
Parathyroid Hormone Related Protein (PTHrP)-Associated Molecular Signatures in Tissue Differentiation and Non-Tumoral Diseases

 ,
,  and
and 
Simple Summary
Abstract
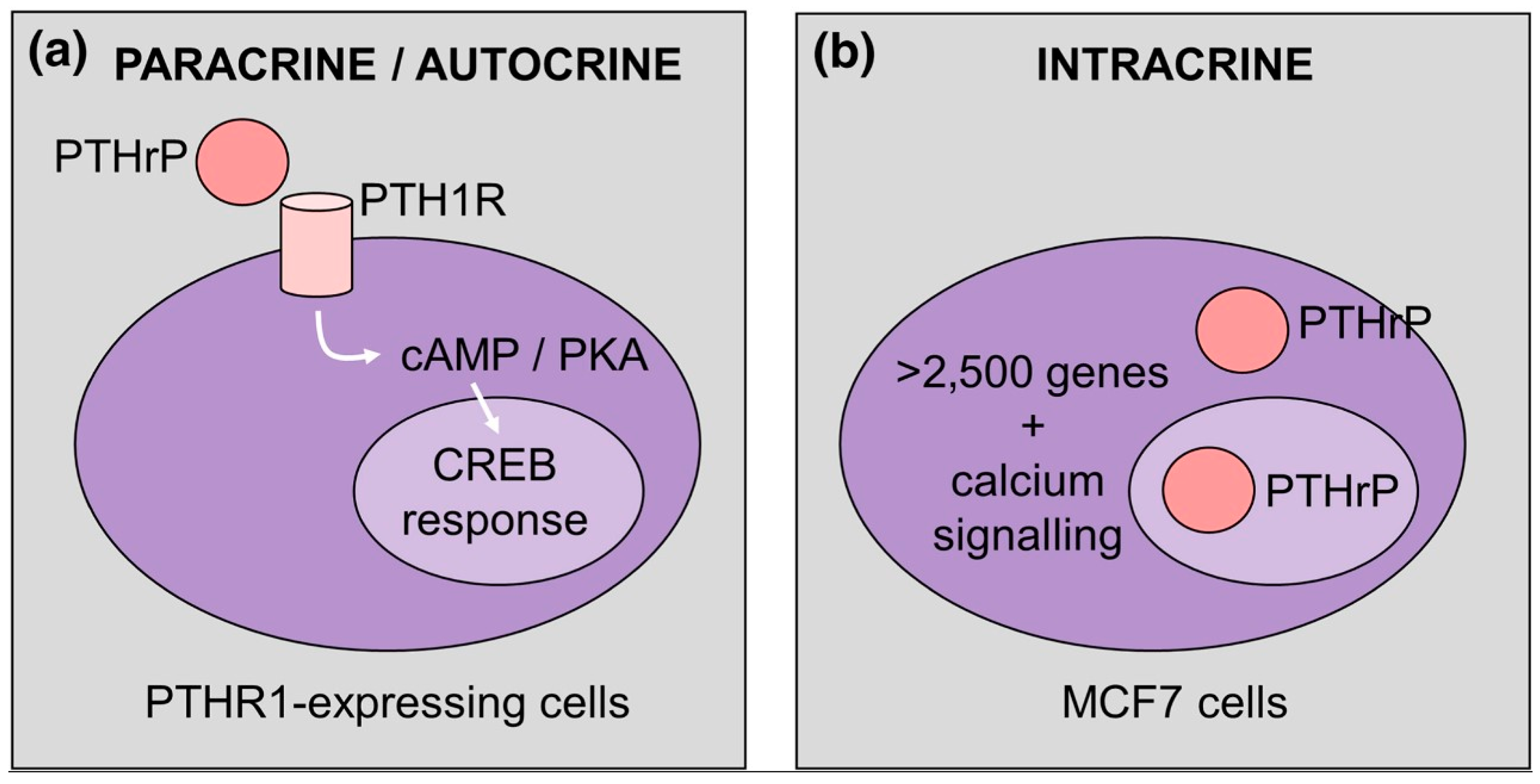
1. A Brief Note about Parathyroid Hormone-Related Protein (PTHrP) Structure and Function
2. Adipose Tissue: PTHrP-Related Signatures in Adipogenesis and Transdifferentiation
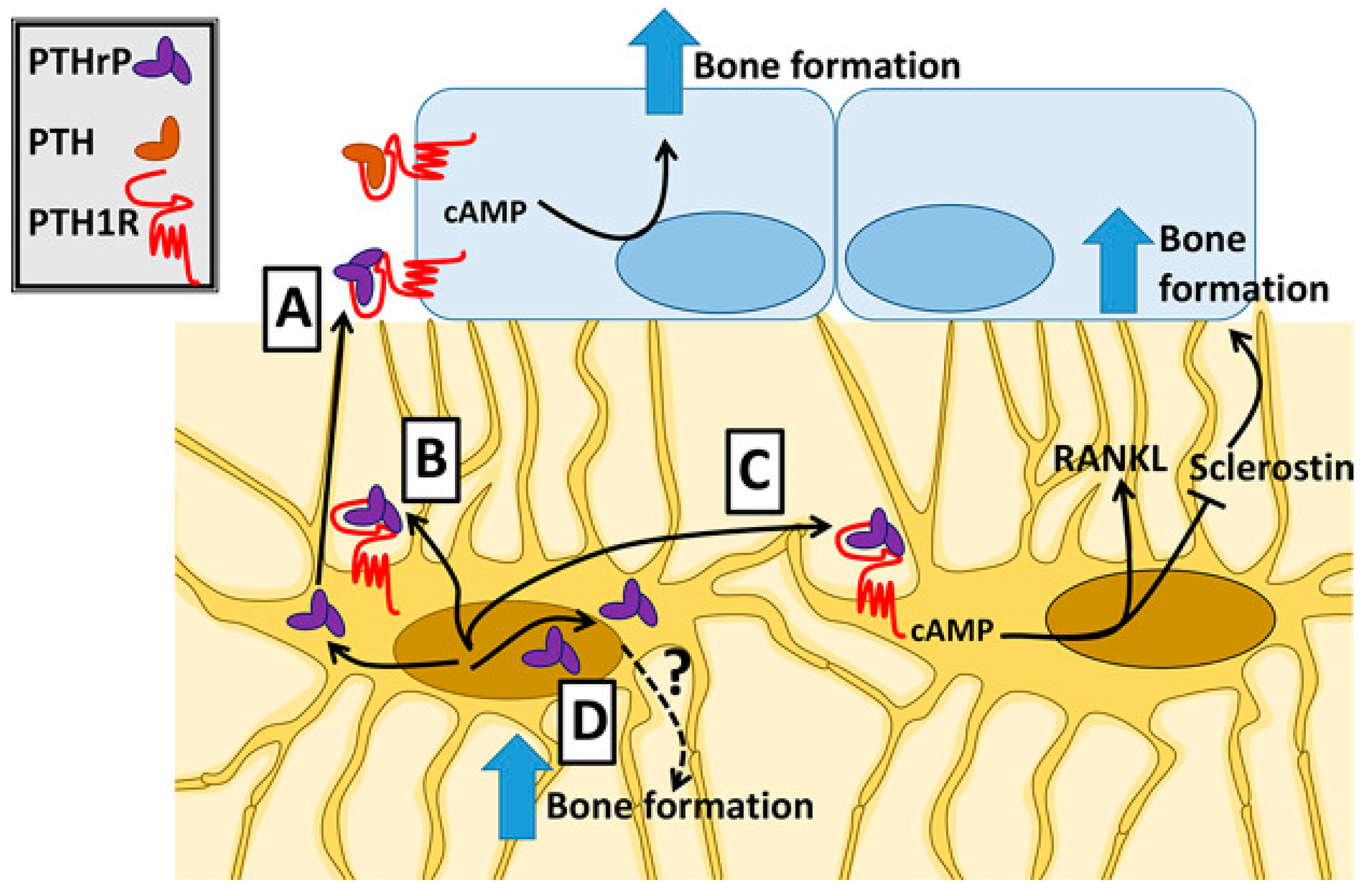
3. Bone and Dental Tissues: PTHrP-Related Signatures in Osteoblatogenesis, Osteoclastogenesis, and Ossification
4. Cartilaginous Tissue: PTHrP-Related Signatures in the Repression of Chondrocyte Differentiation
5. Intestinal Epithelium: PTHrP-Related Signatures in Calcium Uptake by Enterocytes
6. Liver Parenchyma: PTHrP-Related Signatures in the Fibrotic Reaction of Hepatic Stellate Cells
7. Lung Epithelium: PTHrP-Related Signatures in Pulmonary Surfactant Production
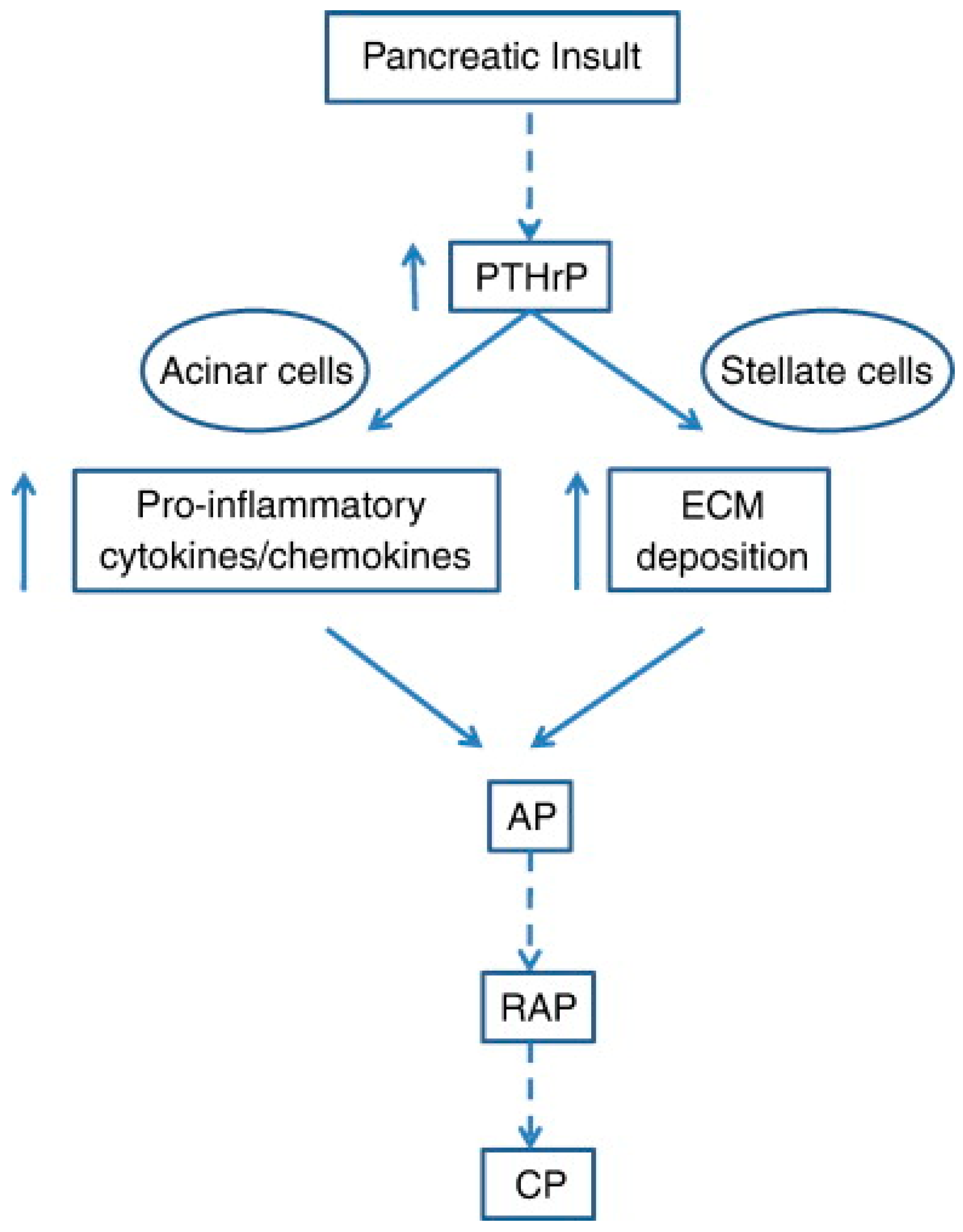
8. Exocrine and Endocrine Pancreas: PTHrP-Related Signatures in β-Cell Biology and in the Onset of Pancreatitis
9. Renal Parenchyma: PTHrP-Related Signatures in the Promotion of Kidney Cell Survival and in Diabetes-Related Changes
10. Skin: PTHrP-Related Signatures in Keratinocyte Development and Antiaging Effect
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luparello, C. Parathyroid Hormone-Related Protein (PTHrP): A Key Regulator of Life/Death Decisions by Tumor Cells with Potential Clinical Applications. Cancers 2011, 3, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Longo, A.; Librizzi, M.; Naselli, F.; Caradonna, F.; Tobiasch, E.; Luparello, C. PTHrP in Differentiating Human Mesenchymal Stem Cells: Transcript Isoform Expression, Promoter Methylation, and Protein Accumulation. Biochimie 2013, 95, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.J.; Johnson, R.W. Multiple actions of parathyroid hormone-related protein in breast cancer bone metastasis. Br. J. Pharmacol. 2021, 178, 1923–1935. [Google Scholar] [CrossRef]
- Martin, T.J. PTH1R Actions on Bone Using the cAMP/Protein Kinase A Pathway. Front Endocrinol. 2022, 12, 833221. [Google Scholar] [CrossRef]
