Medical Biology of Cancer-Associated Fibroblasts in Pancreatic Cancer


Abstract
Simple Summary
Abstract
1. Introduction
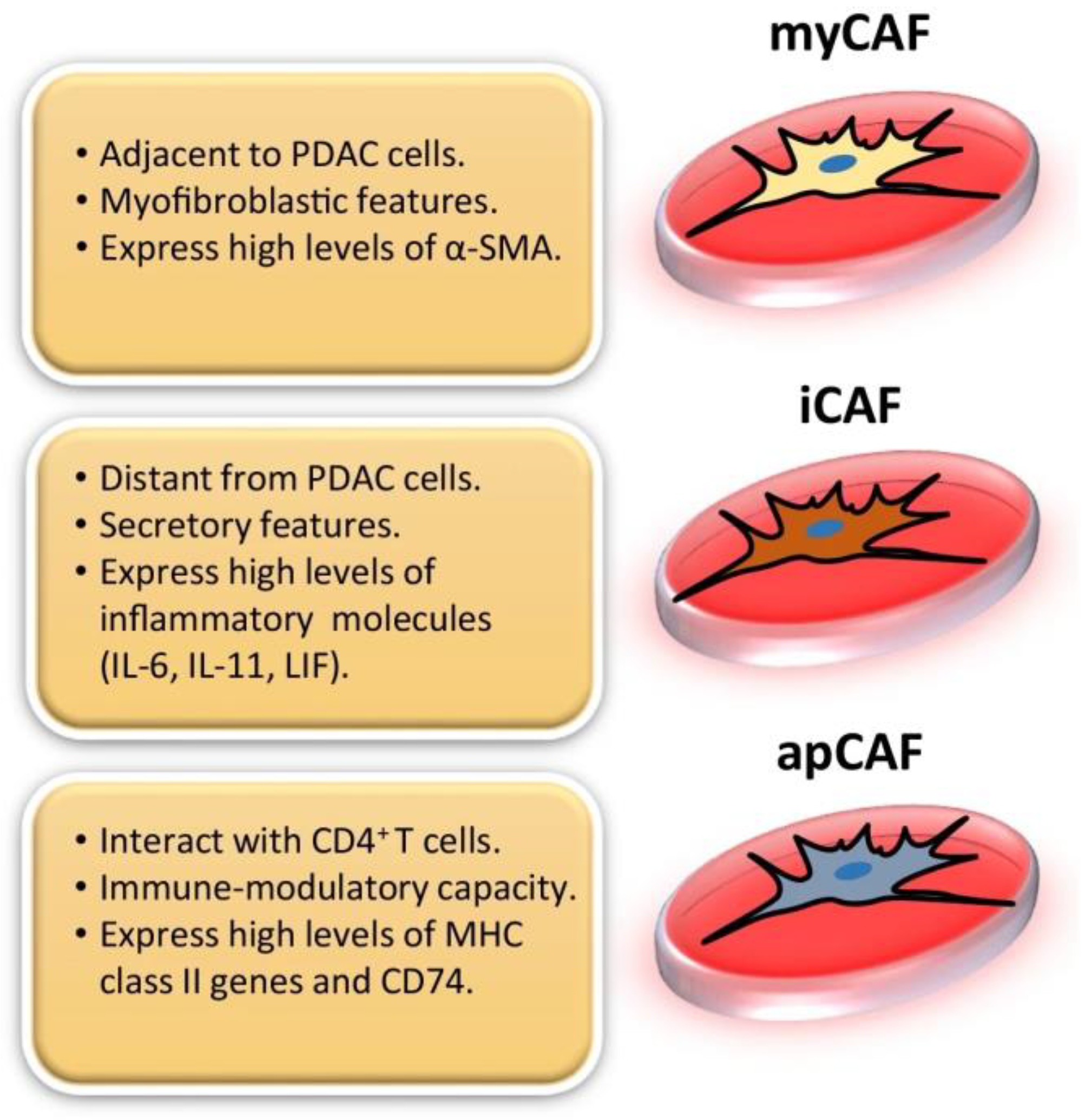
2. CAF and Fibroblast Heterogeneity
3. Cellular Origins of Pancreatic CAFs

4. Cell Surface Markers of CAFs
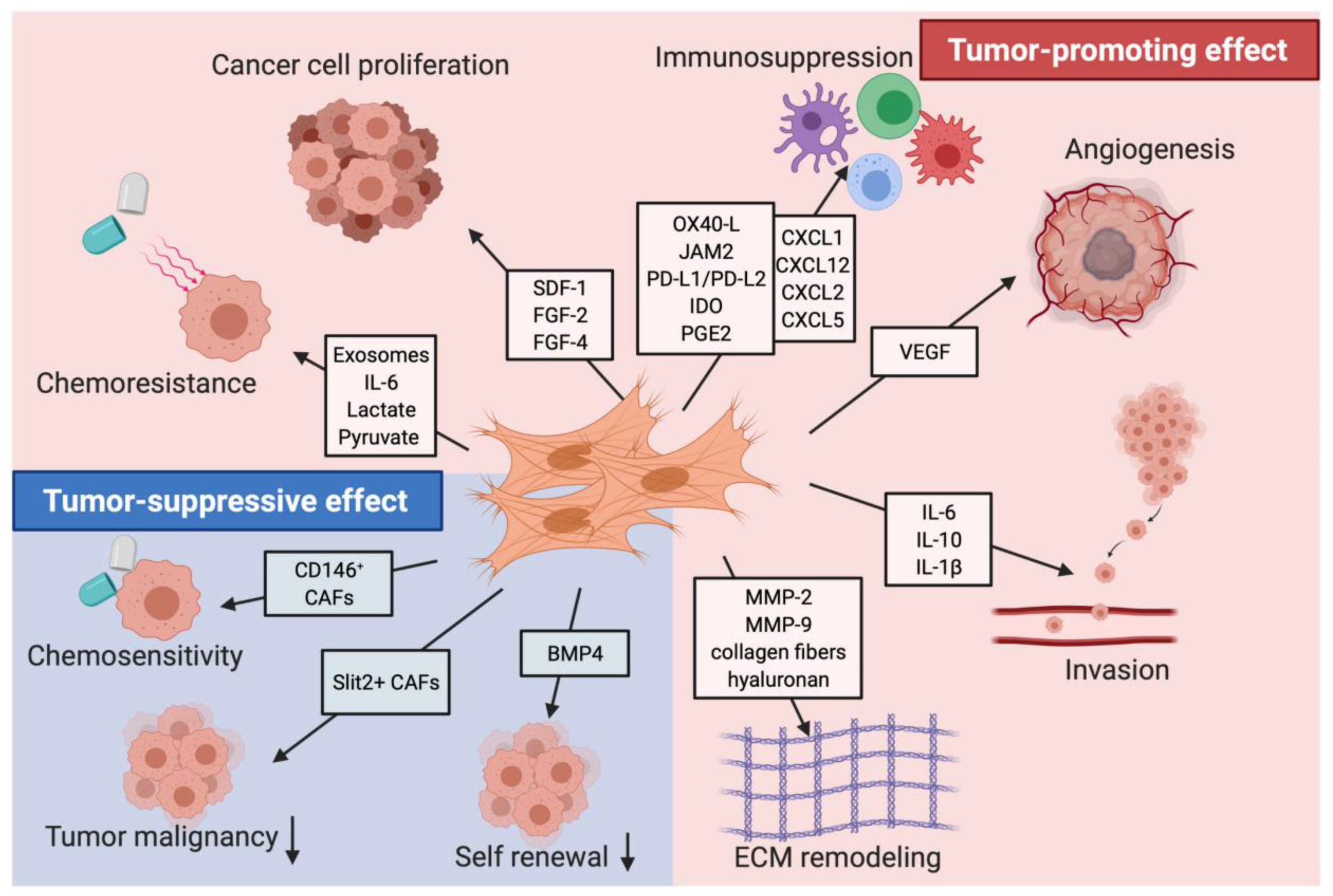
5. CAF Signaling Pathways
6. Immune Suppression of CAFs

7. CAFs as Potential Anti-PDAC Treatment
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cancer of the Pancreas—Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/pancreas.html (accessed on 15 April 2023).
