
Cell Cycle Control by Optogenetically Regulated Cell Cycle Inhibitor Protein p21

 ,
,  and
and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. The Cell Culture and Transfection
2.2. Plasmids
2.3. Transfection and Western Blot
2.4. Illumination
2.5. Flow Cytometry
2.6. SEAP Assay
2.7. Fixation of Cells for Microscopy
2.8. Live Cell Imaging
2.9. Statistical Analysis
3. Results
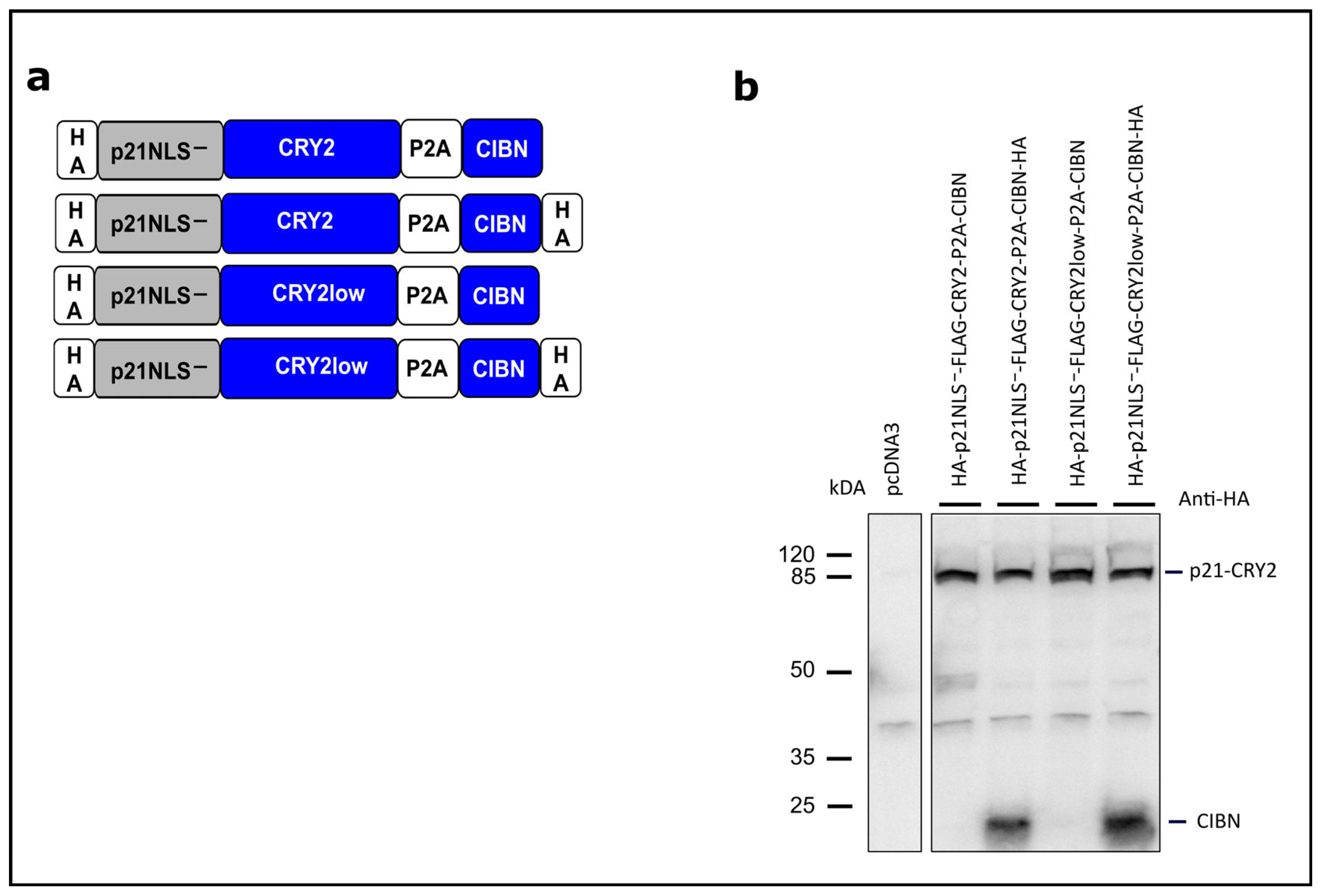
3.1. Light-Controlled p21 Applying a Bicistronic CRY2/CIBN System
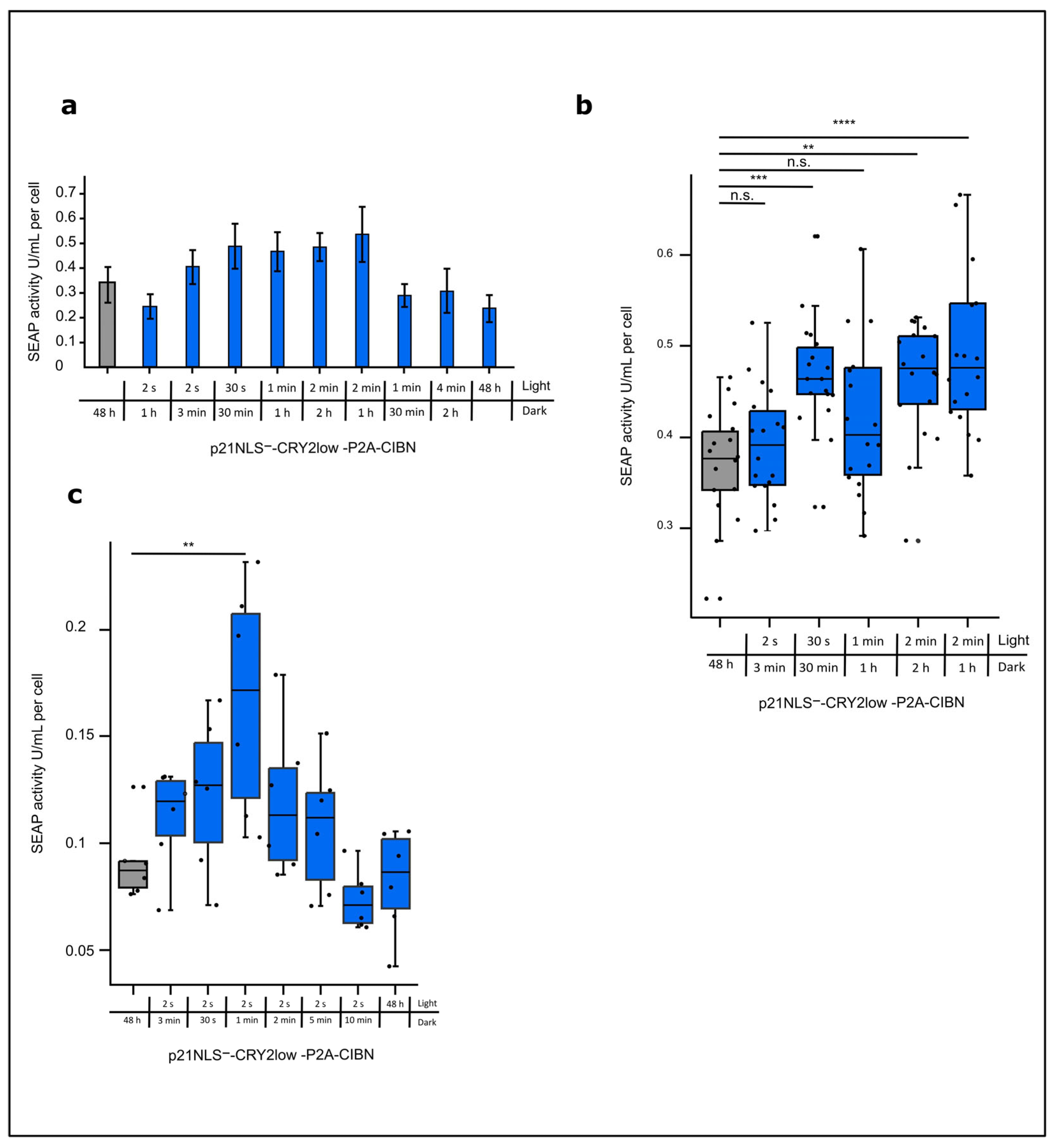
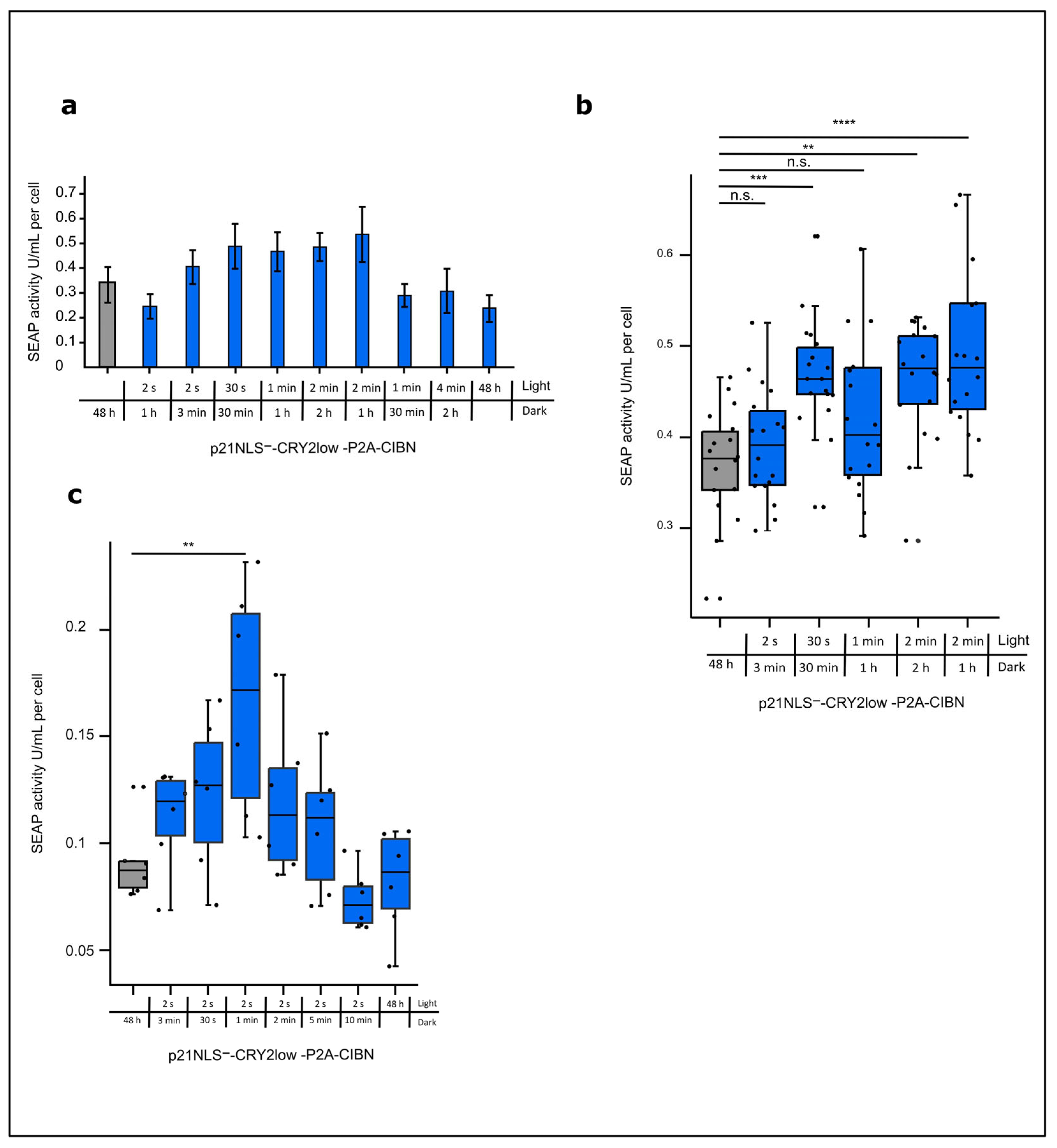
3.2. Functional Characterization of Light-Control p21
Optimizing the Light-Control of p21 by the CRY2/CIBN System
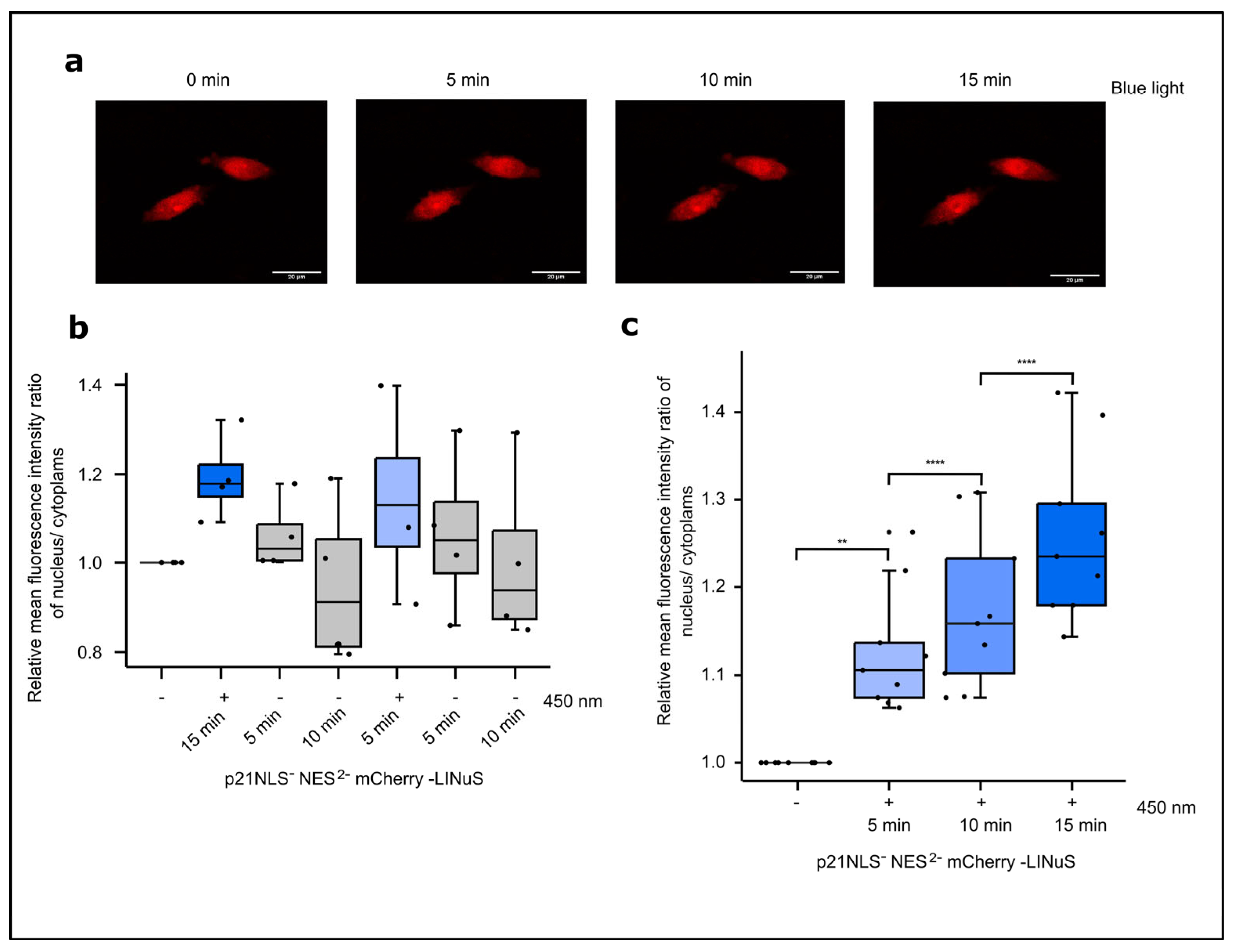
3.3. Light-Controlled p21 Applying the LINuS System
3.3.1. Light-Controlled G1 Cell Cycle Arrest by p21-LINuS
3.3.2. Light-Controlled Increase in Cell-Specific Productivity with p21-LINuS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Łukasik, P.; Załuski, M.; Gutowska, I. Cyclin-Dependent Kinases (CDK) and Their Role in Diseases Development-Review. Int. J. Mol. Sci. 2021, 22, 2935. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Schieke, S.M.; McCoy, J.P.; Finkel, T. Coordination of mitochondrial bioenergetics with G1 phase cell cycle progression. Cell Cycle 2008, 7, 1782–1787. