
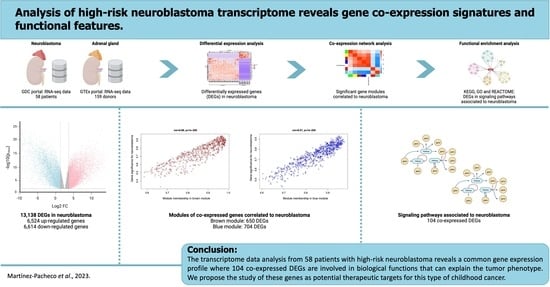
Analysis of High-Risk Neuroblastoma Transcriptome Reveals Gene Co-Expression Signatures and Functional Features


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
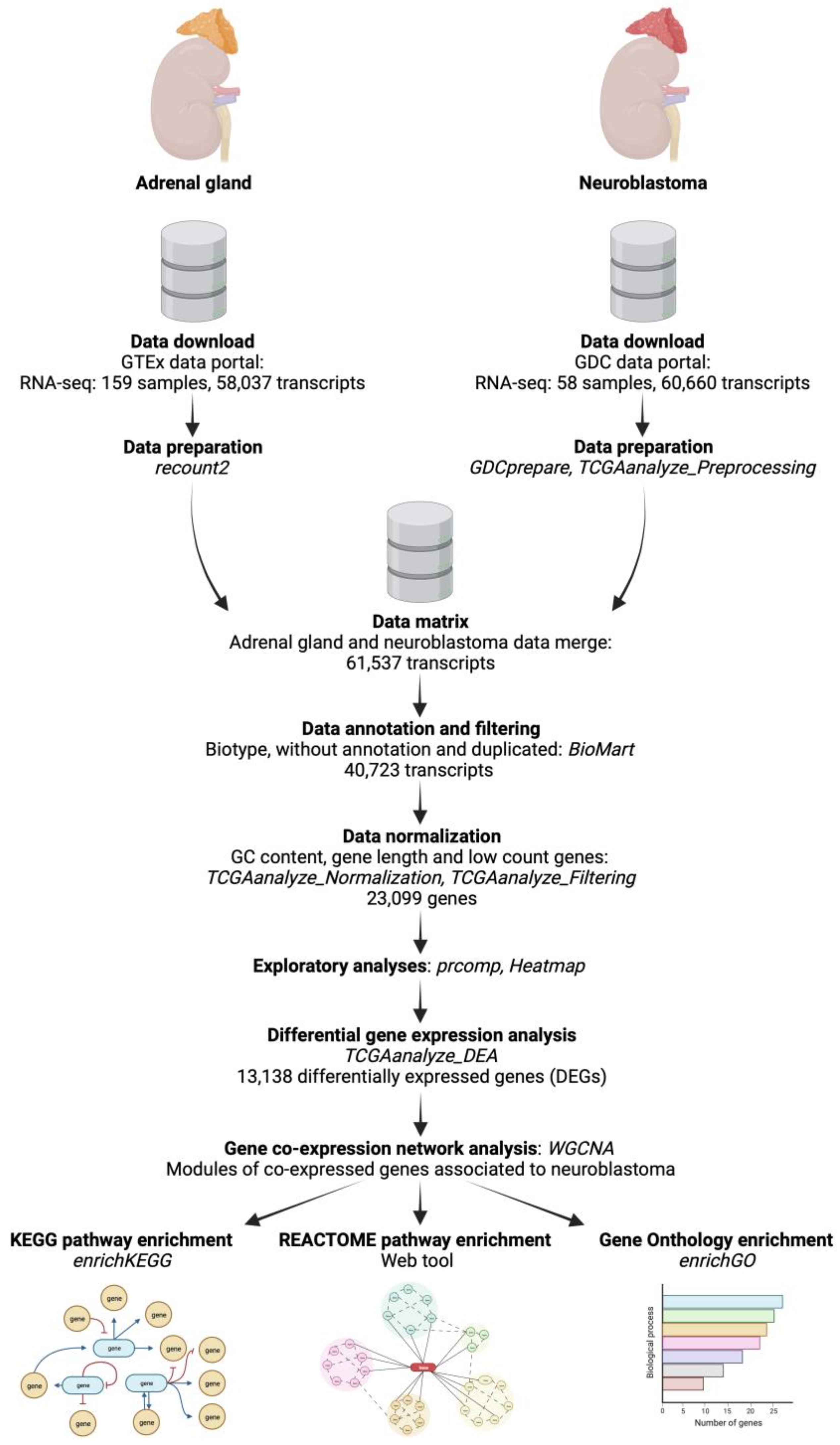
2.1. Data Retrieval and Preparation
2.2. Exploratory Analyses of the Features of Neuroblastoma and Normal Adrenal Gland Data
2.3. Analysis of Differentially Expressed Genes (DEGs) between Neuroblastoma and Normal Adrenal Gland Samples
2.4. Weighted Correlation Network Analysis (WGCNA) of DEGs and Identification of Modules Associated with Neuroblastoma
2.5. Functional Enrichment Analyses of Genes from the Significantly Correlated Modules
2.6. Data Processing and Statistical Analysis
3. Results
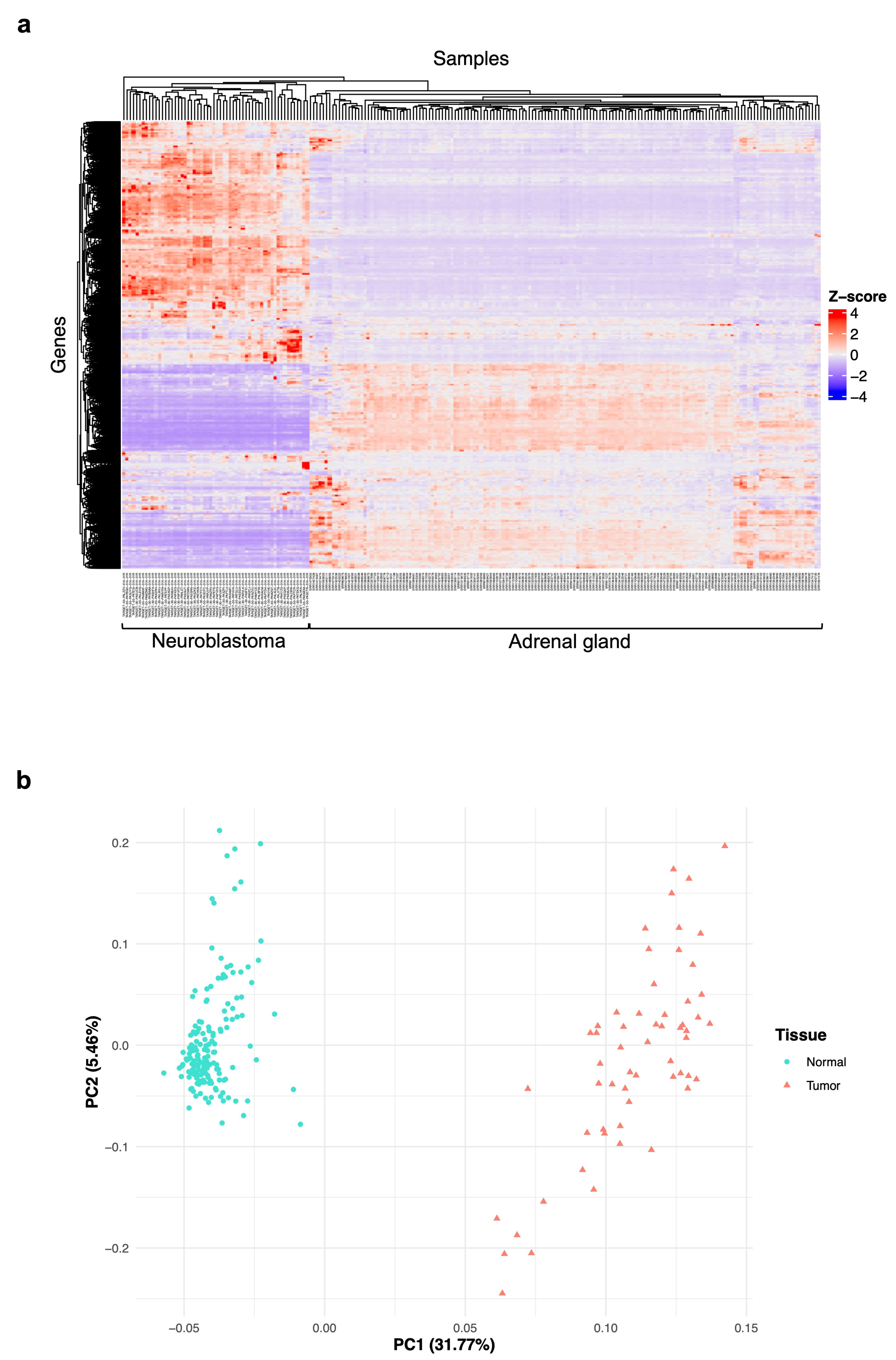
3.1. Exploration of the Gene Expression Data Features of Neuroblastoma and Normal Adrenal Gland
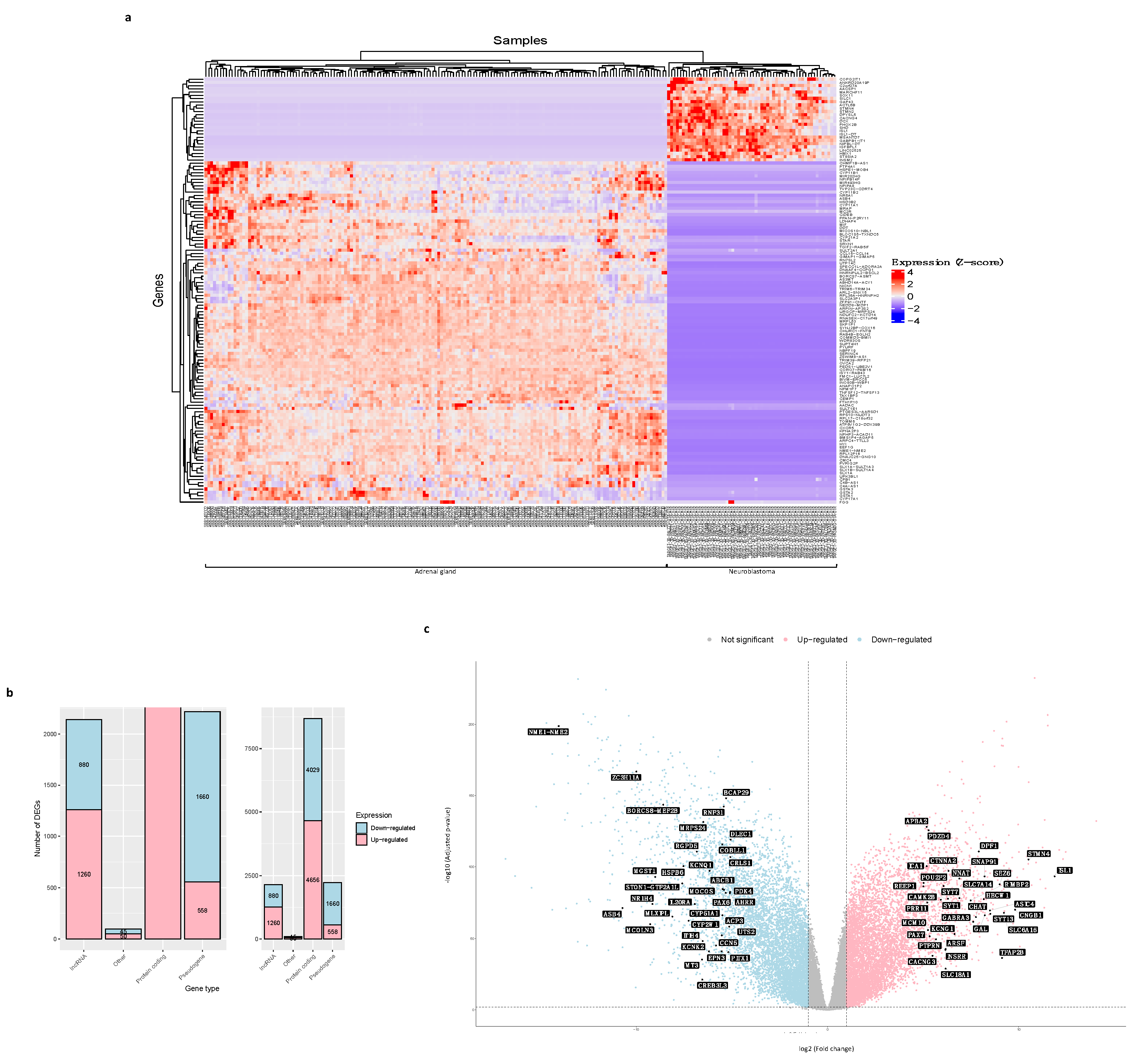
3.2. DEGs between Neuroblastoma and Normal Adrenal Gland Samples
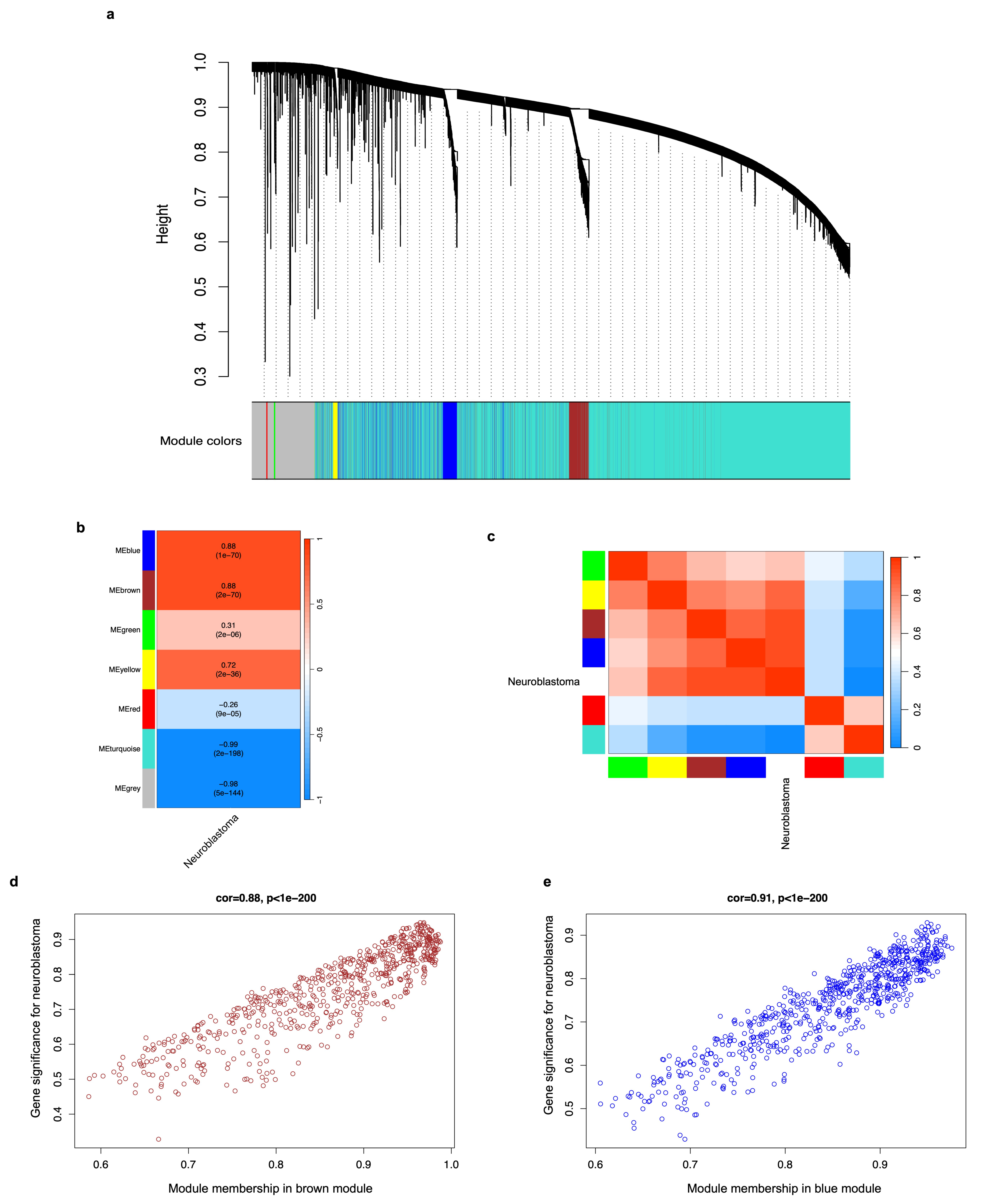
3.3. DEG Co-Expression Analysis (WGCNA) and Identification of Gene Modules Associated with Neuroblastoma
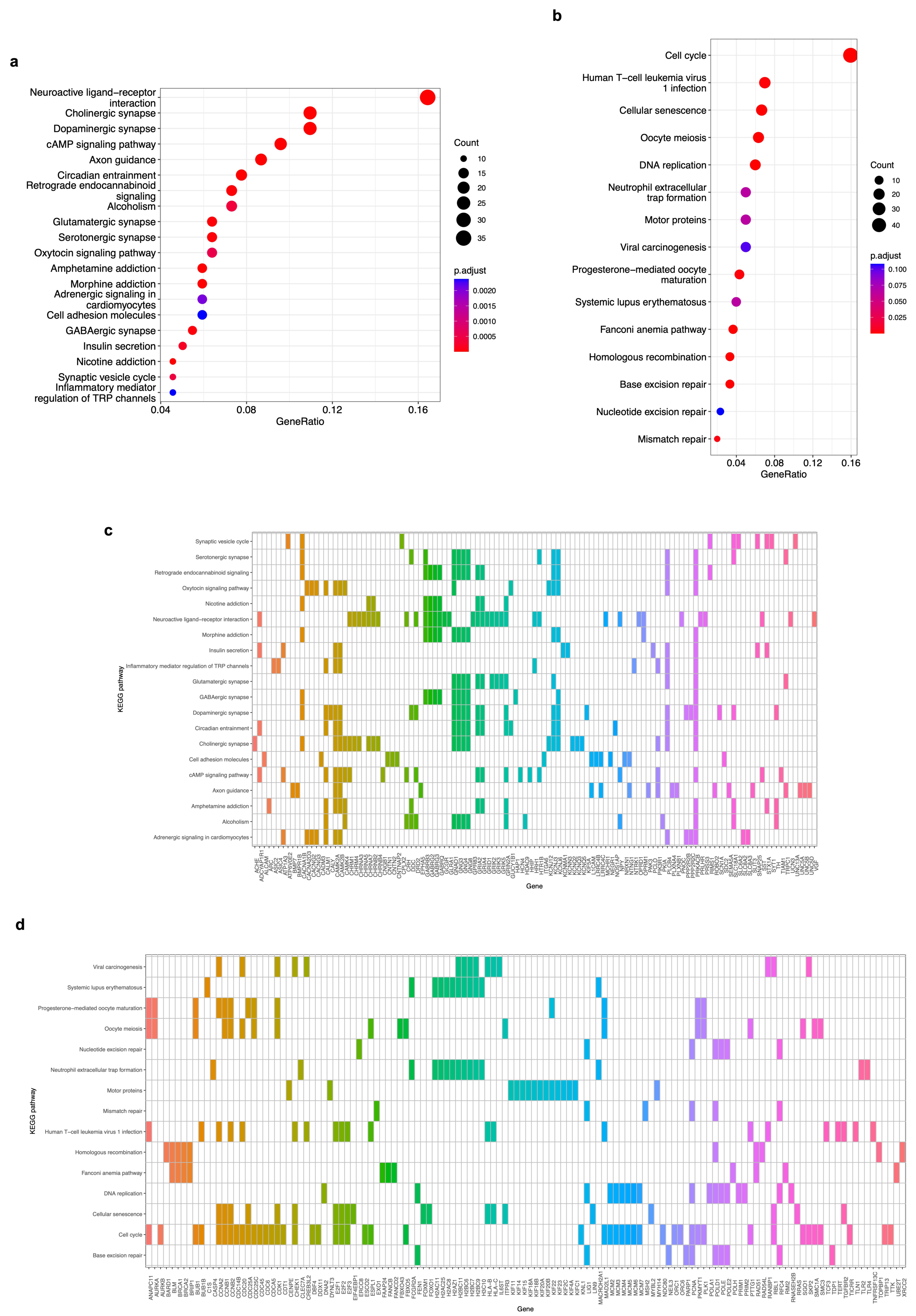
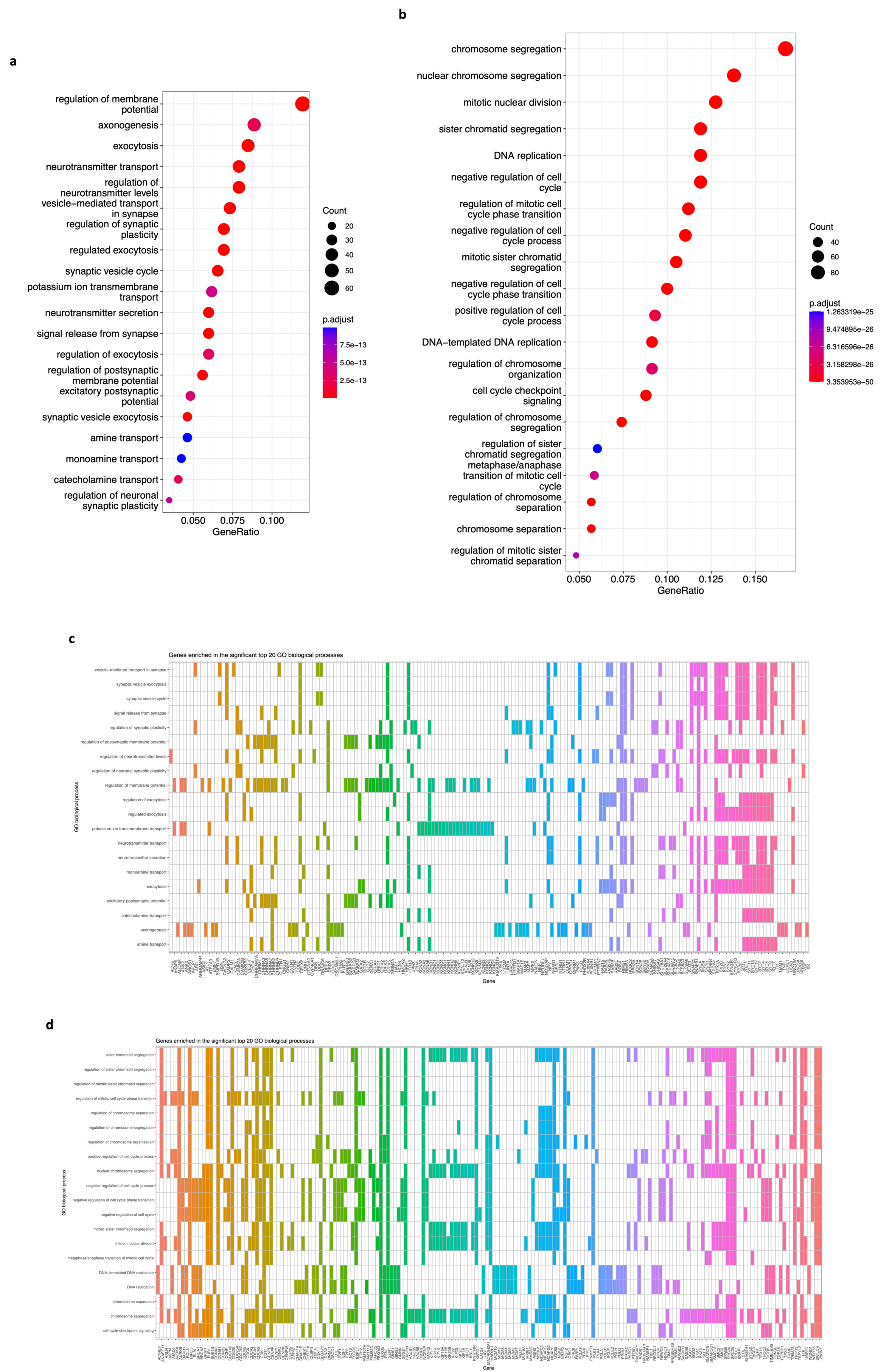
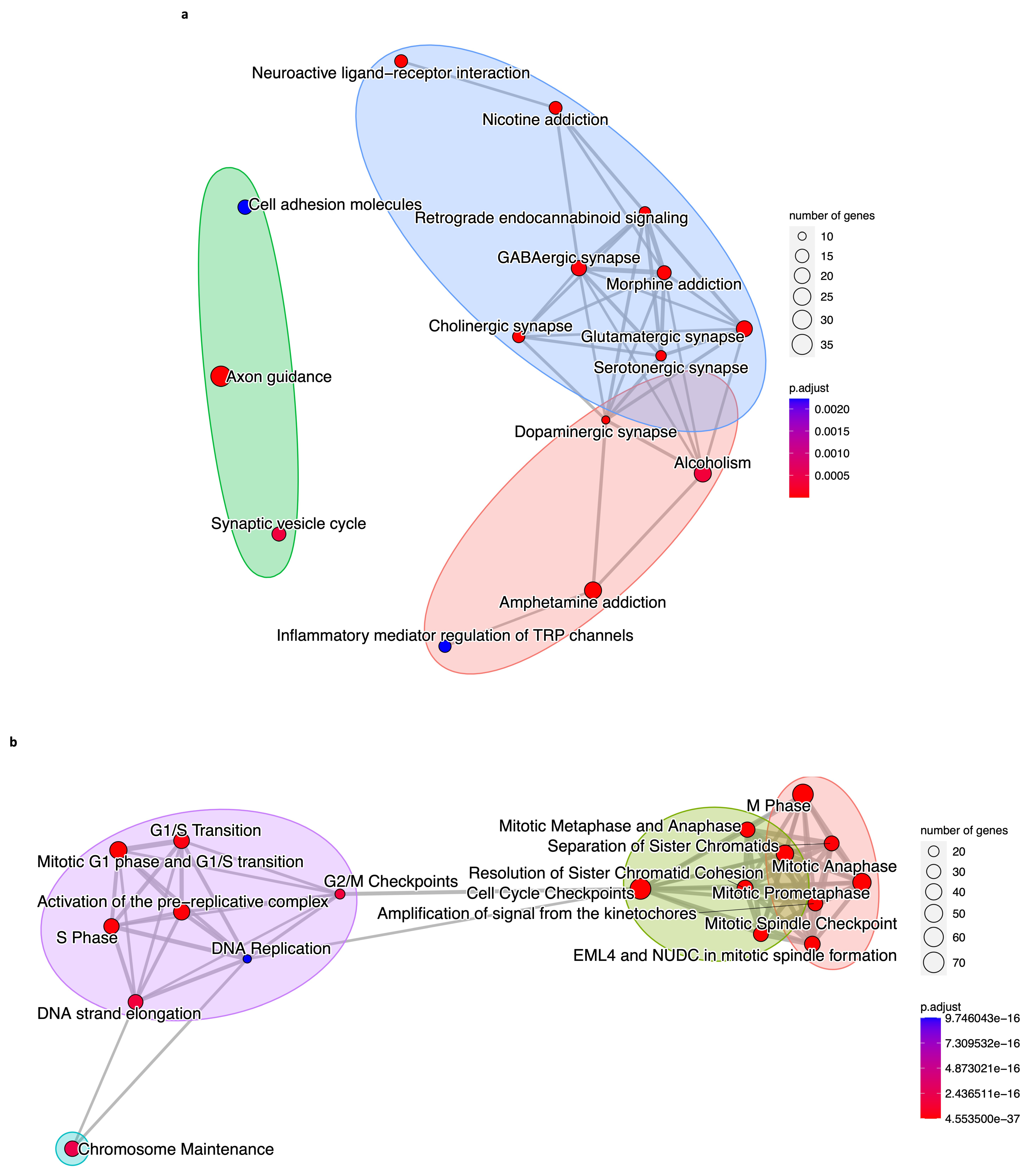