- Suva, L.J.; Friedman, P.A. Structural pharmacology of PTH and PTHrP. Vitam. Horm. 2022, 120, 1–21. [Google Scholar]
- Luparello, C.; Librizzi, M. Parathyroid hormone-related protein (PTHrP)-dependent modulation of gene expression signatures in cancer cells. Vitam. Horm. 2022, 120, 179–214. [Google Scholar]
- Lagathu, C.; Christodoulides, C.; Tan, C.Y.; Virtue, S.; Laudes, M.; Campbell, M.; Ishikawa, K.; Ortega, F.; Tinahones, F.J.; Fernández-Real, J.-M.; et al. Secreted Frizzled-Related Protein 1 Regulates Adipose Tissue Expansion and Is Dysregulated in Severe Obesity. Int. J. Obes. 2010, 34, 1695–1705. [Google Scholar] [CrossRef]
- Roca-Rodríguez, M.M.; el Bekay, R.; Garrido-Sanchez, L.; Gómez-Serrano, M.; Coin-Aragüez, L.; Oliva-Olivera, W.; Lhamyani, S.; Clemente-Postigo, M.; García-Santos, E.; de Luna Diaz, R.; et al. Parathyroid Hormone-Related Protein, Human Adipose-Derived Stem Cells Adipogenic Capacity and Healthy Obesity. J. Clin. Endocrinol. Metab. 2015, 100, E826–E835. [Google Scholar] [CrossRef]
- Chan, G.K.; Miao, D.; Deckelbaum, R.; Bolivar, I.; Karaplis, A.; Goltzman, D. Parathyroid Hormone-Related Peptide Interacts with Bone Morphogenetic Protein 2 to Increase Osteoblastogenesis and Decrease Adipogenesis in Pluripotent C3H10T½ Mesenchymal Cells. Endocrinology 2003, 144, 5511–5520. [Google Scholar] [CrossRef]
- Zhang, X.; Cheng, Q.; Wang, Y.; Leung, P.S.; Mak, K.K. Hedgehog Signaling in Bone Regulates Whole-Body Energy Metabolism through a Bone–Adipose Endocrine Relay Mediated by PTHrP and Adiponectin. Cell Death Differ. 2017, 24, 225–237. [Google Scholar] [CrossRef]
- Combs, T.P.; Berg, A.H.; Obici, S.; Scherer, P.E.; Rossetti, L. Endogenous Glucose Production Is Inhibited by the Adipose-Derived Protein Acrp30. J. Clin. Investig. 2001, 108, 1875–1881. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Ito, Y.; Tsuchida, A.; Yokomizo, T.; Kita, S.; Sugiyama, T.; Miyagishi, M.; Hara, K.; Tsunoda, M.; et al. Cloning of Adiponectin Receptors That Mediate Antidiabetic Metabolic Effects. Nature 2003, 423, 762–769. [Google Scholar] [CrossRef]