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Becker, A.E.; Hernandez, Y.G.; Frucht, H.; Lucas, A.L. Pancreatic ductal adenocarcinoma: Risk factors, screening, and early detection. World J. Gastroenterol. 2014, 20, 11182–11198. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef] [PubMed]
- Ilic, M.; Ilic, I. Epidemiology of pancreatic cancer. World J. Gastroenterol. 2016, 22, 9694–9705. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Louault, K.; Li, R.R.; DeClerck, Y.A. Cancer-Associated Fibroblasts: Understanding Their Heterogeneity. Cancers 2020, 12, 3108. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Wei, W.F.; Wu, H.Z.; Fan, L.S.; Wang, W. Cancer-Associated Fibroblast Heterogeneity: A Factor That Cannot Be Ignored in Immune Microenvironment Remodeling. Front. Immunol. 2021, 12, 671595. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. myCAFs are better than yours: Targeting myofibroblasts potentiates immunotherapy. Trends Cancer 2023, 9, 1–2. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Wu, Y.; Fang, K.; Sweeney, S.; Roesner, U.K.; Parrish, M.; Patel, K.; Walter, T.; Piermattei, J.; Trimboli, A.; et al. The splanchnic mesenchyme is the tissue of origin for pancreatic fibroblasts during homeostasis and tumorigenesis. Nat. Commun. 2023, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.S.; Januszyk, M.; Delitto, D.; Yost, K.E.; Griffin, M.; Guo, J.; Guardino, N.; Delitto, A.E.; Chinta, M.; Burcham, A.R.; et al. Multiomic analysis reveals conservation of cancer-associated fibroblast phenotypes across species and tissue of origin. Cancer Cell 2022, 40, 1392–1406.e1397. [Google Scholar] [CrossRef] [PubMed]
- Manoukian, P.; Bijlsma, M.; van Laarhoven, H. The Cellular Origins of Cancer-Associated Fibroblasts and Their Opposing Contributions to Pancreatic Cancer Growth. Front. Cell Dev. Biol. 2021, 9, 743907. [Google Scholar] [CrossRef] [PubMed]
- Barrera, L.N.; Ridley, P.M.; Bermejo-Rodriguez, C.; Costello, E.; Perez-Mancera, P.A. The role of microRNAs in the modulation of cancer-associated fibroblasts activity during pancreatic cancer pathogenesis. J. Physiol. Biochem. 2023, 79, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Chen, H.; Zhao, L.; Hu, J.; Yang, W.; Li, G.; Cheng, C.; Zhao, Z.; Zhang, T.; Li, L.; et al. Cancer-Associated Fibroblast (CAF) Heterogeneity and Targeting Therapy of CAFs in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 655152. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Huang, H.; Wang, Z.; Zhang, Y.; Pradhan, R.N.; Ganguly, D.; Chandra, R.; Murimwa, G.; Wright, S.; Gu, X.; Maddipati, R.; et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell 2022, 40, 656–673.e657. [Google Scholar] [CrossRef] [PubMed]
- Vaish, U.; Jain, T.; Are, A.C.; Dudeja, V. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma: An Update on Heterogeneity and Therapeutic Targeting. Int. J. Mol. Sci. 2021, 22, 13408. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef]
- González-Silva, L.; Quevedo, L.; Varela, I. Tumor Functional Heterogeneity Unraveled by scRNA-seq Technologies. Trends Cancer 2020, 6, 13–19. [Google Scholar] [CrossRef]
- Shinkawa, T.; Ohuchida, K.; Nakamura, M. Heterogeneity of Cancer-Associated Fibroblasts and the Tumor Immune Microenvironment in Pancreatic Cancer. Cancers 2022, 14, 3994. [Google Scholar] [CrossRef]
- Chijimatsu, R.; Kobayashi, S.; Takeda, Y.; Kitakaze, M.; Tatekawa, S.; Arao, Y.; Nakayama, M.; Tachibana, N.; Saito, T.; Ennishi, D.; et al. Establishment of a reference single-cell RNA sequencing dataset for human pancreatic adenocarcinoma. iScience 2022, 25, 104659. [Google Scholar] [CrossRef]