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, J.S.; Dale, M.P.; Rosser, S.J. Decoupling Growth and Protein Production in CHO Cells: A Targeted Approach. Front. Bioeng. Biotechnol. 2021, 9, 658325. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.A.; Jeffrey, P.D.; Patten, A.K.; Massagué, J.; Pavletich, N.P. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature 1996, 382, 325–331. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Are p27 and p21 cytoplasmic oncoproteins? Cell Cycle 2002, 1, 391–393. [Google Scholar] [CrossRef]
- Dutto, I.; Tillhon, M.; Cazzalini, O.; Stivala, L.A.; Prosperi, E. Biology of the cell cycle inhibitor p21(CDKN1A): Molecular mechanisms and relevance in chemical toxicology. Arch. Toxicol. 2015, 89, 155–178. [Google Scholar] [CrossRef]
- Xiong, Y.; Hannon, G.J.; Zhang, H.; Casso, D.; Kobayashi, R.; Beach, D. p21 is a universal inhibitor of cyclin kinases. Nature 1993, 366, 701–704. [Google Scholar] [CrossRef]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef]
- Cmielová, J.; Rezáčová, M. p21Cip1/Waf1 protein and its function based on a subcellular localization corrected. J. Cell. Biochem. 2011, 112, 3502–3506. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Vilarrupla, A.; Díaz, C.; Canela, N.; Rahn, H.P.; Bachs, O.; Agell, N. Identification of the nuclear localization signal of p21(cip1) and consequences of its mutation on cell proliferation. FEBS Lett. 2002, 531, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.Y.; Kim, I.Y.; Kwon, K.