 18, 1060–1067. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, T.; Chen, H.X.; Xie, Y.Y.; Chen, L.Q.; Zhao, Y.L.; Liu, B.D.; Jin, L.; Zhang, W.; Liu, C.; et al. Systematic calibration of epitranscriptomic maps using a synthetic modification-free RNA library. Nat. Methods 2021, 18, 1213–1222. [Google Scholar] [CrossRef]
- Hu, L.; Liu, S.; Peng, Y.; Ge, R.; Su, R.; Senevirathne, C.; Harada, B.T.; Dai, Q.; Wei, J.; Zhang, L.; et al. m(6)A RNA modifications are measured at single-base resolution across the mammalian transcriptome. Nat. Biotechnol 2022, 40, 1210–1219. [Google Scholar] [CrossRef]
- Cheng, W.; Liu, F.; Ren, Z.; Chen, W.; Chen, Y.; Liu, T.; Ma, Y.; Cao, N.; Wang, J. Parallel functional assessment of m(6)A sites in human endodermal differentiation with base editor screens. Nat. Commun. 2022, 13, 478. [Google Scholar] [CrossRef]
- Liu, B.; Liu, F.; Wang, X.; Chen, J.; Fang, L.; Chou, K.C. Pse-in-One: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015, 43, W65–W71. [Google Scholar] [CrossRef]
- Chen, W.; Lei, T.Y.; Jin, D.C.; Lin, H.; Chou, K.C. PseKNC: A flexible web server for generating pseudo K-tuple nucleotide composition. Anal. Biochem. 2014, 456, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, X.; Brooker, J.; Lin, H.; Zhang, L.; Chou, K.C. PseKNC-General: A cross-platform package for generating various modes of pseudo nucleotide compositions. Bioinformatics 2015, 31, 119–120. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, P.; Li, F.; Marquez-Lago, T.T.; Leier, A.; Revote, J.; Zhu, Y.; Powell, D.R.; Akutsu, T.; Webb, G.I.; et al. iLearn: An integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data. Brief Bioinform. 2020, 21, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, P.; Li, C.; Li, F.; Xiang, D.; Chen, Y.Z.; Akutsu, T.; Daly, R.J.; Webb, G.I.; Zhao, Q.; et al. iLearnPlus: A comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res. 2021, 49, e60. [Google Scholar] [CrossRef]
- Chen, W.; Yang, H.; Feng, P.; Ding, H.; Lin, H. iDNA4mC: Identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 2017, 33, 3518–3523. [Google Scholar] [CrossRef]
- Xu, Z.C.; Feng, P.M.; Yang, H.; Qiu, W.R.; Chen, W.; Lin, H. iRNAD: A computational tool for identifying D modification sites in RNA sequence. Bioinformatics 2019, 35, 4922–4929. [Google Scholar] [CrossRef]
- Chen, W.; Song, X.; Lv, H.; Lin, H. iRNA-m2G: Identifying N(2)-methylguanosine Sites Based on Sequence-Derived Information. Mol. Ther. Nucleic Acids 2019, 18, 253–258. [Google Scholar] [CrossRef]
- Yang, H.; Lv, H.; Ding, H.; Chen, W.; Lin, H. iRNA-2OM: A Sequence-Based Predictor for Identifying 2′-O-Methylation Sites in Homo sapiens. J. Comput. Biol. 2018, 25, 1266–1277. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol 2011, 6, 26. [Google Scholar] [CrossRef]
- Long, H.; Liao, B.; Xu, X.; Yang, J. A Hybrid Deep Learning Model for Predicting Protein Hydroxylation Sites. Int. J. Mol. Sci. 2018, 19, 2817. [Google Scholar] [CrossRef]
