

Exploring Longitudinal Gut Microbiome towards Metabolic Functional Changes Associated in Atopic Dermatitis in Early Childhood
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Fecal Sample Collection
2.2. Clinical Data Collection
2.3. Fecal Sample Preparation
2.4. Microbial DNA Extraction and 16S rRNA Gene Sequencing
2.5. Microbiome Data Processing
2.6. Microbial Taxonomy, Functional Composition, and Integrative Meta-Omics Analysis
3. Results and Discussion
3.1. Assessment of Participant Characteristics
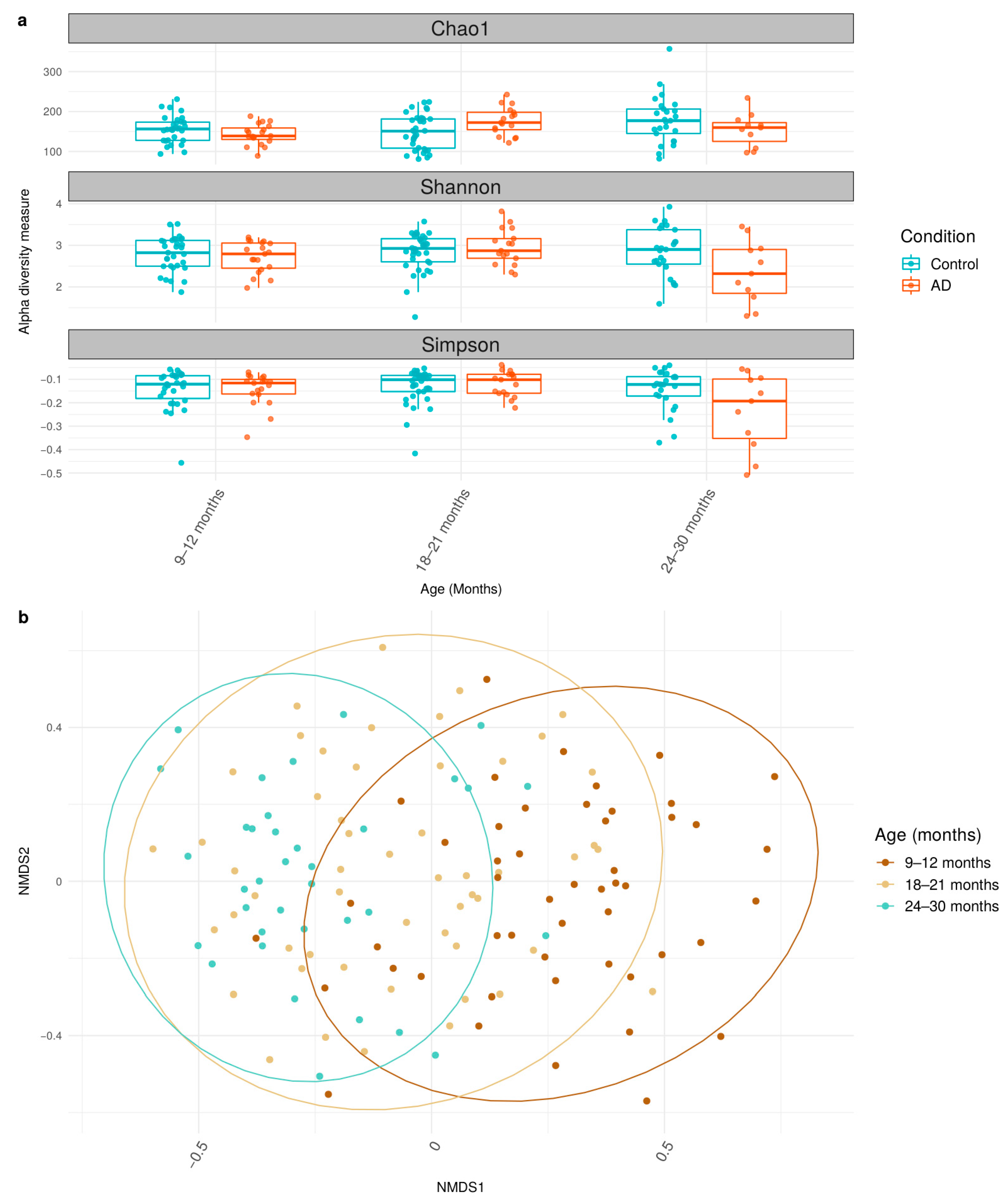
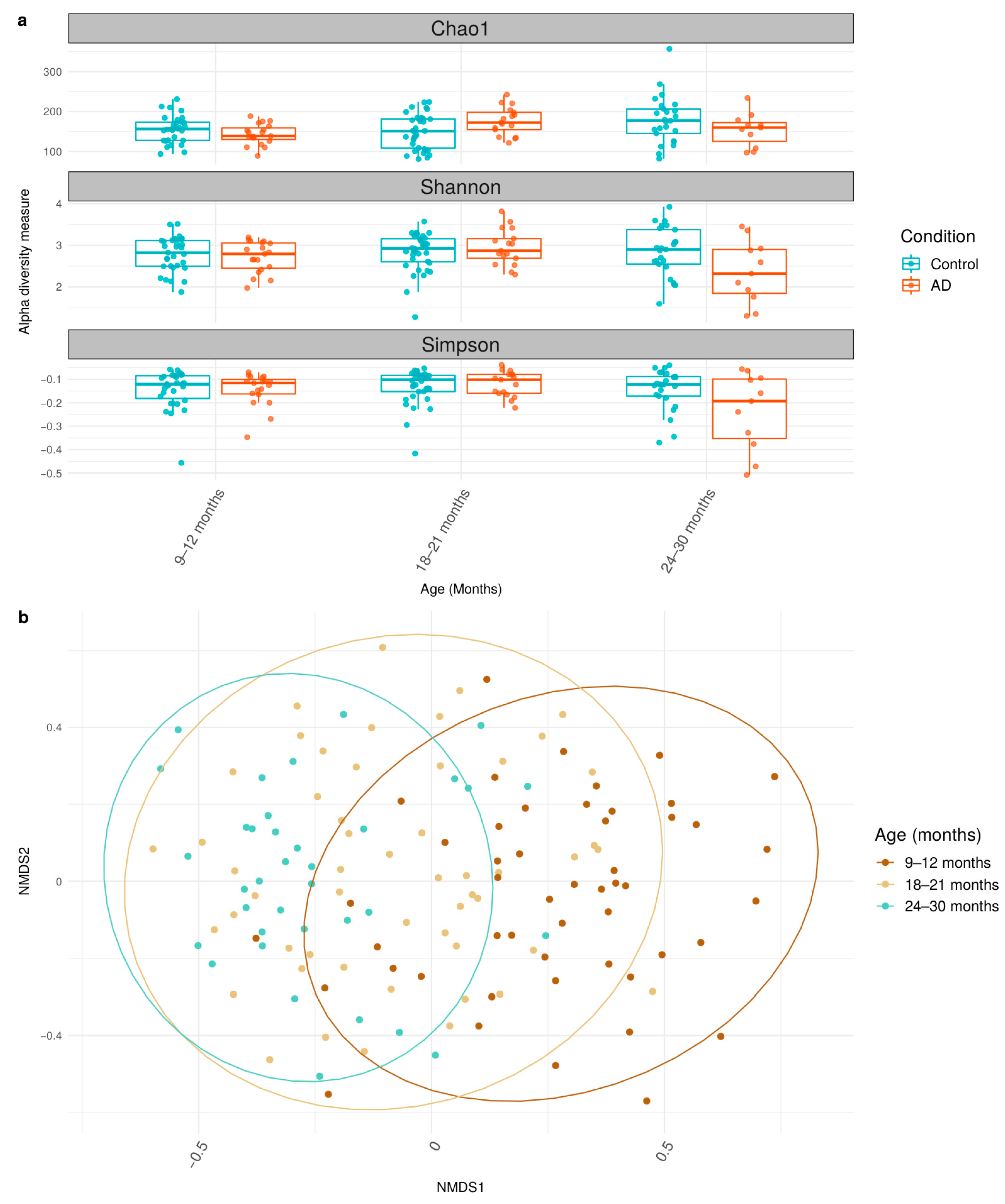
3.2. Differential Shifts in Gut Microbial Diversity Trajectories over Time between Healthy and AD Participants
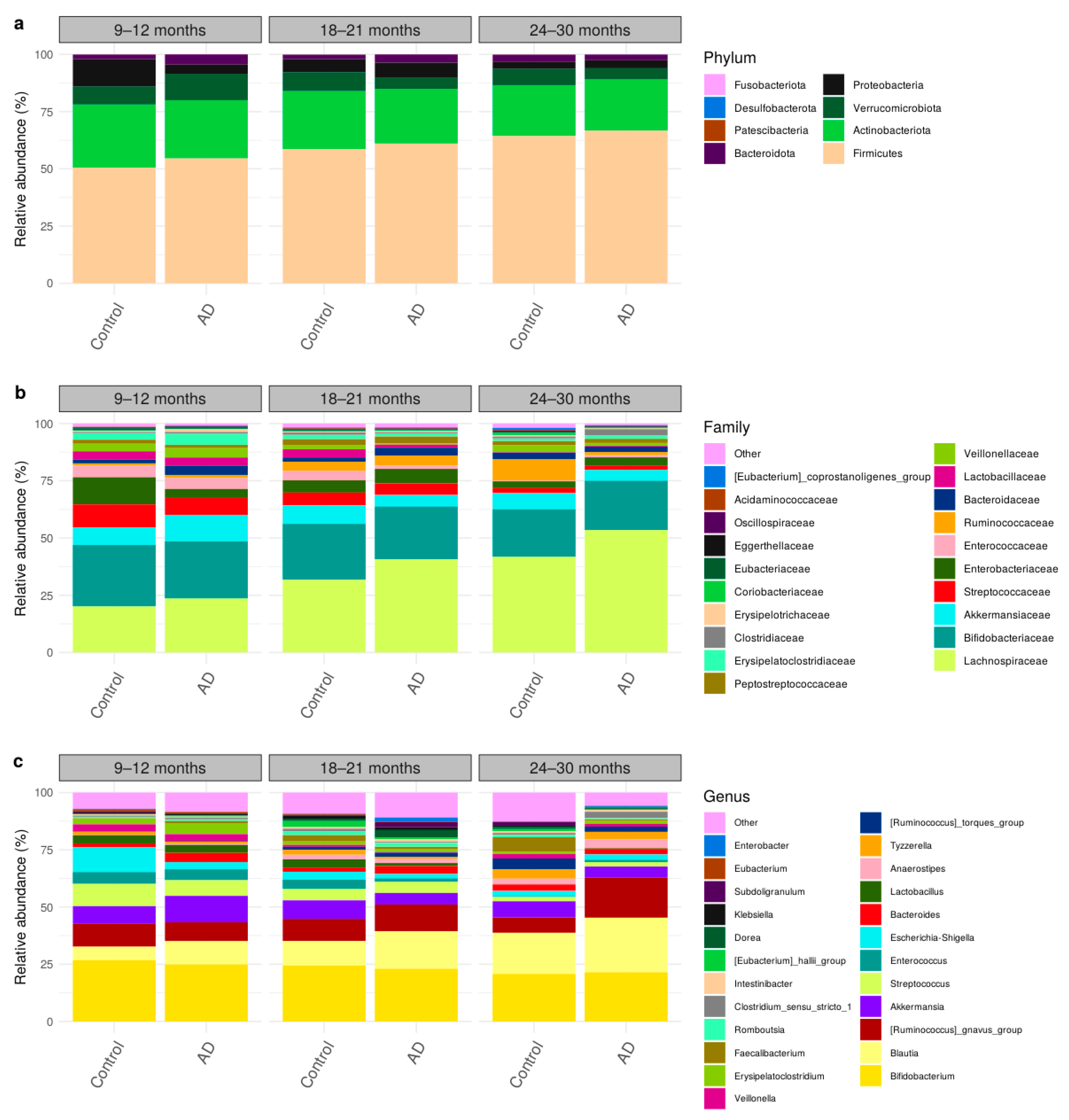
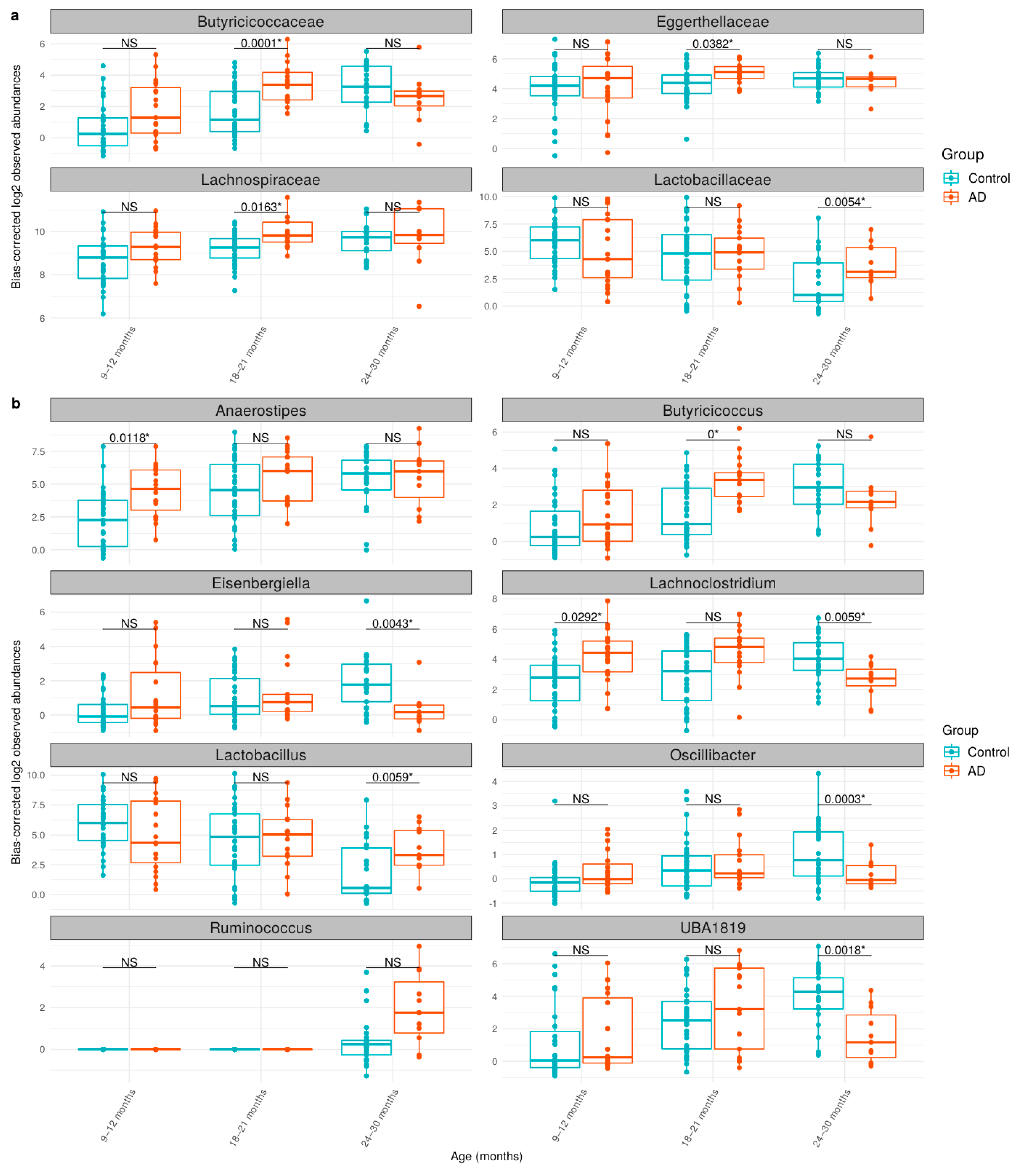
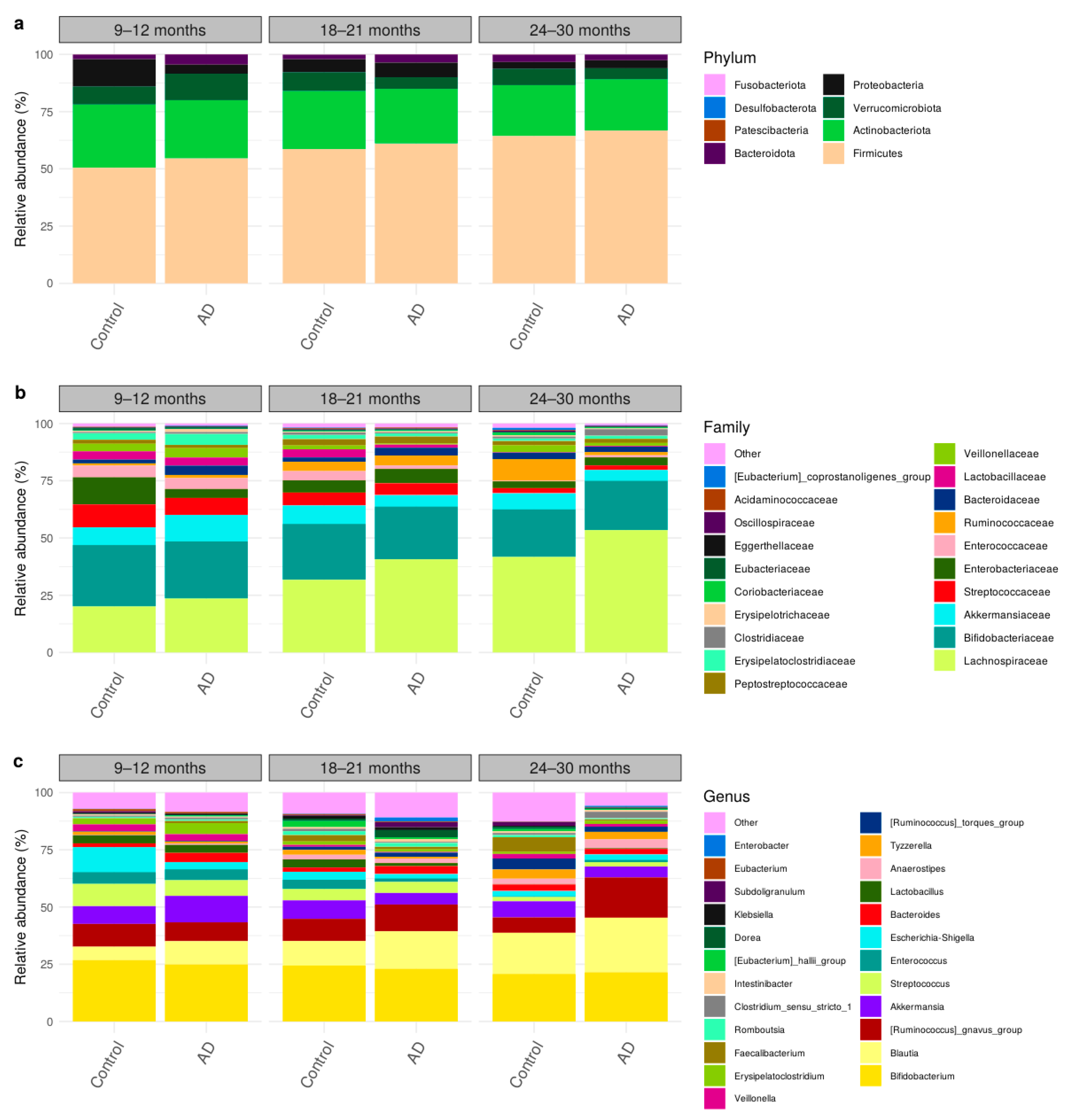
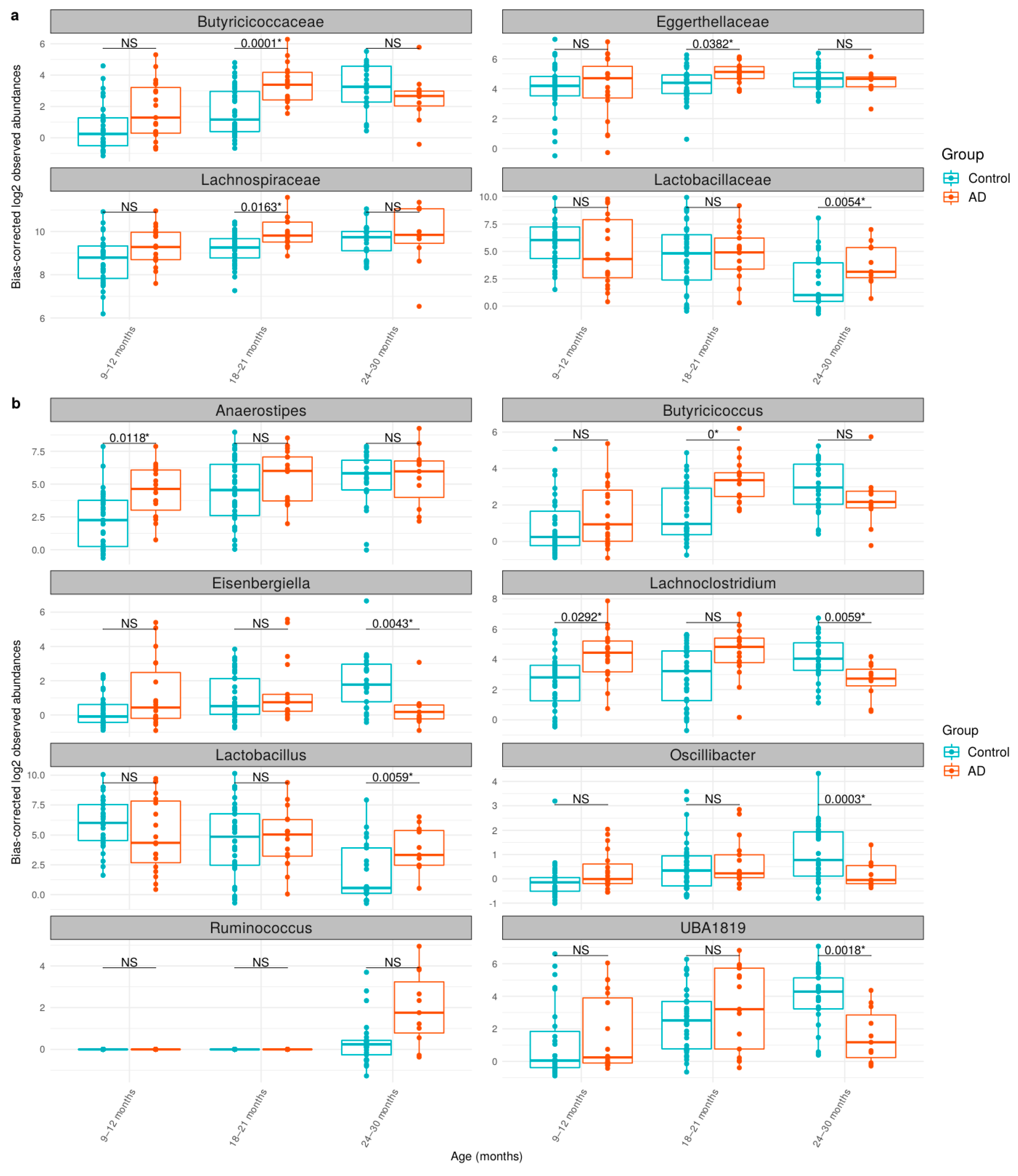
3.3. Comparison of Bacteria Abundances in Healthy and AD Participants
3.4. Metabolic Functional Compositions of Longitudinal Gut Microbiome
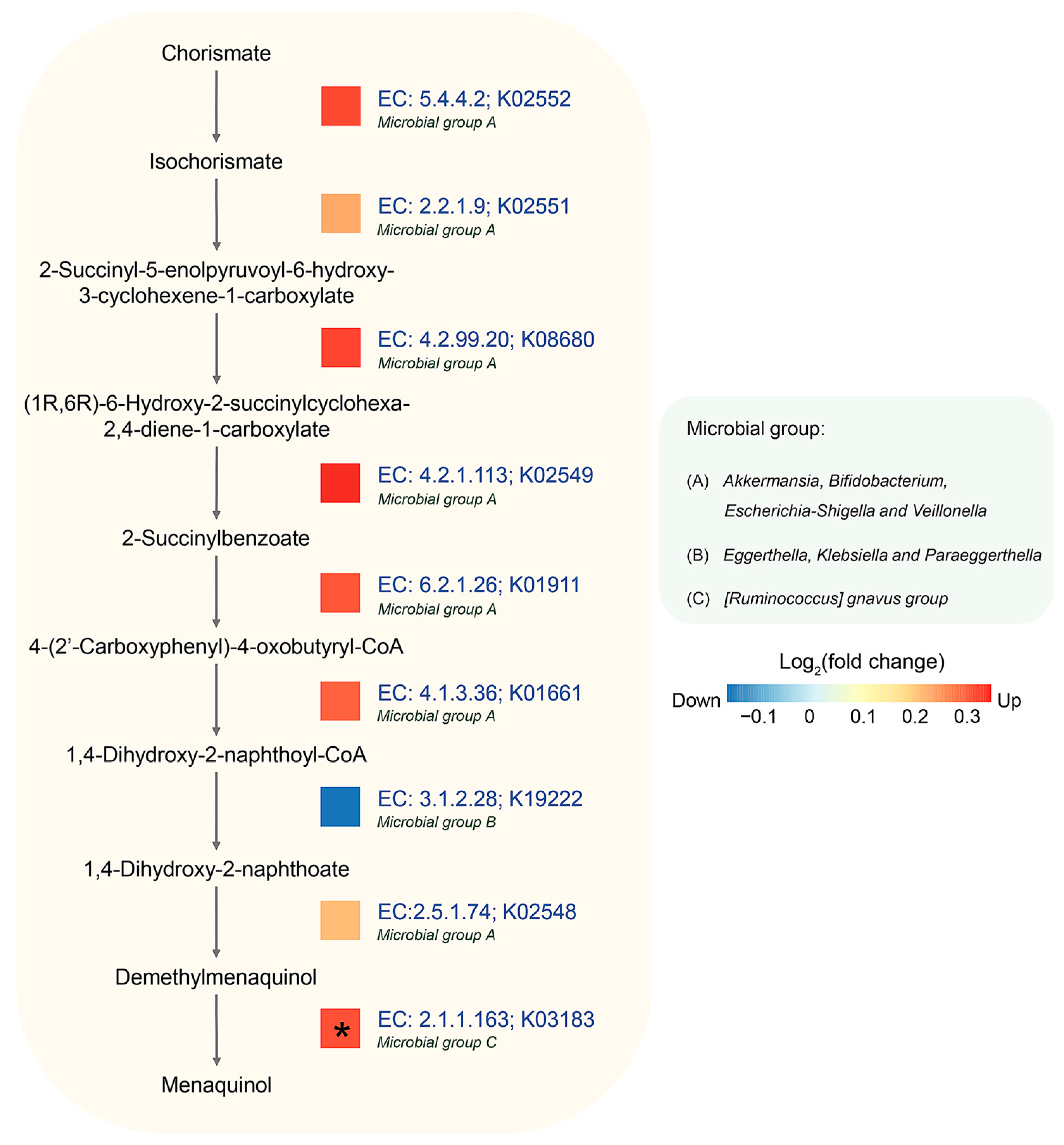
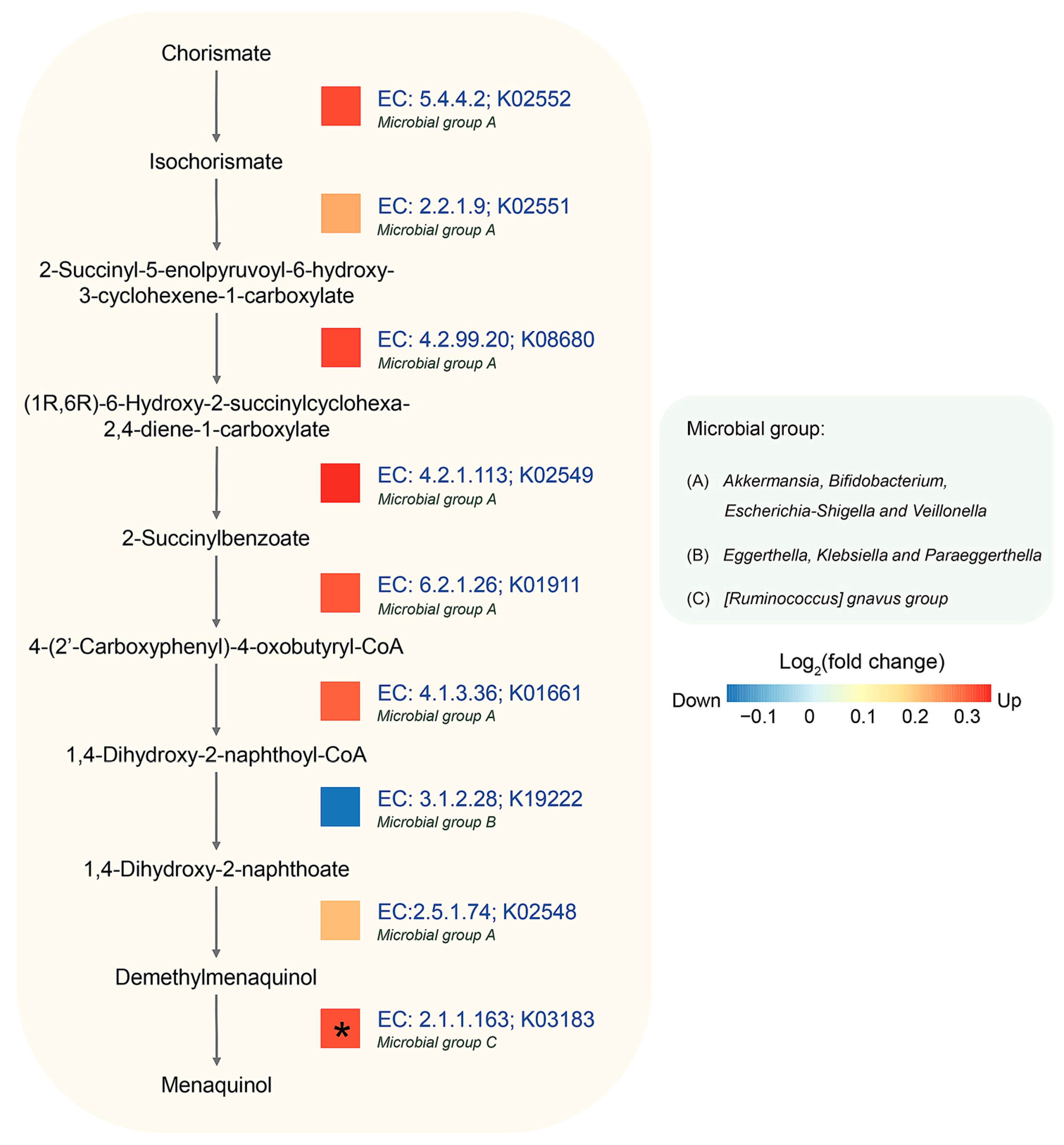
3.5. Identification of Potential Metabolic Routes and Associated Bacteria Genera in AD Using Integrated Metagenomic and Metaproteomic Approaches
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bieber, T. Atopic dermatitis: An expanding therapeutic pipeline for a complex disease. Nat. Rev. Drug Discov. 2022, 21, 21–40. [Google Scholar] [CrossRef]
- Liu, Y.; Du, X.; Zhai, S.; Tang, X.; Liu, C.; Li, W. Gut microbiota and atopic dermatitis in children: A scoping review. BMC Pediatr. 2022, 22, 323. [Google Scholar] [CrossRef]
- Williams, H.; Stewart, A.; von Mutius, E.; Cookson, W.; Anderson, H.R.; International Study of Asthma and Allergies in Childhood (ISAAC) Phase One and Three Study Groups. Is eczema really on the increase worldwide? J. Allergy Clin. Immunol. 2008, 121, 947–954.e915. [Google Scholar] [CrossRef] [PubMed]
- Deckers, I.A.; McLean, S.; Linssen, S.; Mommers, M.; van Schayck, C.P.; Sheikh, A. Investigating international time trends in the incidence and prevalence of atopic eczema 1990-2010: A systematic review of epidemiological studies. PLoS ONE 2012, 7, e39803. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Cai, J.; Sun, C.; Zou, Z.; Zhang, J.; Huang, C. Time-trends for eczema prevalences among children and adults from 1985 to 2015 in China: A systematic review. BMC Public Health 2022, 22, 1294. [Google Scholar] [CrossRef] [PubMed]
- Pugliarello, S.; Cozzi, A.; Gisondi, P.; Girolomoni, G. Phenotypes of atopic dermatitis. J. Dtsch. Dermatol. Ges. 2011, 9, 12–20. [Google Scholar] [CrossRef]