-S. Cytoplasmic localization and ubiquitination of p21(Cip1) by reactive oxygen species. Biochem. Biophys. Res. Commun. 2007, 358, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.Y.; Lee, C.; Kwon, K.-S. Extracellular signal-regulated kinase 2-dependent phosphorylation induces cytoplasmic localization and degradation of p21Cip1. Mol. Cell. Biol. 2009, 29, 3379–3389. [Google Scholar] [CrossRef]
- Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Kreis, N.-N.; Louwen, F.; Yuan, J. The Multifaceted p21 (Cip1/Waf1/CDKN1A) in Cell Differentiation, Migration and Cancer Therapy. Cancers 2019, 11, 1220. [Google Scholar] [CrossRef]
- Fischer, A.A.M.; Kramer, M.M.; Radziwill, G.; Weber, W. Shedding light on current trends in molecular optogenetics. Curr. Opin. Chem. Biol. 2022, 70, 102196. [Google Scholar] [CrossRef]
- Zhu, H.; Nie, L.; Maki, C.G. Cdk2-dependent Inhibition of p21 stability via a C-terminal cyclin-binding motif. J. Biol. Chem. 2005, 280, 29282–29288. [Google Scholar] [CrossRef]
- Rössig, L.; Jadidi, A.S.; Urbich, C.; Badorff, C.; Zeiher, A.M.; Dimmeler, S. Akt-dependent phosphorylation of p21(Cip1) regulates PCNA binding and proliferation of endothelial cells. Mol. Cell. Biol. 2001, 21, 5644–5657. [Google Scholar] [CrossRef]