3.4. Functional Enrichment Analyses (KEGG, GO, and REACTOME) of DEGs of Modules Associated with Neuroblastoma
3.5. Data Availability
4. Discussion
Scope and Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Park, J.R.; Eggert, A.; Caron, H. Neuroblastoma: Biology, Prognosis, and Treatment. Hematol. Oncol. Clin. N. Am. 2010, 24, 65–86. [Google Scholar] [CrossRef]
- Otte, J.; Dyberg, C.; Pepich, A.; Johnsen, J.I. MYCN Function in Neuroblastoma Development. Front. Oncol. 2021, 10, 624079. [Google Scholar] [CrossRef] [PubMed]
- Deyell, R.J.; Attiyeh, E.F. Advances in the Understanding of Constitutional and Somatic Genomic Alterations in Neuroblastoma. Cancer Genet. 2011, 204, 113–121. [Google Scholar] [CrossRef] [PubMed]
- De Preter, K.; Vandesompele, J.; Heimann, P.; Yigit, N.; Beckman, S.; Schramm, A.; Eggert, A.; Stallings, R.L.; Benoit, Y.; Renard, M.; et al. Human Fetal Neuroblast and Neuroblastoma Transcriptome Analysis Confirms Neuroblast Origin and Highlights Neuroblastoma Candidate Genes. Genome Biol. 2006, 7, R84. [Google Scholar] [CrossRef]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; McGrady, P.W.; et al. ALK Mutations Confer Differential Oncogenic Activation and Sensitivity to ALK Inhibition Therapy in Neuroblastoma. Cancer Cell 2014, 26, 682–694. [Google Scholar] [CrossRef]