- Dainichi, T.; Kitoh, A.; Otsuka, A.; Nakajima, S.; Nomura, T.; Kaplan, D.H.; Kabashima, K. The epithelial immune microenvironment (EIME) in atopic dermatitis and psoriasis. Nat. Immunol. 2018, 19, 1286–1298. [Google Scholar] [CrossRef]
- Alam, M.J.; Xie, L.; Yap, Y.A.; Marques, F.Z.; Robert, R. Manipulating Microbiota to Treat Atopic Dermatitis: Functions and Therapies. Pathogens 2022, 11, 642. [Google Scholar] [CrossRef]
- Balboul, S.; Kahn, J.; Tracy, A.; Peacock, A.; Cline, A. The Application of Dupilumab to Pediatric Patients Aged 6–11yrs with Moderate-to-Severe Atopic Dermatitis Whose Disease is Not Adequately Controlled: The Clinical Data so Far. Drug Des. Dev. Ther. 2023, 17, 1323–1327. [Google Scholar] [CrossRef]
- Wang, C.; Kraus, C.N.; Patel, K.G.; Ganesan, A.K.; Grando, S.A. Real-world experience of dupilumab treatment for atopic dermatitis in adults: A retrospective analysis of patients’ records. Int. J. Dermatol. 2020, 59, 253–256. [Google Scholar] [CrossRef]
- Napolitano, M.; Fabbrocini, G.; Genco, L.; Martora, F.; Potestio, L.; Patruno, C. Rapid improvement in pruritus in atopic dermatitis patients treated with upadacitinib: A real-life experience. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1497–1498. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Teixeira, H.D.; Simpson, E.L.; Papp, K.A.; Pangan, A.L.; Blauvelt, A.; Thaci, D.; Chu, C.Y.; Hong, H.C.; Katoh, N.; et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): Results from two replicate double-blind, randomised controlled phase 3 trials. Lancet 2021, 397, 2151–2168. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Teixeira, H.D.; de Bruin-Weller, M.; Bieber, T.; Soong, W.; Kabashima, K.; Werfel, T.; Zeng, J.; Huang, X.; Hu, X.; et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis (AD Up): Results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2021, 397, 2169–2181. [Google Scholar] [CrossRef] [PubMed]
- Melli, L.; Carmo-Rodrigues, M.S.D.; Araujo-Filho, H.B.; Mello, C.S.; Tahan, S.; Pignatari, A.C.C.; Sole, D.; Morais, M.B. Gut microbiota of children with atopic dermatitis: Controlled study in the metropolitan region of Sao Paulo, Brazil. Allergol. Immunopathol. 2020, 48, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Deng, Y.-H.; Huang, Y.-H.; Ke, H.-J.; Guo, Y.; Wu, J.-L. Comparison of gut microbiota between infants with atopic dermatitis and healthy controls in Guangzhou, China. J. Asthma Allergy 2021, 14, 493–500. [Google Scholar] [CrossRef]
- Penders, J.; Gerhold, K.; Stobberingh, E.E.; Thijs, C.; Zimmermann, K.; Lau, S.; Hamelmann, E. Establishment of the intestinal microbiota and its role for atopic dermatitis in early childhood. J. Allergy Clin. Immunol. 2013, 132, 601–607.e8. [Google Scholar] [CrossRef]