- Kramer, M.M.; Lataster, L.; Weber, W.; Radziwill, G. Optogenetic Approaches for the Spatiotemporal Control of Signal Transduction Pathways. Int. J. Mol. Sci. 2021, 22, 5300. [Google Scholar] [CrossRef]
- Kumar, S.; Khammash, M. Platforms for Optogenetic Stimulation and Feedback Control. Front. Bioeng. Biotechnol. 2022, 10, 918917. [Google Scholar] [CrossRef] [PubMed]
- Kolar, K.; Knobloch, C.; Stork, H.; Žnidarič, M.; Weber, W. OptoBase: A Web Platform for Molecular Optogenetics. ACS Synth. Biol. 2018, 7, 1825–1828. [Google Scholar] [CrossRef] [PubMed]
- Huala, E.; Oeller, P.W.; Liscum, E.; Han, I.S.; Larsen, E.; Briggs, W.R. Arabidopsis NPH1: A protein kinase with a putative redox-sensing domain. Science 1997, 278, 2120–2123. [Google Scholar] [CrossRef]
- Niopek, D.; Benzinger, D.; Roensch, J.; Draebing, T.; Wehler, P.; Eils, R.; Di Ventura, B. Engineering light-inducible nuclear localization signals for precise spatiotemporal control of protein dynamics in living cells. Nat.Commun. 2014, 5, 4404. [Google Scholar] [CrossRef]
- Niopek, D.; Wehler, P.; Roensch, J.; Eils, R.; Di Ventura, B. Optogenetic control of nuclear protein export. Nat. Commun. 2016, 7, 10624. [Google Scholar] [CrossRef]
- Kennedy, M.J.; Hughes, R.M.; Peteya, L.A.; Schwartz, J.W.; Ehlers, M.D.; Tucker, C.L. Rapid blue-light-mediated induction of protein interactions in living cells. Nat. Methods 2010, 7, 973–975. [Google Scholar] [CrossRef]