- Schulte, J.H.; Marschall, T.; Martin, M.; Rosenstiel, P.; Mestdagh, P.; Schlierf, S.; Thor, T.; Vandesompele, J.; Eggert, A.; Schreiber, S.; et al. Deep Sequencing Reveals Differential Expression of MicroRNAs in Favorable versus Unfavorable Neuroblastoma. Nucleic Acids Res. 2010, 38, 5919–5928. [Google Scholar] [CrossRef]
- Misiak, D.; Hagemann, S.; Bell, J.L.; Busch, B.; Lederer, M.; Bley, N.; Schulte, J.H.; Hüttelmaier, S. The MicroRNA Landscape of MYCN-Amplified Neuroblastoma. Front. Oncol. 2021, 11, 647737. [Google Scholar] [CrossRef]
- Zhang, W.; Yu, Y.; Hertwig, F.; Thierry-Mieg, J.; Zhang, W.; Thierry-Mieg, D.; Wang, J.; Furlanello, C.; Devanarayan, V.; Cheng, J.; et al. Comparison of RNA-Seq and Microarray-Based Models for Clinical Endpoint Prediction. Genome Biol. 2015, 16, 133. [Google Scholar] [CrossRef]
- Bedoya-Reina, O.C.; Li, W.; Arceo, M.; Plescher, M.; Bullova, P.; Pui, H.; Kaucka, M.; Kharchenko, P.; Martinsson, T.; Holmberg, J.; et al. Single-Nuclei Transcriptomes from Human Adrenal Gland Reveal Distinct Cellular Identities of Low and High-Risk Neuroblastoma Tumors. Nat. Commun. 2021, 12, 5309. [Google Scholar] [CrossRef]