- Widhiati, S.; Purnomosari, D.; Wibawa, T.; Soebono, H. The role of gut microbiome in inflammatory skin disorders: A systematic review. Dermatol. Rep. 2022, 14, 9188. [Google Scholar] [CrossRef]
- Lee, M.J.; Park, Y.M.; Kim, B.; Tae, I.H.; Kim, N.E.; Pranata, M.; Kim, T.; Won, S.; Kang, N.J.; Lee, Y.K.; et al. Disordered development of gut microbiome interferes with the establishment of the gut ecosystem during early childhood with atopic dermatitis. Gut Microbes 2022, 14, 2068366. [Google Scholar] [CrossRef]
- Galazzo, G.; van Best, N.; Bervoets, L.; Dapaah, I.O.; Savelkoul, P.H.; Hornef, M.W.; The GI-MDH Consortium; Lau, S.; Hamelmann, E.; Penders, J. Development of the Microbiota and Associations with Birth Mode, Diet, and Atopic Disorders in a Longitudinal Analysis of Stool Samples, Collected From Infancy Through Early Childhood. Gastroenterology 2020, 158, 1584–1596. [Google Scholar] [CrossRef]
- Simonyte Sjodin, K.; Hammarstrom, M.L.; Ryden, P.; Sjodin, A.; Hernell, O.; Engstrand, L.; West, C.E. Temporal and long-term gut microbiota variation in allergic disease: A prospective study from infancy to school age. Allergy 2019, 74, 176–185. [Google Scholar] [CrossRef]
- Niu, J.; Xu, L.; Qian, Y.; Sun, Z.; Yu, D.; Huang, J.; Zhou, X.; Wang, Y.; Zhang, T.; Ren, R.; et al. Evolution of the Gut Microbiome in Early Childhood: A Cross-Sectional Study of Chinese Children. Front. Microbiol. 2020, 11, 439. [Google Scholar] [CrossRef] [PubMed]
- Ta, L.D.H.; Chan, J.C.Y.; Yap, G.C.; Purbojati, R.W.; Drautz-Moses, D.I.; Koh, Y.M.; Tay, C.J.X.; Huang, C.H.; Kioh, D.Y.Q.; Woon, J.Y.; et al. A compromised developmental trajectory of the infant gut microbiome and metabolome in atopic eczema. Gut Microbes 2020, 12, 1801964. [Google Scholar] [CrossRef] [PubMed]
- Cait, A.; Cardenas, E.; Dimitriu, P.A.; Amenyogbe, N.; Dai, D.; Cait, J.; Sbihi, H.; Stiemsma, L.; Subbarao, P.; Mandhane, P.J.; et al. Reduced genetic potential for butyrate fermentation in the gut microbiome of infants who develop allergic sensitization. J. Allergy Clin. Immunol. 2019, 144, 1638–1647. [Google Scholar] [CrossRef] [PubMed]
- Wopereis, H.; Sim, K.; Shaw, A.; Warner, J.O.; Knol, J.; Kroll, J.S. Intestinal microbiota in infants at high risk for allergy: Effects of prebiotics and role in eczema development. J. Allergy Clin. Immunol. 2018, 141, 1334–1342.e5. [Google Scholar] [CrossRef]
- Patumcharoenpol, P.; Nakphaichit, M.; Panagiotou, G.; Senavonge, A.; Suratannon, N.; Vongsangnak, W. MetGEMs Toolbox: Metagenome-scale models as integrative toolbox for uncovering metabolic functions and routes of human gut microbiome. PLoS Comput. Biol. 2021, 17, e1008487. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Kingkaw, A.; Nakphaichit, M.; Suratannon, N.; Nitisinprasert, S.; Wongoutong, C.; Chatchatee, P.; Krobthong, S.; Charoenlappanit, S.; Roytrakul, S.; Vongsangnak, W. Analysis of the infant gut microbiome reveals metabolic functional roles associated with healthy infants and infants with atopic dermatitis using metaproteomics. PeerJ 2020, 8, e9988. [Google Scholar] [CrossRef]