- Wang, D.; Liang, Y.; Xu, D. Capsule network for protein post-translational modification site prediction. Bioinformatics 2019, 35, 2386–2394. [Google Scholar] [CrossRef]
- Rao, R.S.P.; Zhang, N.; Xu, D.; Møller, I.M. CarbonylDB: A curated data-resource of protein carbonylation sites. Bioinformatics 2018, 34, 2518–2520. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Wang, J.; Deng, L. SDN2GO: An Integrated Deep Learning Model for Protein Function Prediction. Front. Bioeng. Biotechnol. 2020, 8, 391. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, J.; Cai, Y.; Deng, L. DeepMiR2GO: Inferring Functions of Human MicroRNAs Using a Deep Multi-Label Classification Model. Int. J. Mol. Sci. 2019, 20, 6046. [Google Scholar] [CrossRef] [PubMed]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. Imagenet classification with deep convolutional neural networks. Adv. Neural Inf. Process. Syst. 2012, 25. [Google Scholar] [CrossRef]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef]
- Ståhl, N.; Falkman, G.; Karlsson, A.; Mathiason, G.; Boström, J. Deep Reinforcement Learning for Multiparameter Optimization in de novo Drug Design. J. Chem. Inf. Model. 2019, 59, 3166–3176. [Google Scholar] [CrossRef]
- Deng, L.; Cai, Y.; Zhang, W.; Yang, W.; Gao, B.; Liu, H. Pathway-Guided Deep Neural Network toward Interpretable and Predictive Modeling of Drug Sensitivity. J. Chem. Inf. Model. 2020, 60, 4497–4505. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Fast, E.; Liu, C.L.; Muftuoglu, Y.; Sworder, B.J.; Diehn, M.; Levy, R.; et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat. Biotechnol. 2019, 37, 1332–1343. [Google Scholar] [CrossRef]
- Racle, J.; Michaux, J.; Rockinger, G.A.; Arnaud, M.; Bobisse, S.; Chong, C.; Guillaume, P.; Coukos, G.; Harari, A.; Jandus, C.; et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat. Biotechnol. 2019, 37, 1283–1286. [Google Scholar] [CrossRef]
- Xu, Z.; Luo, M.; Lin, W.; Xue, G.; Wang, P.; Jin, X.; Xu, C.; Zhou, W.; Cai, Y.; Yang, W.; et al. DLpTCR: An ensemble deep learning framework for predicting immunogenic peptide recognized by T cell receptor. Brief Bioinform. 2021, 22, bbab335. [Google Scholar] [CrossRef]
- Jiang, Q.; Wang, G.; Jin, S.; Li, Y.; Wang, Y. Predicting human microRNA-disease associations based on support vector machine. Int. J. Data Min. Bioinform. 2013, 8, 282–293. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, Z.C.; Su, W.; Yang, Y.H.; Lv, H.; Yang, H.; Lin, H. iCarPS: A computational tool for identifying protein carbonylation sites by novel encoded features. Bioinformatics 2021, 37, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xiao, X.; Xu, Z.C. iPromoter-5mC: A Novel Fusion Decision Predictor for the Identification of 5-Methylcytosine Sites in Genome-Wide DNA Promoters. Front. Cell Dev. Biol. 2020, 8, 614. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Xu, Z.C.; Qiu, W.R.; Wang, P.; Ge, H.T.; Chou, K.C. iPSW(2L)-PseKNC: A two-layer predictor for identifying promoters and their strength by hybrid features via pseudo K-tuple nucleotide composition. Genomics 2019, 111, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.C.; Wang, P.; Qiu, W.R.; Xiao, X. iSS-PC: Identifying Splicing Sites via Physical-Chemical Properties Using Deep Sparse Auto-Encoder. Sci. Rep. 2017, 7, 8222. [Google Scholar] [CrossRef]