- Hosein, A.N.; Huang, H.; Wang, Z.; Parmar, K.; Du, W.; Huang, J.; Maitra, A.; Olson, E.; Verma, U.; Brekken, R.A. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Hu, B.; Wu, C.; Mao, H.; Gu, H.; Dong, H.; Yan, J.; Qi, Z.; Yuan, L.; Dong, Q.; Long, J. Subpopulations of cancer-associated fibroblasts link the prognosis and metabolic features of pancreatic ductal adenocarcinoma. Ann. Transl. Med. 2022, 10, 262. [Google Scholar] [CrossRef] [PubMed]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; Diprete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef]
- Grapa, C.M.; Mocan, T.; Gonciar, D.; Zdrehus, C.; Mosteanu, O.; Pop, T.; Mocan, L. Epidermal Growth Factor Receptor and Its Role in Pancreatic Cancer Treatment Mediated by Nanoparticles. Int. J. Nanomed. 2019, 14, 9693–9706. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef] [PubMed]
- Bachem, M.G.; Schünemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Omary, M.B.; Lugea, A.; Lowe, A.W.; Pandol, S.J. The pancreatic stellate cell: A star on the rise in pancreatic diseases. J. Clin. Investig. 2007, 117, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Sunami, Y.; Rebelo, A.; Kleeff, J. Lipid Metabolism and Lipid Droplets in Pancreatic Cancer and Stellate Cells. Cancers 2017, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Adjuto-Saccone, M.; Soubeyran, P.; Garcia, J.; Audebert, S.; Camoin, L.; Rubis, M.; Roques, J.; Binétruy, B.; Iovanna, J.L.; Tournaire, R. TNF-α induces endothelial-mesenchymal transition promoting stromal development of pancreatic adenocarcinoma. Cell Death Dis. 2021, 12, 649. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef]
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. (Landmark Ed.) 2010, 15, 166–179. [Google Scholar] [CrossRef]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef]
- Rosty, C.; Ueki, T.; Argani, P.; Jansen, M.; Yeo, C.J.; Cameron, J.L.; Hruban, R.H.; Goggins, M. Overexpression of S100A4 in pancreatic ductal adenocarcinomas is associated with poor differentiation and DNA hypomethylation. Am. J. Pathol. 2002, 160, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Bughda, R.; Dimou, P.; D’Souza, R.R.; Klampatsa, A. Fibroblast Activation Protein (FAP)-Targeted CAR-T Cells: Launching an Attack on Tumor Stroma. Immunotargets Ther. 2021, 10, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.N.; Magiera, L.; Kraman, M.; Fearon, D.T. Tumoral immune suppression by macrophages expressing fibroblast activation protein-α and heme oxygenase-1. Cancer Immunol. Res. 2014, 2, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.W.; Deonarine, A.; Jones, J.O.; Denton, A.E.; Feig, C.; Lyons, S.K.; Espeli, M.; Kraman, M.; McKenna, B.; Wells, R.J.; et al. Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J. Exp. Med. 2013, 210, 1137–1151. [Google Scholar] [CrossRef]
- Griffin, M.F.; Talbott, H.E.; Guardino, N.J.; Guo, J.L.; Spielman, A.F.; Chen, K.; Parker, J.B.L.; Mascharak, S.; Henn, D.; Liang, N.; et al. Piezo inhibition prevents and rescues scarring by targeting the adipocyte to fibroblast transition. bioRxiv 2023. [Google Scholar] [CrossRef]
- Jotzu, C.; Alt, E.; Welte, G.; Li, J.; Hennessy, B.T.; Devarajan, E.; Krishnappa, S.; Pinilla, S.; Droll, L.; Song, Y.H. Adipose tissue derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor derived factors. Cell. Oncol. 2011, 34, 55–67. [Google Scholar] [CrossRef]
- Kidd, S.; Spaeth, E.; Watson, K.; Burks, J.; Lu, H.; Klopp, A.; Andreeff, M.; Marini, F.C. Origins of the tumor microenvironment: Quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS ONE 2012, 7, e30563. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Agorku, D.J.; Langhammer, A.; Heider, U.; Wild, S.; Bosio, A.; Hardt, O. CD49b, CD87, and CD95 Are Markers for Activated Cancer-Associated Fibroblasts Whereas CD39 Marks Quiescent Normal Fibroblasts in Murine Tumor Models. Front. Oncol. 2019, 9, 716. [Google Scholar] [CrossRef]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Pahler, J.; Bergers, G.; Hanahan, D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008, 5, e19. [Google Scholar] [CrossRef]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2020, 146, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Helms, E.; Onate, M.K.; Sherman, M.H. Fibroblast Heterogeneity in the Pancreatic Tumor Microenvironment. Cancer Discov. 2020, 10, 648–656. [Google Scholar] [CrossRef]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther. 2021, 6, 218. [Google Scholar] [CrossRef]