- Kimura, S.; Sekiguchi, M.; Watanabe, K.; Hiwatarai, M.; Seki, M.; Yoshida, K.; Isobe, T.; Shiozawa, Y.; Suzuki, H.; Hoshino, N.; et al. Association of High-Risk Neuroblastoma Classification Based on Expression Profiles with Differentiation and Metabolism. PLoS ONE 2021, 16, e0245526. [Google Scholar] [CrossRef]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor Package for Integrative Analysis of TCGA Data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Silva, T.C.; Colaprico, A.; Olsen, C.; D’Angelo, F.; Bontempi, G.; Ceccarelli, M.; Noushmehr, H. TCGA Workflow: Analyze Cancer Genomics and Epigenomics Data Using Bioconductor Packages. F1000Research 2016, 5, 1542. [Google Scholar] [CrossRef] [PubMed]
- Collado-Torres, L.; Nellore, A.; Kammers, K.; Ellis, S.E.; Taub, M.A.; Hansen, K.D.; Jaffe, A.E.; Langmead, B.; Leek, J.T. Reproducible RNA-Seq Analysis Using Recount2. Nat. Biotechnol. 2017, 35, 319–321. [Google Scholar] [CrossRef] [PubMed]
- Mounir, M.; Lucchetta, M.; Silva, T.C.; Olsen, C.; Bontempi, G.; Chen, X.; Noushmehr, H.; Colaprico, A.; Papaleo, E. New Functionalities in the TCGAbiolinks Package for the Study and Integration of Cancer Data from GDC and GTEx. PLoS Comput. Biol. 2019, 15, e1006701. [Google Scholar] [CrossRef]