- Xu, Z.-C.; Qiu, W.-R.; Xiao, X. iRSpotH-TNCPseAAC: Identifying recombination spots in human by using pseudo trinucleotide composition with an ensemble of support vector machine classifiers. Lett. Org. Chem. 2017, 14, 703–713. [Google Scholar] [CrossRef]
- Xu, Z.-C.; Jiang, S.-Y.; Qiu, W.-R.; Liu, Y.-C.; Xiao, X. iDHSs-PseTNC: Identifying DNase I hypersensitive sites with pseuo trinucleotide component by deep sparse auto-encoder. Lett. Org. Chem. 2017, 14, 655–664. [Google Scholar] [CrossRef]
- Huang, Y.; Zhou, D.; Wang, Y.; Zhang, X.; Su, M.; Wang, C.; Sun, Z.; Jiang, Q.; Sun, B.; Zhang, Y. Prediction of transcription factors binding events based on epigenetic modifications in different human cells. Epigenomics 2020, 12, 1443–1456. [Google Scholar] [CrossRef]
- Su, Z.D.; Huang, Y.; Zhang, Z.Y.; Zhao, Y.W.; Wang, D.; Chen, W.; Chou, K.C.; Lin, H. iLoc-lncRNA: Predict the subcellular location of lncRNAs by incorporating octamer composition into general PseKNC. Bioinformatics 2018, 34, 4196–4204. [Google Scholar] [CrossRef]





| Tool | Species | Experimental Method | Sequence Length (nt) | Features a | Algorithm d | Evaluation Strategy | Year | Webserver b | Data Size c |
|---|---|---|---|---|---|---|---|---|---|
| iRNA-Methyl [26] | SC | m6A-Seq | 51 | PseDNC | SVM | Jackknife | 2015 | http://lin-group.cn/server/iRNA-Methyl (accessed on 12 September 2022) | Smet1307 |
| m6Apred [37] | SC | m6A-Seq | 21 | CPD | SVM | Jackknife independent test | 2015 | http://lin-group.cn/server/m6Apred (accessed on 12 September 2022) | Smet1307sub |
| pRNAm-PC [27] | SC | m6A-Seq | 51 | PseDNC, AC, CC | SVM | Jackknife | 2016 | Decommissioned | Smet1307 |
| RNA-MethylPred [28] | SC | m6A-Seq | 51 | DNC, KNN scores | SVM | Jackknife | 2016 | No | Smet1307 |
| M6A-HPCS [29] | SC | m6A-Seq | 51 | HPCS | SVM | 10-fold CV | 2016 | Decommissioned | Smet1307 |
| TargetM6A [38] | SC | m6A-Seq | 21 | PSNP, PSDP, NC | SVM | Jackknife, independent test | 2016 | Decommissioned | Smet1307sub |
| RAM-ESVM [35] | SC | m6A-Seq | 51 | PseDNC | Ensemble SVM | 10-fold CV | 2017 | Decommissioned | Smet1307 |
| iRNA(m6A)-PseDNC [30] | SC | m6A-Seq | 51 | PseDNC | SVM | 10-fold CV | 2018 | http://lin-group.cn/server/iRNA(m6A)-PseDNC.php (accessed on 12 September 2022) | Smet1307 |
| M6APred-EL [36] | SC | m6A-Seq | 51 | PS(k-mer)NP, RFHC-GACs, AC, CC | Ensemble (SVM) | 10-fold CV | 2018 | Decommissioned | Smet1307 |
| DeepM6APred [31] | SC | m6A-Seq | 51 | Deep features, NPPS | SVM | 10-fold CV | 2018 | Decommissioned | Smet1307 |