- Eichenfield, L.F.; Ahluwalia, J.; Waldman, A.; Borok, J.; Udkoff, J.; Boguniewicz, M. Current guidelines for the evaluation and management of atopic dermatitis–a comparison of the Joint Task Force Practice Parameter and American Academy of Dermatology Guidelines. Alergol. Pol. -Pol. J. Allergol. 2017, 4, 158–168. [Google Scholar] [CrossRef]
- Kisuse, J.; La-ongkham, O.; Nakphaichit, M.; Therdtatha, P.; Momoda, R.; Tanaka, M.; Fukuda, S.; Popluechai, S.; Kespechara, K.; Sonomoto, K. Urban diets linked to gut microbiome and metabolome alterations in children: A comparative cross-sectional study in Thailand. Front. Microbiol. 2018, 9, 1345. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Peddada, S.D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020, 11, 3514. [Google Scholar] [CrossRef] [PubMed]
- Pelucchi, C.; Galeone, C.; Bach, J.F.; La Vecchia, C.; Chatenoud, L. Pet exposure and risk of atopic dermatitis at the pediatric age: A meta-analysis of birth cohort studies. J. Allergy Clin. Immunol. 2013, 132, 616–622.e7. [Google Scholar] [CrossRef] [PubMed]
- Roduit, C.; Wohlgensinger, J.; Frei, R.; Bitter, S.; Bieli, C.; Loeliger, S.; Buchele, G.; Riedler, J.; Dalphin, J.C.; Remes, S.; et al. Prenatal animal contact and gene expression of innate immunity receptors at birth are associated with atopic dermatitis. J. Allergy Clin. Immunol. 2011, 127, 179–185. [Google Scholar] [CrossRef]
- Batool, T.; Reece, P.L.; Schulze, K.M.; Morrison, K.M.; Atkinson, S.A.; Anand, S.S.; Teo, K.K.; Denburg, J.A.; Cyr, M.M.; Investigators, F.S. Prenatal and early-life predictors of atopy and allergic disease in Canadian children: Results of the Family Atherosclerosis Monitoring In earLY life (FAMILY) Study. J. Dev. Orig. Health Dis. 2016, 7, 665–671. [Google Scholar] [CrossRef]
- Ismail, I.H.; Oppedisano, F.; Joseph, S.J.; Boyle, R.J.; Licciardi, P.V.; Robins-Browne, R.M.; Tang, M.L. Reduced gut microbial diversity in early life is associated with later development of eczema but not atopy in high-risk infants. Pediatr. Allergy Immunol. 2012, 23, 674–681. [Google Scholar] [CrossRef]
- Nakayama, J.; Watanabe, K.; Jiang, J.; Matsuda, K.; Chao, S.H.; Haryono, P.; La-Ongkham, O.; Sarwoko, M.A.; Sujaya, I.N.; Zhao, L.; et al. Diversity in gut bacterial community of school-age children in Asia. Sci. Rep. 2015, 5, 8397. [Google Scholar] [CrossRef]