- Tarazona, S.; Furió-Tarí, P.; Turrà, D.; Di Pietro, A.; Nueda, M.J.; Ferrer, A.; Conesa, A. Data Quality Aware Analysis of Differential Expression in RNA-Seq with NOISeq R/Bioc Package. Nucleic Acids Res. 2015, 43, e140. [Google Scholar] [CrossRef]
- Tarazona, S.; García-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential Expression in RNA-Seq: A Matter of Depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef]
- Nueda, M.J.; Ferrer, A.; Conesa, A. ARSyN: A Method for the Identification and Removal of Systematic Noise in Multifactorial Time Course Microarray Experiments. Biostatistics 2012, 13, 553–566. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Gu, Z. Complex Heatmap Visualization. iMeta 2022, 1, e43. [Google Scholar] [CrossRef]
- Mardia, K.V.; Kent, J.T.; Bibby, J.M. Multivariate Analysis, 1st ed.; Academic Press: London, UK, 1970. [Google Scholar]
- Becker, R.A.; Chambers, J.M.; Wilks, A.R. The New S Language, 1st ed.; Chapman and Hall/CRC: Boca Raton, FL, USA, 2008; ISBN 9781351074988. [Google Scholar]
- Venables, W.N.; Ripley, B.D. Statistical Analysis of Financial Data in S-Plus, 4th ed.; Springer: New York, NY, USA, 2004; ISBN 0-387-20286-2. [Google Scholar]
- R Core Team, R. A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2002. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Softw. 2012, 46, i11. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. Toward Understanding the Origin and Evolution of Cellular Organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for Taxonomy-Based Analysis of Pathways and Genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.; Mi, H. PANTHER: Making Genome-scale Phylogenetics Accessible to All. Protein Sci. 2022, 31, 8–22. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium the Gene Ontology Knowledgebase in 2023. Genetics 2023, 224, iyad031. [CrossRef]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The Reactome Pathway Knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M.; Falcon, S.; Pages, H.; Li, N. Org.Hs.Eg.Db: Genome Wide Annotation for Human. R Package Version 2019, 3, 1–3. [Google Scholar] [CrossRef]
- Yu, G.; He, Q.-Y. ReactomePA: An R/Bioconductor Package for Reactome Pathway Analysis and Visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Yan, G.-R.; He, Q.-Y. DOSE: An R/Bioconductor Package for Disease Ontology Semantic and Enrichment Analysis. Bioinformatics 2015, 31, 608–609. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development for R; RStudio, PBC: Boston, MA, USA, 2020; Available online: http://www.rstudio.com (accessed on 1 April 2023).