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Löhr, M.; Schmidt, C.; Ringel, J.; Kluth, M.; Müller, P.; Nizze, H.; Jesnowski, R. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001, 61, 550–555. [Google Scholar]
- Aoyagi, Y.; Oda, T.; Kinoshita, T.; Nakahashi, C.; Hasebe, T.; Ohkohchi, N.; Ochiai, A. Overexpression of TGF-beta by infiltrated granulocytes correlates with the expression of collagen mRNA in pancreatic cancer. Br. J. Cancer 2004, 91, 1316–1326. [Google Scholar] [CrossRef]
- Heldin, C.H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal. 2013, 11, 97. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.; Khan, A.W.; Kim, M.S.; Choi, S. The Role of Fibroblast Growth Factor (FGF) Signaling in Tissue Repair and Regeneration. Cells 2021, 10, 3242. [Google Scholar] [CrossRef]
- Lam, A.P.; Gottardi, C.J. β-catenin signaling: A novel mediator of fibrosis and potential therapeutic target. Curr. Opin. Rheumatol. 2011, 23, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Ogana, H.; Gang, E.; Kim, H.N.; Kim, Y.M. Wnt Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2021, 1270, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2023–2037. [Google Scholar] [CrossRef]
- Diehl, J.N.; Hibshman, P.S.; Ozkan-Dagliyan, I.; Goodwin, C.M.; Howard, S.V.; Cox, A.D.; Der, C.J. Targeting the ERK mitogen-activated protein kinase cascade for the treatment of KRAS-mutant pancreatic cancer. Adv. Cancer Res. 2022, 153, 101–130. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Bugg, D.; Ghearing, N.; Dorn, L.E.; Kim, P.; Sargent, M.A.; Gunaje, J.; Otsu, K.; Davis, J. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation 2017, 136, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Roux, D.; Lenormand, P.; Dowd, S.; Keyse, S.; Pouysségur, J. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J. 1999, 18, 664–674. [Google Scholar] [CrossRef]
- Drosten, M.; Barbacid, M. Targeting the MAPK Pathway in KRAS-Driven Tumors. Cancer Cell 2020, 37, 543–550. [Google Scholar] [CrossRef]
- Pei, L.; Liu, Y.; Liu, L.; Gao, S.; Gao, X.; Feng, Y.; Sun, Z.; Zhang, Y.; Wang, C. Roles of cancer-associated fibroblasts (CAFs) in anti- PD-1/PD-L1 immunotherapy for solid cancers. Mol. Cancer 2023, 22, 29. [Google Scholar] [CrossRef]
- Gok Yavuz, B.; Gunaydin, G.; Gedik, M.E.; Kosemehmetoglu, K.; Karakoc, D.; Ozgur, F.; Guc, D. Cancer associated fibroblasts sculpt tumour microenvironment by recruiting monocytes and inducing immunosuppressive PD-1. Sci. Rep. 2019, 9, 3172. [Google Scholar] [CrossRef] [PubMed]
- Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Flint, T.R.; Janowitz, T.; Connell, C.M.; Roberts, E.W.; Denton, A.E.; Coll, A.P.; Jodrell, D.I.; Fearon, D.T. Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-tumor Immunity. Cell Metab. 2016, 24, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.; Elkord, E. FoxP3. Cancer Lett. 2020, 490, 174–185. [Google Scholar] [CrossRef]
- Mhaidly, R.; Mechta-Grigoriou, F. Role of cancer-associated fibroblast subpopulations in immune infiltration, as a new means of treatment in cancer. Immunol. Rev. 2021, 302, 259–272. [Google Scholar] [CrossRef]