- Taslimi, A.; Vrana, J.D.; Chen, D.; Borinskaya, S.; Mayer, B.J.; Kennedy, M.J.; Tucker, C.L. An optimized optogenetic clustering tool for probing protein interaction and function. Nat. Commun. 2014, 5, 4925. [Google Scholar] [CrossRef]
- Duan, L.; Hope, J.; Ong, Q.; Lou, H.-Y.; Kim, N.; McCarthy, C.; Acero, V.; Lin, M.Z.; Cui, B. Understanding CRY2 interactions for optical control of intracellular signaling. Nat. Commun. 2017, 8, 547. [Google Scholar] [CrossRef] [PubMed]
- Wend, S.; Wagner, H.J.; Müller, K.; Zurbriggen, M.D.; Weber, W.; Radziwill, G. Optogenetic control of protein kinase activity in mammalian cells. ACS Synth.Biol. 2014, 3, 280–285. [Google Scholar] [CrossRef]
- Fischer, A.; Warscheid, B.; Weber, W.; Radziwill, G. Optogenetic clustering of CNK1 reveals mechanistic insights in RAF and AKT signalling controlling cell fate decisions. Sci.Rep. 2016, 6, 38155. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, S.-R.; Li, L.-H.; Park, H.-J.; Park, J.-H.; Lee, K.Y.; Kim, M.-K.; Shin, B.A.; Choi, S.-Y. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS ONE 2011, 6, e18556. [Google Scholar] [CrossRef] [PubMed]
- Beyer, H.M.; Gonschorek, P.; Samodelov, S.L.; Meier, M.; Weber, W.; Zurbriggen, M.D. AQUA Cloning: A Versatile and Simple Enzyme-Free Cloning Approach. PLoS ONE 2015, 10, e0137652. [Google Scholar] [CrossRef] [PubMed]
- Bugaj, L.J.; Lim, W.A. High-throughput multicolor optogenetics in microwell plates. Nat. Protoc. 2019, 14, 2205–2228. [Google Scholar] [CrossRef]