- Shimada, H.; Ikegaki, N. Genetic and Histopathological Heterogeneity of Neuroblastoma and Precision Therapeutic Approaches for Extremely Unfavorable Histology Subgroups. Biomolecules 2022, 12, 79. [Google Scholar] [CrossRef]
- Chiu, S.-L.; Cline, H.T. Insulin Receptor Signaling in the Development of Neuronal Structure and Function. Neural Dev. 2010, 5, 7. [Google Scholar] [CrossRef]
- Shao, Z.; Yang, Y.; Hu, Z. Editorial: Regulation of Synaptic Structure and Function. Front. Mol. Neurosci. 2022, 15, 1060367. [Google Scholar] [CrossRef]
- Yuasa-Kawada, J.; Kinoshita-Kawada, M.; Tsuboi, Y.; Wu, J.Y. Neuronal Guidance Genes in Health and Diseases. Protein Cell 2023, 14, 238–261. [Google Scholar] [CrossRef]
- Delloye-Bourgeois, C.; Bertin, L.; Thoinet, K.; Jarrosson, L.; Kindbeiter, K.; Buffet, T.; Tauszig-Delamasure, S.; Bozon, M.; Marabelle, A.; Combaret, V.; et al. Microenvironment-Driven Shift of Cohesion/Detachment Balance within Tumors Induces a Switch toward Metastasis in Neuroblastoma. Cancer Cell 2017, 32, 427–443.e8. [Google Scholar] [CrossRef]
- Sandoval, J.A.; Grosfeld, J.L.; Hickey, R.J.; Malkas, L.H. Structural Analysis of the Human Neuroblastoma DNA Replication Complex: Insights into Faulty Proliferation. J. Pediatr. Surg. 2006, 41, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Stafman, L.; Beierle, E. Cell Proliferation in Neuroblastoma. Cancers 2016, 8, 13. [Google Scholar] [CrossRef]
- Southgate, H.E.D.; Chen, L.; Curtin, N.J.; Tweddle, D.A. Targeting the DNA Damage Response for the Treatment of High Risk Neuroblastoma. Front. Oncol. 2020, 10, 371. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, S.; Decaesteker, B.; Vanhauwaert, S.; De Wilde, B.; De Vos, W.H.; Speleman, F. Cellular Senescence in Neuroblastoma. Br. J. Cancer 2022, 126, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Williams, V.; Filippova, M.; Filippov, V.; Duerksen-Hughes, P. Viral Carcinogenesis: Factors Inducing DNA Damage and Virus Integration. Cancers 2014, 6, 2155–2186. [Google Scholar] [CrossRef]
- Torgovnick, A.; Schumacher, B. DNA Repair Mechanisms in Cancer Development and Therapy. Front. Genet. 2015, 6, 157. [Google Scholar] [CrossRef]
- Wang, M.; Chen, S.; Ao, D. Targeting DNA Repair Pathway in Cancer: Mechanisms and Clinical Application. MedComm 2021, 2, 654–691. [Google Scholar] [CrossRef]
- da Costa, A.A.B.A.; Chowdhury, D.; Shapiro, G.I.; D’Andrea, A.D.; Konstantinopoulos, P.A. Targeting Replication Stress in Cancer Therapy. Nat. Rev. Drug Discov. 2023, 22, 38–58. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Wang, B.; Demaria, M. Senescence and Cancer—Role and Therapeutic Opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 619–636. [Google Scholar] [CrossRef]
- Leung, Y.-M.; Huang, C.-F.; Chao, C.-C.; Lu, D.-Y.; Kuo, C.-S.; Cheng, T.-H.; Chang, L.-Y.; Chou, C.-H. Voltage-Gated K+ Channels Play a Role in CAMP-Stimulated Neuritogenesis in Mouse Neuroblastoma N2A Cells. J. Cell Physiol. 2011, 226, 1090–1098. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, P.; Su, H.; Cai, L.; Zhao, L.; Zhou, H. Bioinformatics Analysis of Neuroblastoma MiRNA Based on GEO Data. Pharmgenomics Pers. Med. 2021, 14, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Wienke, J.; Dierselhuis, M.P.; Tytgat, G.A.M.; Künkele, A.; Nierkens, S.; Molenaar, J.J. The Immune Landscape of Neuroblastoma: Challenges and Opportunities for Novel Therapeutic Strategies in Pediatric Oncology. Eur. J. Cancer 2021, 144, 123–150. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the Immune System in Cancer: From Tumor Initiation to Metastatic Progression. Genes. Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell Cycle Control in Cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Barnes, A.P.; Polleux, F. Establishment of Axon-Dendrite Polarity in Developing Neurons. Annu. Rev. Neurosci. 2009, 32, 347–381. [Google Scholar] [CrossRef]
- Focke, P.J.; Wang, X.; Larsson, H.P. Neurotransmitter Transporters: Structure Meets Function. Structure 2013, 21, 694–705. [Google Scholar] [CrossRef]