- Iida, T.; Mizutani, Y.; Esaki, N.; Ponik, S.M.; Burkel, B.M.; Weng, L.; Kuwata, K.; Masamune, A.; Ishihara, S.; Haga, H.; et al. Pharmacologic conversion of cancer-associated fibroblasts from a protumor phenotype to an antitumor phenotype improves the sensitivity of pancreatic cancer to chemotherapeutics. Oncogene 2022, 41, 2764–2777. [Google Scholar] [CrossRef]
- Gunaydin, G. CAFs Interacting With TAMs in Tumor Microenvironment to Enhance Tumorigenesis and Immune Evasion. Front. Oncol. 2021, 11, 668349. [Google Scholar] [CrossRef]
- Ohshio, Y.; Teramoto, K.; Hanaoka, J.; Tezuka, N.; Itoh, Y.; Asai, T.; Daigo, Y.; Ogasawara, K. Cancer-associated fibroblast-targeted strategy enhances antitumor immune responses in dendritic cell-based vaccine. Cancer Sci. 2015, 106, 134–142. [Google Scholar] [CrossRef]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef]
- Norton, J.; Foster, D.; Chinta, M.; Titan, A.; Longaker, M. Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment. Cancers 2020, 12, 1347. [Google Scholar] [CrossRef]
- Huang, T.X.; Guan, X.Y.; Fu, L. Therapeutic targeting of the crosstalk between cancer-associated fibroblasts and cancer stem cells. Am. J. Cancer Res. 2019, 9, 1889–1904. [Google Scholar] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Borriello, L.; Nakata, R.; Sheard, M.A.; Fernandez, G.E.; Sposto, R.; Malvar, J.; Blavier, L.; Shimada, H.; Asgharzadeh, S.; Seeger, R.C.; et al. Cancer-Associated Fibroblasts Share Characteristics and Protumorigenic Activity with Mesenchymal Stromal Cells. Cancer Res. 2017, 77, 5142–5157. [Google Scholar] [CrossRef]
- Nicolas, A.M.; Pesic, M.; Engel, E.; Ziegler, P.K.; Diefenhardt, M.; Kennel, K.B.; Buettner, F.; Conche, C.; Petrocelli, V.; Elwakeel, E.; et al. Inflammatory fibroblasts mediate resistance to neoadjuvant therapy in rectal cancer. Cancer Cell 2022, 40, 168–184.e113. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, M.; Diefenhardt, M.; Nicolas, A.M.; Rödel, F.; Ghadimi, M.; Hofheinz, R.D.; Greten, F.R.; Rödel, C.; Fokas, E.; Group, G.R.C.S. ACO/ARO/AIO-21—Capecitabine-based chemoradiotherapy in combination with the IL-1 receptor antagonist anakinra for rectal cancer Patients: A phase I trial of the German rectal cancer study group. Clin. Transl. Radiat. Oncol. 2022, 34, 99–106. [Google Scholar] [CrossRef]
- Dominguez, C.X.; Müller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Almhanna, K. Pancreatic cancer and immune checkpoint inhibitors-still a long way to go. Transl. Gastroenterol. Hepatol. 2021, 6, 6. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| CAF Subtypes | Markers | Functions | References |
|---|---|---|---|
| myCAFs | αSMA, Col1a1, TAGLN, MYL9, TPM1, TPM2, MMP11, POSTN | ECM remodeling, Tumor invasion, proliferation, and metastasis | [10,14,23,29,30] |
| iCAFs | IL-6, IL-8, CXCL1, CXCL2, CXCL12, PDGFRα, CFD, CCL2, | Immune suppression, Chemoresistance | [11,14,15,17,23] |
| apCAFs | MHC II, CD74 | Antigen presentation, Immune modulation | [15,17,22,23] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgan, A.; Griffin, M.; Kameni, L.; Wan, D.C.; Longaker, M.T.; Norton, J.A. Medical Biology of Cancer-Associated Fibroblasts in Pancreatic Cancer. Biology 2023, 12, 1044. https://doi.org/10.3390/biology12081044
Morgan A, Griffin M, Kameni L, Wan DC, Longaker MT, Norton JA. Medical Biology of Cancer-Associated Fibroblasts in Pancreatic Cancer. Biology. 2023; 12(8):1044. https://doi.org/10.3390/biology12081044
Chicago/Turabian StyleMorgan, Annah, Michelle Griffin, Lionel Kameni, Derrick C. Wan, Michael T. Longaker, and Jeffrey A. Norton. 2023. "Medical Biology of Cancer-Associated Fibroblasts in Pancreatic Cancer" Biology 12, no. 8: 1044. https://doi.org/10.3390/biology12081044
APA StyleMorgan, A., Griffin, M., Kameni, L., Wan, D. C., Longaker, M. T., & Norton, J. A. (2023). Medical Biology of Cancer-Associated Fibroblasts in Pancreatic Cancer. Biology, 12(8), 1044. https://doi.org/10.3390/biology12081044