- Kir, S.; White, J.P.; Kleiner, S.; Kazak, L.; Cohen, P.; Baracos, V.E.; Spiegelman, B.M. Tumour-Derived PTH-Related Protein Triggers Adipose Tissue Browning and Cancer Cachexia. Nature 2014, 513, 100–104. [Google Scholar] [CrossRef]
- De Jesus, L.A.; Carvalho, S.D.; Ribeiro, M.O.; Schneider, M.; Kim, S.-W.; Harney, J.W.; Larsen, P.R.; Bianco, A.C. The Type 2 Iodothyronine Deiodinase Is Essential for Adaptive Thermogenesis in Brown Adipose Tissue. J. Clin. Investig. 2001, 108, 1379–1385. [Google Scholar] [CrossRef]
- Qin, B.; Qincao, L.; He, S.; Liao, Y.; Shi, J.; Xie, F.; Diao, N.; Bai, L. Parathyroid Hormone-Related Protein Prevents High-Fat-Diet-Induced Obesity, Hepatic Steatosis and Insulin Resistance in Mice. Endocr. J. 2022, 69, 55–65. [Google Scholar] [CrossRef]
- Tezze, C.; Romanello, V.; Sandri, M. FGF21 as Modulator of Metabolism in Health and Disease. Front. Physiol. 2019, 10, 419. [Google Scholar] [CrossRef]
- De Miguel, F.; Martinez-Fernandez, P.; Guillen, C.; Valin, A.; Rodrigo, A.; Martinez, M.E.; Esbrit, P. Parathyroid hormone-related protein (107–139) stimulates interleukin-6 expression in human osteoblastic cells. J. Am. Soc. Nephrol. 1999, 10, 796–803. [Google Scholar] [CrossRef]
- Esbrit, P.; Alvarez-Arroyo, M.V.; DE Miguel, F.; Martin, O.; Martinez, M.E.; Caramelo, C. C-terminal parathyroid hormone-related protein increases vascular endothelial growth factor in human osteoblastic cells. J. Am. Soc. Nephrol. 2000, 11, 1085–1092. [Google Scholar] [CrossRef]
- Alonso, V.; de Gortázar, A.R.; Ardura, J.A.; Andrade-Zapata, I.; Alvarez-Arroyo, M.V.; Esbrit, P. Parathyroid hormone-related protein (107–139) increases human osteoblastic cell survival by activation of vascular endothelial growth factor receptor-2. J. Cell. Physiol. 2008, 217, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Lozano, D.; Sánchez-Salcedo, S.; Portal-Núñez, S.; Vila, M.; López-Herradón, A.; Ardura, J.A.; Mulero, F.; Gómez-Barrena, E.; Vallet-Regí, M.; Esbrit, P. Parathyroid hormone-related protein (107–111) improves the bone regeneration potential of gelatin-glutaraldehyde biopolymer-coated hydroxyapatite. Acta Biomater. 2014, 10, 3307–3316. [Google Scholar] [CrossRef]