- Wernroth, M.L.; Peura, S.; Hedman, A.M.; Hetty, S.; Vicenzi, S.; Kennedy, B.; Fall, K.; Svennblad, B.; Andolf, E.; Pershagen, G.; et al. Development of gut microbiota during the first 2 years of life. Sci. Rep. 2022, 12, 9080. [Google Scholar] [CrossRef]
- Ryan, F.J.; Ahern, A.M.; Fitzgerald, R.S.; Laserna-Mendieta, E.J.; Power, E.M.; Clooney, A.G.; O’Donoghue, K.W.; McMurdie, P.J.; Iwai, S.; Crits-Christoph, A.; et al. Colonic microbiota is associated with inflammation and host epigenomic alterations in inflammatory bowel disease. Nat. Commun. 2020, 11, 1512. [Google Scholar] [CrossRef] [PubMed]
- Henke, M.T.; Kenny, D.J.; Cassilly, C.D.; Vlamakis, H.; Xavier, R.J.; Clardy, J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc. Natl. Acad. Sci. USA 2019, 116, 12672–12677. [Google Scholar] [CrossRef]
- De Filippis, F.; Paparo, L.; Nocerino, R.; Della Gatta, G.; Carucci, L.; Russo, R.; Pasolli, E.; Ercolini, D.; Berni Canani, R. Specific gut microbiome signatures and the associated pro-inflamatory functions are linked to pediatric allergy and acquisition of immune tolerance. Nat. Commun. 2021, 12, 5958. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.R.; Lee, S.H.; Kim, B.; Nam, M.H.; Ahn, Y.K.; Park, Y.M.; Jeong, S.M.; Park, M.J.; Song, K.B.; Lee, S.Y.; et al. Ruminococcus gnavus ameliorates atopic dermatitis by enhancing Treg cell and metabolites in BALB/c mice. Pediatr. Allergy Immunol. 2022, 33, e13678. [Google Scholar] [CrossRef] [PubMed]
- Chua, H.H.; Chou, H.C.; Tung, Y.L.; Chiang, B.L.; Liao, C.C.; Liu, H.H.; Ni, Y.H. Intestinal Dysbiosis Featuring Abundance of Ruminococcus gnavus Associates with Allergic Diseases in Infants. Gastroenterology 2018, 154, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hou, J.; Tsui, J.C.-C.; Wang, L.; Zhou, J.; Chan, U.K.; Lo, C.J.Y.; Siu, P.L.K.; Loo, S.K.F.; Tsui, S.K.W. Unique gut microbiome signatures among adult patients with moderate to severe atopic dermatitis in southern Chinese. Int. J. Mol. Sci. 2023, 24, 12856. [Google Scholar] [CrossRef]
- Sasaki, M.; Schwab, C.; Ramirez Garcia, A.; Li, Q.; Ferstl, R.; Bersuch, E.; Akdis, C.A.; Lauener, R.; CK-CARE Study Group; Frei, R.; et al. The abundance of Ruminococcus bromii is associated with faecal butyrate levels and atopic dermatitis in infancy. Allergy 2022, 77, 3629–3640. [Google Scholar] [CrossRef]
- Nogal, A.; Louca, P.; Zhang, X.; Wells, P.M.; Steves, C.J.; Spector, T.D.; Falchi, M.; Valdes, A.M.; Menni, C. Circulating Levels of the Short-Chain Fatty Acid Acetate Mediate the Effect of the Gut Microbiome on Visceral Fat. Front. Microbiol. 2021, 12, 711359. [Google Scholar] [CrossRef]
- Opiyo, S.O.; Pardy, R.L.; Moriyama, H.; Moriyama, E.N. Evolution of the Kdo2-lipid A biosynthesis in bacteria. BMC Evol. Biol. 2010, 10, 362. [Google Scholar] [CrossRef]