| M6A-PXGB [34] | SC | m6A-Seq | 51 | PSNP, PSDP, NC | XGBoost | 10-fold CV | 2019 | No | Smet1307 |
| m6A-pred [33] | SC | m6A-Seq | 51 | CPD, DNC, TNC | RF | 10-fold CV | 2020 | No | Smet1307 |
| m6A-Finder [32] | SC | m6A-Seq | 51 | AC, NC | SVM | Jackknife | 2022 | No | Smet1307 |
| iMethyl-Deep [39] | SC | m6A-Seq, m6A-CLIP, miCLIP | 51 | One-hot | CNN | 10-fold CV, independent test | 2020 | https://github.com/abdul-bioinfo/iMethyl-deep (accessed on 12 September 2022) | Smet1307; Smet3270 |
| iMethyl-STTNC [45] | SC, HSA | m6A-Seq | 51-41 | PseDNC, PseTNC, STNC, STTNC | SVM | 10-fold CV | 2018 | No | Smet1307; Hmet1130 |
| iRNA-PseColl [40] | HSA | m6A-Seq | 41 | CPD | SVM | Jackknife | 2017 | http://lin-group.cn/server/iRNA-PseColl.html (accessed on 12 September 2022) | Hmet1130 |
| iRNA-Mod-CNN [41] | HSA | m6A-Seq | 41 | K-Gram | CNN | 5-fold CV | 2021 | No | Hmet1130 |
| HMpre [60] | HSA | miCLIP | 51 | SLRF, FREI, SNP | XGBoost | Independent test | 2018 | No | 26,512:271,214 |
| WHISTLE [59] | HSA | m6A-CLIP, miCLIP | unknown | CPD, Genomic features | SVM | 5-fold CV, independent test | 2019 | No | 37,899 (1:10) |
| m6Aboost [61] | HSA | miCLIP | 21 | Experimental and sequence features | AdaBoost | 5-fold CV, independent test | 2021 | No | 11,701:42,090 |
| MultiRM [58] | HSA | m6A-CLIP, miCLIP | 51 | One-hot | CNN+BiLSTM | Independent test | 2021 | Decommissioned | 65,178 (1:1) |
| DeepM6ASeq-EL [69] | HSA | m6A-CLIP, miCLIP | unknown | One-hot, CPD, Word2vec | Ensemble (CNN+LSTM) | Independent test | 2022 | No | 37,899 (1:10) |
| ConsRM [65] | HSA | m6A-CLIP, m6A-REF-Seq, miCLIP | 11 | CPD, Genomic features | SVM | 5-fold CV, independent test | 2021 | http://180.208.58.19/conservation/ (accessed on 12 September 2022) | 177,998 (1:1) |
| MethyRNA [46] | HSA, MMU | m6A-Seq, MeRIP-Seq | 41 | CPD | SVM | Jackknife | 2016 | http://lin-group.cn/server/MethyRNA (accessed on 12 September 2022) | Hmet1130 Mmet725 |
| SRAMP [63] | HSA, MMU | miCLIP | W | One-hot, SPE, KNN scores, PSSP | RF | 5-fold CV, independent test | 2016 | http://www.cuilab.cn/sramp/ (accessed on 12 September 2022) | 57,433, mRNA; 68,083, full transcripts (1:10) |
| RNAMethPre [62] | HSA, MMU | MiCLIP-seq, m6A-CLIP | 101 | One-hot, NC, SLS | SVM | 5-fold CV, independent test | 2016 | Decommissioned | HSA: 29,547, mRNA; 31,728, full transcripts (1:1) MMU: 22,740, mRNA; 24,705, full transcripts (1:1) |
| iRNA-3typeA [47] | HSA, MMU | m6A-Seq, MeRIP-Seq | 41 | CPD | SVM | Jackknife | 2018 | http://lin-group.cn/server/iRNA-3typeA.php (accessed on 12 September 2022) | Hmet1130; Mmet725 |
| Gene2vec [67] | HSA, MMU | MiCLIP-seq, m6A-CLIP | 1001 | One-hot, NMSE, word embedding | CNN+ensemble | 10-fold CV, independent test | 2019 | Decommissioned | 495,572 (1:10) |
| M6ATH [42] | At | m6A-seq | 25 | CPD | SVM | Jackknife | 2016 | http://lin-group.cn/server/M6ATH (accessed on 12 September 2022) | Amet394 |