- Niida, A.; Hiroko, T.; Kasai, M.; Furukawa, Y.; Nakamura, Y.; Suzuki, Y.; Sugano, S.; Akiyama, T. DKK1, a negative regulator of Wnt signaling, is a target of the β-catenin/TCF pathway. Oncogene 2004, 23, 8520–8526. [Google Scholar] [CrossRef] [PubMed]
- Semënov, M.; Tamai, K.; He, X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J. Biol. Chem. 2005, 80, 26770–26775. [Google Scholar] [CrossRef] [PubMed]
- Rutkovskiy, A.; Stensløkken, K.O.; Vaage, I.J. Osteoblast Differentiation at a Glance. Med. Sci. Monit. Basic Res. 2016, 22, 95–106. [Google Scholar] [CrossRef]
- Fukushima, H.; Jimi, E.; Kajiya, H.; Motokawa, W.; Okabe, K. Parathyroid-hormone-related protein induces expression of receptor activator of NF-κB ligand in human periodontal ligament cells via a cAMP/protein kinase A-independent pathway. J. Dent. Res. 2005, 84, 329–334. [Google Scholar] [CrossRef]
- Sun, H.; Li, Q.; Zhang, Y.; Bi, Y.; Li, X.; Shu, Y.; Chen, X.; Jin, Z.; Ge, C. Regulation of OPG and RANKL expressed by human dental follicle cells in osteoclastogenesis. Cell Tissue Res. 2015, 362, 399–405. [Google Scholar] [CrossRef]
- Mak, K.K.; Bi, Y.; Wan, C.; Chuang, P.T.; Clemens, T.; Young, M.; Yang, Y. Hedgehog signaling in mature osteoblasts regulates bone formation and resorption by controlling PTHrP and RANKL expression. Dev. Cell 2008, 14, 674–688. [Google Scholar] [CrossRef]
- Ricarte, F.R.; Le Henaff, C.; Kolupaeva, V.G.; Gardella, T.J.; Partridge, N.C. Parathyroid hormone (1–34) and its analogs differentially modulate osteoblastic Rankl expression via PKA/SIK2/SIK3 and PP1/PP2A-CRTC3 signaling. J. Biol. Chem. 2018, 293, 20200–20213. [Google Scholar] [CrossRef]
- Elango, J.; Rahman, S.U.; Henrotin, Y.; de Val, J.E.M.S.; Bao, B.; Wang, S.; Li, B.; Wu, W. Parathyroid Hormone-Related Protein (PTHrP) Accelerates Soluble RANKL Signals for Downregulation of Osteogenesis of Bone Mesenchymal Stem Cells. J. Clin. Med. 2019, 8, 836. [Google Scholar] [CrossRef]