- Portincasa, P.; Bonfrate, L.; Vacca, M.; De Angelis, M.; Farella, I.; Lanza, E.; Khalil, M.; Wang, D.Q.; Sperandio, M.; Di Ciaula, A. Gut Microbiota and Short Chain Fatty Acids: Implications in Glucose Homeostasis. Int. J. Mol. Sci. 2022, 23, 1105. [Google Scholar] [CrossRef]
- Tsukuda, N.; Yahagi, K.; Hara, T.; Watanabe, Y.; Matsumoto, H.; Mori, H.; Higashi, K.; Tsuji, H.; Matsumoto, S.; Kurokawa, K.; et al. Key bacterial taxa and metabolic pathways affecting gut short-chain fatty acid profiles in early life. ISME J. 2021, 15, 2574–2590. [Google Scholar] [CrossRef] [PubMed]
- Nogal, A.; Valdes, A.M.; Menni, C. The role of short-chain fatty acids in the interplay between gut microbiota and diet in cardio-metabolic health. Gut Microbes 2021, 13, 1897212. [Google Scholar] [CrossRef] [PubMed]
- Kingkaw, A.; Raethong, N.; Patumcharoenpol, P.; Suratannon, N.; Nakphaichit, M.; Keawsompong, S.; Roytrakul, S.; Vongsangnak, W. Analyzing Predominant Bacterial Species and Potential Short-Chain Fatty Acid-Associated Metabolic Routes in Human Gut Microbiome Using Integrative Metagenomics. Biology 2022, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Z.; Liu, W.; Pan, L.; Bao, Y.; Yan, Z.; Hong, L. Overabundance of Veillonella parvula promotes intestinal inflammation by activating macrophages via LPS-TLR4 pathway. Cell Death Discov. 2022, 8, 251. [Google Scholar] [CrossRef]
- Karcher, N.; Nigro, E.; Puncochar, M.; Blanco-Miguez, A.; Ciciani, M.; Manghi, P.; Zolfo, M.; Cumbo, F.; Manara, S.; Golzato, D.; et al. Genomic diversity and ecology of human-associated Akkermansia species in the gut microbiome revealed by extensive metagenomic assembly. Genome Biol. 2021, 22, 209. [Google Scholar] [CrossRef]
- Arboleya, S.; Watkins, C.; Stanton, C.; Ross, R.P. Gut Bifidobacteria Populations in Human Health and Aging. Front. Microbiol. 2016, 7, 1204. [Google Scholar] [CrossRef]
- Fujimura, K.E.; Sitarik, A.R.; Havstad, S.; Lin, D.L.; Levan, S.; Fadrosh, D.; Panzer, A.R.; LaMere, B.; Rackaityte, E.; Lukacs, N.W.; et al. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat. Med. 2016, 22, 1187–1191. [Google Scholar] [CrossRef]
- Koike-Takeshita, A.; Koyama, T.; Ogura, K. Identification of a novel gene cluster participating in menaquinone (vitamin K2) biosynthesis. Cloning and sequence determination of the 2-heptaprenyl-1,4-naphthoquinone methyltransferase gene of Bacillus stearothermophilus. J. Biol. Chem. 1997, 272, 12380–12383. [Google Scholar] [CrossRef]
- Karl, J.P.; Fu, X.; Wang, X.; Zhao, Y.; Shen, J.; Zhang, C.; Wolfe, B.E.; Saltzman, E.; Zhao, L.; Booth, S.L. Fecal menaquinone profiles of overweight adults are associated with gut microbiota composition during a gut microbiota-targeted dietary intervention. Am. J. Clin. Nutr. 2015, 102, 84–93. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control (n = 39) | AD (n = 23) | p-Value & | |