- Thomas, O.S.; Hörner, M.; Weber, W. A graphical user interface to design high-throughput optogenetic experiments with the optoPlate-96. Nat. Protoc. 2020, 15, 2785–2787. [Google Scholar] [CrossRef]
- Schlatter, S.; Rimann, M.; Kelm, J.; Fussenegger, M. SAMY, a novel mammalian reporter gene derived from Bacillus stearothermophilus alpha-amylase. Gene 2002, 282, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Wehler, P.; Niopek, D.; Eils, R.; Di Ventura, B. Optogenetic Control of Nuclear Protein Import in Living Cells Using Light-Inducible Nuclear Localization Signals (LINuS). Curr. Protoc. Chem.Biol. 2016, 8, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.R.; Eleftheriou, A. A comparison of the activity, sequence specificity, and CRM1-dependence of different nuclear export signals. Exp. Cell Res. 2000, 256, 213–224. [Google Scholar] [CrossRef]
- Bi, J.-X.; Shuttleworth, J.; Al-Rubeai, M. Uncoupling of cell growth and proliferation results in enhancement of productivity in p21CIP1-arrested CHO cells. Biotechnol. Bioeng. 2004, 85, 741–749. [Google Scholar] [CrossRef]
- Watanabe, S.; Shuttleworth, J.; Al-Rubeai, M. Regulation of cell cycle and productivity in NS0 cells by the over-expression of p21CIP1. Biotechnol. Bioeng. 2002, 77, 1–7. [Google Scholar] [CrossRef]