- Kim, H.J.; Minashima, T.; McCarthy, E.F.; Winkles, J.A.; Kirsch, T. Progressive ankylosis protein (ANK) in osteoblasts and osteoclasts controls bone formation and bone remodeling. J. Bone Min. Res. 2010, 25, 1771–1783. [Google Scholar] [CrossRef]
- Kamel, S.A.; Yee, J.A. Continuous and intermittent exposure of neonatal rat calvarial cells to PTHrP (1–36) inhibits bone nodule mineralization in vitro by downregulating bone sialoprotein expression via the cAMP signaling pathway. F1000Research 2013, 2, 77. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, X.; Wang, C.; Ding, C.; Lin, H.; Liu, A.; Wang, L.; Cao, Y. Exogenous PTHrP Repairs the Damaged Fracture Healing of PTHrP+/− Mice and Accelerates Fracture Healing of Wild Mice. Int. J. Mol. Sci. 2017, 18, 337. [Google Scholar] [CrossRef]
- Zhu, M.; Zhang, J.; Dong, Z.; Zhang, Y.; Wang, R.; Karaplis, A.; Goltzman, D.; Miao, D. The p27 Pathway Modulates the Regulation of Skeletal Growth and Osteoblastic Bone Formation by Parathyroid Hormone-Related Peptide. J. Bone Min. Res. 2015, 30, 1969–1979. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, G.; Gu, Z.; Sun, H.; Karaplis, A.; Goltzman, D.; Miao, D. DNA damage checkpoint pathway modulates the regulation of skeletal growth and osteoblastic bone formation by parathyroid hormone-related peptide. Int. J. Biol. Sci. 2018, 14, 508–517. [Google Scholar] [CrossRef]
- Martín-Guerrero, E.; Tirado-Cabrera, I.; Buendía, I.; Alonso, V.; Gortázar, A.R.; Ardura, J.A. Primary cilia mediate parathyroid hormone receptor type 1 osteogenic actions in osteocytes and osteoblasts via Gli activation. J. Cell. Physiol. 2020, 235, 7356–7369. [Google Scholar] [CrossRef]
- Platas, J.; Guillén, M.I.; Gomar, F.; Castejón, M.A.; Esbrit, P.; Alcaraz, M.J. Anti-senescence and Anti-inflammatory Effects of the C-terminal Moiety of PTHrP Peptides in OA Osteoblasts. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 624–631. [Google Scholar] [CrossRef]
- Ardura, J.A.; Portal-Núñez, S.; Castelbón-Calvo, I.; Martínez de Toda, I.; De la Fuente, M.; Esbrit, P. Parathyroid Hormone-Related Protein Protects Osteoblastic Cells From Oxidative Stress by Activation of MKP1 Phosphatase. J. Cell. Physiol. 2017, 232, 785–796. [Google Scholar] [CrossRef]