|---|---|---|---|
| Sex | 0.60 | ||
| Male | 23 (59%) | 15 (65%) | |
| Female | 16 (41%) | 8 (35%) | |
| Delivery method | 1.00 | ||
| Caesarean | 14 (36%) | 8 (35%) | |
| Vaginal | 25 (64%) | 15 (65%) | |
| Family income (monthly, THB *) | 0.11 | ||
| ≤50,000 | 33 (85%) | 15 (65%) | |
| ≥50,000 | 6 (15%) | 8 (35%) | |
| Birth weight | 0.52 | ||
| mean ± sd | 3228 ± 412 | 3135 ± 621 | |
| Exposure to pet | 0.04 | ||
| Yes | 15 (38%) | 6 (26%) | |
| No | 24 (62%) | 17 (74%) | |
| Mother history of AD | 1.00 | ||
| Yes | 6 (15%) | 3 (13%) | |
| No | 33 (85%) | 20 (87%) |
| Taxonomic Level | Taxonomic Name | q-Value | Log2FC * |
|---|---|---|---|
| Order | Lachnospirales | 0.0006 | 1.03 |
| Family | Butyricicoccaceae | 0.0036 | 1.56 |
| Family | Lachnospiraceae | 0.0133 | 0.87 |
| Genus | Anaerostipes | 0.0077 | 2.11 |
| Genus | Ruminococcus | 0.0292 | 0.99 |
| Genus | Butyricicoccus | 0.0386 | 1.39 |
| Age | Taxonomic Level | Taxonomic Name | q-Value | Log2FC * |
|---|---|---|---|---|
| 9–12 months | Genus | Anaerostipes | 0.0118 | 3.18 |
| Genus | Lachnoclostridium | 0.0292 | 2.56 | |
| 18–21 months | Family | Butyricicoccaceae | 0.0001 | 2.60 |
| Family | Lachnospiraceae | 0.0163 | 1.02 | |
| Family | Eggerthellaceae | 0.0382 | 1.12 | |
| Genus | Butyricicoccus | 0.0000 | 2.66 | |
| 24–30 months | Family | Lactobacillaceae | 0.0054 | 3.49 |
| Genus | Oscillibacter | 0.0003 | −1.62 | |
| Genus | UBA1819 | 0.0018 | −3.84 | |
| Genus | Eisenbergiella | 0.0043 | −2.26 | |
| Genus | Lactobacillus | 0.0059 | 3.70 | |
| Genus | Lachnoclostridium | 0.0059 | −2.52 |
| Metabolic Pathways-Based MetaCyc | q-Value | Log2FC * |
|---|---|---|
| superpathway of menaquinol-9 biosynthesis (PWY-5845) | 0.0033 | 0.49 |
| superpathway of menaquinol-6 biosynthesis I (PWY-5850) | 0.0033 | 0.49 |
| superpathway of menaquinol-10 biosynthesis (PWY-5896) | 0.0033 | 0.49 |
| superpathway of demethylmenaquinol-6 biosynthesis I (PWY-5860) | 0.0166 | 0.50 |
| superpathway of demethylmenaquinol-9 biosynthesis (PWY-5862) | 0.0166 | 0.50 |
| superpathway of (Kdo)2-lipid A biosynthesis (KDO-NAGLIPASYN-PWY) | 0.0225 | 0.69 |
| superpathway of histidine, purine, and pyrimidine biosynthesis (PRPP-PWY) | 0.0287 | 0.38 |
| TCA cycle IV (2-oxoglutarate decarboxylase) (P105-PWY) | 0.0390 | 0.65 |
| Age | Metabolic Pathways-Based MetaCyc/ KO IDs | q-Value | Log2FC * |
|---|---|---|---|
| PICRUSt2 | |||
| 18–21 months | superpathway of histidine, purine, and pyrimidine biosynthesis (PRPP-PWY) | 0.0461 | 0.53 |