- Venkatesh, H.; Monje, M. Neuronal Activity in Ontogeny and Oncology. Trends Cancer 2017, 3, 89–112. [Google Scholar] [CrossRef] [PubMed]
- Amano, T.; Richelson, E.; Nirenberg, M. Neurotransmitter Synthesis by Neuroblastoma Clones. Proc. Natl. Acad. Sci. USA 1972, 69, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Tonini, G.P. Growth, Progression and Chromosome Instability of Neuroblastoma: A New Scenario of Tumorigenesis? BMC Cancer 2017, 17, 20. [Google Scholar] [CrossRef]
- Jallepalli, P.V.; Lengauer, C. Chromosome Segregation and Cancer: Cutting through the Mystery. Nat. Rev. Cancer 2001, 1, 109–117. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.A.; Lund, S.P.; Pine, P.S.; Binder, H.; Clevert, D.-A.; Conesa, A.; Dopazo, J.; Fasold, M.; Hochreiter, S.; Hong, H.; et al. Assessing Technical Performance in Differential Gene Expression Experiments with External Spike-in RNA Control Ratio Mixtures. Nat. Commun. 2014, 5, 5125. [Google Scholar] [CrossRef]
- SEQC/MAQC-III Consortium. A Comprehensive Assessment of RNA-Seq Accuracy, Reproducibility and Information Content by the Sequencing Quality Control Consortium. Nat. Biotechnol. 2014, 32, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gong, B.; Bushel, P.R.; Thierry-Mieg, J.; Thierry-Mieg, D.; Xu, J.; Fang, H.; Hong, H.; Shen, J.; Su, Z.; et al. The Concordance between RNA-Seq and Microarray Data Depends on Chemical Treatment and Transcript Abundance. Nat. Biotechnol. 2014, 32, 926–932. [Google Scholar] [CrossRef]
- Edgar, R. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Vasudevan, S.A.; Nuchtern, J.G. Gene Profiling of High Risk Neuroblastoma. World J. Surg. 2005, 29, 317–324. [Google Scholar] [CrossRef]
- Stainczyk, S.A.; Westermann, F. Neuroblastoma—Telomere Maintenance, Deregulated Signaling Transduction and Beyond. Int. J. Cancer 2022, 150, 903–915. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Co-Expression Module | Gene Symbol | |||
|---|---|---|---|---|
| Brown module | ACHE | CHRNB4 | GRIK5 | L1CAM |
| ALCAM | CNTN1 | GRIN2A | NRXN1 | |
| ATP1A3 | CNTN2 | HCN4 | RIMS1 | |
| CACNG2 | GABRB3 | KCNJ12 | SCN1A | |
| CALM1 | GABRG2 | KCNJ3 | SLC8A2 | |
| CAMK2A | GABRG3 | KCNJ6 | SLC8A3 | |
| CAMK2B | GABRQ | KCNMA1 | SNAP25 | |
| CHRNA3 | GLRA1 | KCNN3 | STX1A | |
| CHRNA5 | GRIA4 | KCNQ2 | SYT1 | |
| CHRNA7 | GRIK2 | KCNQ3 | ||
| CHRNB2 | GRIK3 | KCNQ5 | ||
| Blue module | ANAPC11 | CDCA5 | LIG1 | POLD1 |
| AURKB | CDK1 | MAD2L1 | POLE | |
| BARD1 | CDT1 | MCM2 | POLE2 | |
| BLM | CENPE | MCM3 | PRIM1 | |
| BRCA1 | CHEK1 | MCM4 | PRIM2 | |
| BRIP1 | DBF4 | MCM5 | PTTG1 | |
| BUB1 | DNA2 | MCM6 | RBL1 | |
| BUB1B | E2F1 | MCM7 | RFC4 | |
| CCNA2 | ESCO2 | MYBL2 | RMI2 | |
| CCNB1 | ESPL1 | NDC80 | SGO1 | |
| CCNB2 | FBXO5 | ORC1 | SMC1A | |
| CDC20 | FEN1 | ORC6 | SMC3 | |
| CDC25A | KIF18A | PCNA | TERT | |
| CDC25C | KIF23 | PKMYT1 | TOPBP1 | |
| CDC45 | KIF2C | PLK1 | ||
| CDC6 | KNL1 | POLA1 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Pacheco, M.L.; Hernández-Lemus, E.; Mejía, C. Analysis of High-Risk Neuroblastoma Transcriptome Reveals Gene Co-Expression Signatures and Functional Features. Biology 2023, 12, 1230. https://doi.org/10.3390/biology12091230
Martínez-Pacheco ML, Hernández-Lemus E, Mejía C. Analysis of High-Risk Neuroblastoma Transcriptome Reveals Gene Co-Expression Signatures and Functional Features. Biology. 2023; 12(9):1230. https://doi.org/10.3390/biology12091230
Chicago/Turabian StyleMartínez-Pacheco, Mónica Leticia, Enrique Hernández-Lemus, and Carmen Mejía. 2023. "Analysis of High-Risk Neuroblastoma Transcriptome Reveals Gene Co-Expression Signatures and Functional Features" Biology 12, no. 9: 1230. https://doi.org/10.3390/biology12091230
APA StyleMartínez-Pacheco, M. L., Hernández-Lemus, E., & Mejía, C. (2023). Analysis of High-Risk Neuroblastoma Transcriptome Reveals Gene Co-Expression Signatures and Functional Features. Biology, 12(9), 1230. https://doi.org/10.3390/biology12091230