- Onan, D.; Allan, E.H.; Quinn, J.M.; Gooi, J.H.; Pompolo, S.; Sims, N.A.; Gillespie, M.T.; Martin, T.J. The chemokine Cxcl1 is a novel target gene of parathyroid hormone (PTH)/PTH-related protein in committed osteoblasts. Endocrinology 2009, 150, 2244–2253. [Google Scholar] [CrossRef]
- Portal-Núñez, S.; Lozano, D.; de Castro, L.F.; de Gortázar, A.R.; Nogués, X.; Esbrit, P. Alterations of the Wnt/beta-catenin pathway and its target genes for the N- and C-terminal domains of parathyroid hormone-related protein in bone from diabetic mice. FEBS Lett. 2010, 584, 3095–3100. [Google Scholar] [CrossRef]
- Maycas, M.; McAndrews, K.A.; Sato, A.Y.; Pellegrini, G.G.; Brown, D.M.; Allen, M.R.; Plotkin, L.I.; Gortazar, A.R.; Esbrit, P.; Bellido, T. PTHrP-Derived Peptides Restore Bone Mass and Strength in Diabetic Mice: Additive Effect of Mechanical Loading. J. Bone Min. Res. 2017, 32, 486–497. [Google Scholar] [CrossRef]
- Makino, A.; Takagi, H.; Takahashi, Y.; Hase, N.; Sugiyama, H.; Yamana, K.; Kobayashi, T. Abaloparatide Exerts Bone Anabolic Effects with Less Stimulation of Bone Resorption-Related Factors: A Comparison with Teriparatide. Calcif. Tissue Int. 2018, 103, 289–297. [Google Scholar] [CrossRef]
- Ansari, N.; Ho, P.W.; Crimeen-Irwin, B.; Poulton, I.J.; Brunt, A.R.; Forwood, M.R.; Divieti Pajevic, P.; Gooi, J.H.; Martin, T.J.; Sims, N.A. Autocrine and Paracrine Regulation of the Murine Skeleton by Osteocyte-Derived Parathyroid Hormone-Related Protein. J. Bone Min. Res. 2018, 33, 137–153. [Google Scholar] [CrossRef]
- Pieles, O.; Reck, A.; Morsczeck, C. High endogenous expression of parathyroid hormone-related protein (PTHrP) supports osteogenic differentiation in human dental follicle cells. Histochem. Cell Biol. 2020, 154, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Nagata, M.; Gupta, A.; Matsushita, Y.; Yamaguchi, T.; Mizuhashi, K.; Maki, K.; Ruellas, A.C.; Cevidanes, L.S.; Kronenberg, H.M.; et al. Autocrine regulation of mesenchymal progenitor cell fates