| AthMethPre [43] | At | m6A-seq, MeRIP-seq | 41 | One-hot, PIkmer | SVM | 5-fold CV, independent test | 2016 | Decommissioned | 5081 (1:1) |
| RAM-NPPS [48] | SC, HSA, At | m6A-Seq, PA-m6A-seq | 51 | NPPS | SVM | 10-fold CV | 2017 | Decommissioned | Smet1307; Hmet8366; Amet394 |
| m6A-word2vec [49] | SC, HSA, At | m6A-Seq, PA-m6A-seq | 51 | Word embedding | CNN | 10-fold CV | 2020 | No | Smet1307; Hmet1130; Amet394 |
| m6AGE [53] | SC, HSA, At | m6A-Seq, PA-m6A-seq | 21-41-25-101 | Graph embedding, sequence-derived features (CTD, PseKNC, NPS, NPPS, CPD, EIIP, BPB) | CatBoost | 5-fold CV | 2021 | https://github.com/bokunoBike/m6AGE (accessed on 12 September 2022) | Smet1307; Hmet1130; Amet394; Amet2518 |
| DeepM6ASeq [66] | HSA, Mouse, ZF | miCLIP-Seq | 101 | One-hot | CNN+BiLSTM | 5-fold CV, independent test | 2018 | https://github.com/rreybeyb/DeepM6ASeq (accessed on 12 September 2022) | HSA: 49,050; Mouse: 37,716; ZF: 22,108 (1:1) |
| iN6-Methyl (5-step) [50] | SC, HSA, MMU | m6A-seq, MeRIP-seq | 51-41-41 | Word embedding | CNN | 10-fold CV | 2019 | decommissioned | Smet1307; Hmet1130; Mmet725 |
| Chong et al. [52] | SC, HSA, MMU | m6A-seq, MeRIP-seq | 51-41-41 | k-mer | CNN | 10-fold CV | 2021 | No | Smet1307; Hmet1130; Mmet725 |
| MM-m6APred [51] | SC, HSA, MMU | m6A-seq, MeRIP-seq | 51-41-41 | Probability matrix | Second-order Markov | 10-fold CV | 2021 | decommissioned | Smet1307; Hmet1130; Mmet725 |
| M6AMRFS [57] | SC, HSA, MMU, At | m6A-seq, MeRIP-seq | 51-41-41-101 | One-hot; LPSDF | XGBoost | 5-fold CV, independent test | 2018 | decommissioned | Smet1307; Hmet1130; Mmet725; Amet2100 |
| bCNN-Methylpred [54] | SC, HSA, MMU, At | m6A-seq, MeRIP-seq, miCLIP-seq | 51-41-41-101 | Circular encoding, one-hot, NCP | CNN | 10-fold CV | 2021 | https://github.com/Naeem-jbnu/RNA_Modification_Sites (accessed on 12 September 2022) | Smet1307; Hmet1130; Mmet725; Amet1000 |
| m6A-NeuralTool [56] | SC, HSA, MMU, At | m6A-seq, MeRIP-seq | 51-41-41-101 | One-hot | Ensemble (CNN, SVM, NB) | 10-fold CV, independent test | 2021 | http://nsclbio.jbnu.ac.kr/tools/m6A-NeuralTool/ (accessed on 12 September 2022) | Smet3270; Hmet1130; Mmet725; Amet2100 |
| TL-Methy [55] | SC, HSA, MMU, Rice | m6A-seq, MeRIP-seq | 51-41-41-41 | NAC, DNC, TNC, PSTNP, BPB, one-hot, CPD | Ensemble (SVM, KNN, LR, DA) | 10-fold CV | 2022 | https://github.com/LDWang-dlmu/N6-methyladenine (accessed on 12 September 2022) | Smet1307; Hmet1130; Mmet725; Rmet880 |
| M6A-BiNP [64] | SC, HSA, MMU, Rat, At | m6A-seq, MeRIP-seq, miCLIP-seq, m6A-REF-seq | 51-41-41-41-25 | PSP-PMI, PSP-PJMI | SVM | 10-fold CV | 2021 | https://github.com/Mingzhao2017/M6A-BiNP (accessed on 12 September 2022) | Smet1307; Hmet1130; Mmet725; Amet394; Species-/tissue-specific datasets; Human51 (1:1) |
| M6A-GSMS [44] | SC, HSA, MMU, At, DM | m6A-seq, MeRIP-seq | 51-41-41-101-41 | NMBAC, PC-PseDNC-General, PseDPC, one-hot, K-mer | Ensemble (RF, ET, SVM, LGBM, Bagging, Adaboost) | 10-fold CV | 2021 | https://github.com/Wang-Jinyue/M6A-GSMS (accessed on 12 September 2022) | Smet1307; HSA: 5100; MMU: 725; At: 2100; DM: 300 (1:1) |