- Fussenegger, M.; Mazur, X.; Bailey, J.E. A novel cytostatic process enhances the productivity of Chinese hamster ovary cells. Biotechnol. Bioeng. 1997, 55, 927–939. [Google Scholar] [CrossRef]
- Farahani, P.E.; Reed, E.H.; Underhill, E.J.; Aoki, K.; Toettcher, J.E. Signaling, Deconstructed: Using Optogenetics to Dissect and Direct Information Flow in Biological Systems. Annu. Rev. Biomed.Eng. 2021, 23, 61–87. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lataster, L.; Huber, H.M.; Böttcher, C.; Föller, S.; Takors, R.; Radziwill, G. Cell Cycle Control by Optogenetically Regulated Cell Cycle Inhibitor Protein p21. Biology 2023, 12, 1194. https://doi.org/10.3390/biology12091194
Lataster L, Huber HM, Böttcher C, Föller S, Takors R, Radziwill G. Cell Cycle Control by Optogenetically Regulated Cell Cycle Inhibitor Protein p21. Biology. 2023; 12(9):1194. https://doi.org/10.3390/biology12091194
Chicago/Turabian StyleLataster, Levin, Hanna Mereth Huber, Christina Böttcher, Stefanie Föller, Ralf Takors, and Gerald Radziwill. 2023. "Cell Cycle Control by Optogenetically Regulated Cell Cycle Inhibitor Protein p21" Biology 12, no. 9: 1194. https://doi.org/10.3390/biology12091194
APA StyleLataster, L., Huber, H. M., Böttcher, C., Föller, S., Takors, R., & Radziwill, G. (2023). Cell Cycle Control by Optogenetically Regulated Cell Cycle Inhibitor Protein p21. Biology, 12(9), 1194. https://doi.org/10.3390/biology12091194