orchestrates tooth eruption. Proc. Natl. Acad. Sci. USA 2019, 116, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Amizuka, N.; Warshawsky, H.; Henderson, J.E.; Goltzman, D.; Karaplis, A.C. Parathyroid hormone-related peptide-depleted mice show abnormal epiphyseal cartilage development and altered endochondral bone formation. J. Cell Biol. 1994, 126, 1611–1623. [Google Scholar] [CrossRef]
- Carey, D.E.; Liu, X. Expression of bone morphogenetic protein-6 messenger RNA in bovine growth plate chondrocytes of different size. J. Bone Min. Res. 1995, 10, 401–405. [Google Scholar] [CrossRef]
- Lanske, B.; Karaplis, A.C.; Lee, K.; Luz, A.; Vortkamp, A.; Pirro, A.; Karperien, M.; Defize, L.H.; Ho, C.; Mulligan, R.C.; et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science 1996, 273, 663–666. [Google Scholar] [CrossRef]
- Ito, H.; Akiyama, H.; Shigeno, C.; Nakamura, T. Parathyroid hormone-related peptide inhibits the expression of bone morphogenetic protein-4 mRNA through a cyclic AMP/protein kinase A pathway in mouse clonal chondrogenic EC cells, ATDC5. Biochim. Biophys. Acta 2000, 1497, 237–243. [Google Scholar] [CrossRef]
- Li, T.F.; Dong, Y.; Ionescu, A.M.; Rosier, R.N.; Zuscik, M.J.; Schwarz, E.M.; O’Keefe, R.J.; Drissi, H. Parathyroid hormone-related peptide (PTHrP) inhibits Runx2 expression through the PKA signaling pathway. Exp. Cell Res. 2004, 299, 128–136. [Google Scholar] [CrossRef]
- Kozhemyakina, E.; Cohen, T.; Yao, T.P.; Lassar, A.B. Parathyroid hormone-related peptide represses chondrocyte hypertrophy through a protein phosphatase 2A/histone deacetylase 4/MEF2 pathway. Mol. Cell. Biol. 2009, 29, 5751–5762. [Google Scholar] [CrossRef]
- Guo, B.; Wang, S.T.; Duan, C.C.; Li, D.D.; Tian, X.C.; Wang, Q.Y.; Yue, Z.P. Effects of PTHrP on chondrocytes of sika deer antler. Cell Tissue Res. 2013, 354, 451–460. [Google Scholar] [CrossRef]
- Wang, S.T.; Gao, Y.J.; Duan, C.C.; Li, D.D.; Tian, X.C.; Zhang, Q.L.; Guo, B.; Yue, Z.P. Effects of PTHrP on expression of MMP9 and MMP13 in sika deer antler chondrocytes. Cell Biol. Int. 2013, 37, 1300–1307. [Google Scholar] [CrossRef]