| MASS [68] | HSA, MMU, Chim, Rhesus, Pig, Rat, ZF | m6A-Seq, MeRIP-Seq, m6A-CLIP, miCLIP-seq | 101 | One-hot, phylogenetic tree | CNN+BiLSTM | 5-fold CV | 2021 | https://github.com/mlcb-thu/MASS (accessed on 12 September 2022) | HSA: 305,644; MMU: 317,702; Chim: 26,248; Rhesus: 27,059; Pig: 81,501; Rat: 41,735; ZF: 19,834 (1:10) |
| iRNA-m6A [70] | HSA, MMU, Rat | m6A-REF-seq | 41 | AC, CC, CPD, one-hot | SVM | 5-fold CV, independent test | 2020 | http://lin-group.cn/server/iRNA-m6A/ (accessed on 12 September 2022) | TSdata |
| im6A-TS-CNN [71] | HSA, MMU, Rat | m6A-REF-seq | 41 | One-hot | CNN | 5-fold CV, independent test | 2020 | No | TSdata |
| Jia et al. [72] | HSA, MMU, Rat | m6A-REF-seq | 41 | One-hot, sequence feature, KNFR | Ensemble (CNN+capsule+BiGRU) | 5-fold CV, independent test | 2022 | No | TSdata |
| DNN-m6A [73] | HSA, MMU, Rat | m6A-REF-seq | 41 | One-hot, TNC, ENAC, KSNPFs, CPD, PseDNC, PSNP, PSDP | DNN | 5-fold CV, independent test | 2021 | https://github.com/GD818/DNN-m6A (accessed on 12 September 2022) | TSdata |
| TS-m6A-DL [74] | HSA, MMU, Rat | m6A-REF-seq | 41 | One-hot | CNN | 5-fold CV, independent test | 2021 | http://nsclbio.jbnu.ac.kr/tools/TS-m6A-DL/ (accessed on 12 September 2022) | TSdata |
| Deepm6A-MT [80] | HSA, MMU, Rat | m6A-REF-seq | 41 | Word embedding, one-hot, NCP, CPD | CNN+BiGRU | 5-fold CV, independent test | 2024 | http://www.biolscience.cn/Deepm6A-MT/ (accessed on 12 September 2022) | TSdata |
| MTTLm6A [81] | SC | m6A-CLIP, miCLIP-seq | 601 | One-hot | CNN | 5-fold CV, independent test | 2023 | http://47.242.23.141/MTTLm6A/index.php (accessed on 12 September 2022) | 49,338 (1:1) |
| m6A-TCPred [82] | HSA | PA-m6A-seq, miCLIP, m6A-REF-seq | Nucleotide position | NCP, EIIP | SVM | 5-fold CV, independent test | 2024 | www.rnamd.org/m6ATCPred (accessed on 12 September 2022) | 10,424:54,949 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, Z.; Yu, L.; Xu, Z.; Liu, K.; Gu, L. Comprehensive Review and Assessment of Computational Methods for Prediction of N6-Methyladenosine Sites. Biology 2024, 13, 777. https://doi.org/10.3390/biology13100777
Luo Z, Yu L, Xu Z, Liu K, Gu L. Comprehensive Review and Assessment of Computational Methods for Prediction of N6-Methyladenosine Sites. Biology. 2024; 13(10):777. https://doi.org/10.3390/biology13100777
Chicago/Turabian StyleLuo, Zhengtao, Liyi Yu, Zhaochun Xu, Kening Liu, and Lichuan Gu. 2024. "Comprehensive Review and Assessment of Computational Methods for Prediction of N6-Methyladenosine Sites" Biology 13, no. 10: 777. https://doi.org/10.3390/biology13100777
APA StyleLuo, Z., Yu, L., Xu, Z., Liu, K., & Gu, L. (2024). Comprehensive Review and Assessment of Computational Methods for Prediction of N6-Methyladenosine Sites. Biology, 13(10), 777. https://doi.org/10.3390/biology13100777