| 24–30 months | allantoin degradation IV (anaerobic) (PWY0-41) | 0.0027 | 2.01 |
| alpha-galactosidase (K07406) | 0.0195 | 1.20 | |
| uncharacterized protein (K07033) | 0.0468 | 1.28 | |
| MetGEMs | |||
| 18–21 months | purine ribonucleosides degradation (PWY0-1296) | 0.0275 | −0.58 |
| superpathway of pyrimidine deoxyribonucleoside salvage (PWY-7200) | 0.0303 | 0.48 | |
| adenine and adenosine salvage III (PWY-6609) | 0.0390 | −0.63 | |
| undecaprenyl diphosphate synthase (K00806) | 0.0132 | 0.74 | |
| 24–30 months | hydroxymethylpyrimidine kinase/phosphomethylpyrimidine kinase/thiamine-phosphate diphosphorylase(K14153) | 0.0023 | 0.32 |
| hydroxymethylpyrimidine/phosphomethylpyrimidine kinase/thiaminase (K00877) | 0.0179 | 0.38 | |
| hydroxymethylpyrimidine/phosphomethylpyrimidine kinase (K00941) | 0.0179 | 0.38 | |
| succinate dehydrogenase flavoprotein subunit (K00239) | 0.0270 | 0.46 | |
| succinate dehydrogenase iron-sulfur subunit (K00240) | 0.0270 | 0.46 | |
| succinate dehydrogenase cytochrome b subunit (K00241) | 0.0270 | 0.46 | |
| succinate dehydrogenase membrane anchor subunit (K00242) | 0.0270 | 0.46 | |
| fumarate reductase flavoprotein subunit (K00244) | 0.0270 | 0.46 | |
| fumarate reductase iron-sulfur subunit (K00245) | 0.0270 | 0.46 | |
| fumarate reductase subunit C (K00246) | 0.0270 | 0.46 | |
| fumarate reductase subunit D (K00247) | 0.0270 | 0.46 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patumcharoenpol, P.; Kingkaw, A.; Nakphaichit, M.; Chatchatee, P.; Suratannon, N.; Panagiotou, G.; Vongsangnak, W. Exploring Longitudinal Gut Microbiome towards Metabolic Functional Changes Associated in Atopic Dermatitis in Early Childhood. Biology 2023, 12, 1262. https://doi.org/10.3390/biology12091262
Patumcharoenpol P, Kingkaw A, Nakphaichit M, Chatchatee P, Suratannon N, Panagiotou G, Vongsangnak W. Exploring Longitudinal Gut Microbiome towards Metabolic Functional Changes Associated in Atopic Dermatitis in Early Childhood. Biology. 2023; 12(9):1262. https://doi.org/10.3390/biology12091262
Chicago/Turabian StylePatumcharoenpol, Preecha, Amornthep Kingkaw, Massalin Nakphaichit, Pantipa Chatchatee, Narissara Suratannon, Gianni Panagiotou, and Wanwipa Vongsangnak. 2023. "Exploring Longitudinal Gut Microbiome towards Metabolic Functional Changes Associated in Atopic Dermatitis in Early Childhood" Biology 12, no. 9: 1262. https://doi.org/10.3390/biology12091262
APA StylePatumcharoenpol, P., Kingkaw, A., Nakphaichit, M., Chatchatee, P., Suratannon, N., Panagiotou, G., & Vongsangnak, W. (2023). Exploring Longitudinal Gut Microbiome towards Metabolic Functional Changes Associated in Atopic Dermatitis in Early Childhood. Biology, 12(9), 1262. https://doi.org/10.3390/biology12091262