- Pelosi, M.; Lazzarano, S.; Thoms, B.L.; Murphy, C.L. Parathyroid hormone-related protein is induced by hypoxia and promotes expression of the differentiated phenotype of human articular chondrocytes. Clin. Sci. 2013, 125, 461–470. [Google Scholar] [CrossRef]
- Browe, D.C.; Coleman, C.M.; Barry, F.P.; Elliman, S.J. Hypoxia Activates the PTHrP-MEF2C Pathway to Attenuate Hypertrophy in Mesenchymal Stem Cell Derived Cartilage. Sci. Rep. 2019, 9, 13274. [Google Scholar] [CrossRef]
- Fischer, J.; Aulmann, A.; Dexheimer, V.; Grossner, T.; Richter, W. Intermittent PTHrP(1–34) exposure augments chondrogenesis and reduces hypertrophy of mesenchymal stromal cells. Stem Cells Dev. 2014, 23, 2513–2523. [Google Scholar] [CrossRef]
- Karaplis, A.C.; Luz, A.; Glowacki, J.; Bronson, R.T.; Tybulewicz, V.L.; Kronenberg, H.M.; Mulligan, R.C. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 1994, 8, 277–289. [Google Scholar] [CrossRef]
- Nishimori, S.; Lai, F.; Shiraishi, M.; Kobayashi, T.; Kozhemyakina, E.; Yao, T.P.; Lassar, A.B.; Kronenberg, H.M. PTHrP targets HDAC4 and HDAC5 to repress chondrocyte hypertrophy. JCI Insight 2019, 4, e97903. [Google Scholar] [CrossRef]
- Zhou, L.X.; Nemere, I.; Norman, A.W. A parathyroid-related peptide induces transcaltachia (the rapid, hormonal stimulation of intestinal Ca2+ transport). Biochem. Biophys. Res. Commun. 1992, 186, 69–73. [Google Scholar] [CrossRef]
- Larsson, D.; Nemere, I. Vectorial Transcellular Calcium Transport in Intestine: Integration of Current Models. J. Biomed. Biotechnol. 2002, 2, 117–119. [Google Scholar] [CrossRef]
- Beggs, M.R.; Alexander, R.T. Intestinal absorption and renal reabsorption of calcium throughout postnatal development. Exp. Biol. Med. (Maywood) 2017, 242, 840–849. [Google Scholar] [CrossRef]
- Liu, C.; Shao, G.; Lu, Y.; Xue, M.; Liang, F.; Zhang, Z.; Bai, L. Parathyroid Hormone-Related Protein (1–40) Enhances Calcium Uptake in Rat Enterocytes Through PTHR1 Receptor and Protein Kinase Cα/β Signaling. Cell Physiol. Biochem. 2018, 51, 1695–1709. [Google Scholar] [CrossRef]
- He, S.; Xue, M.; Liu, C.; Xie, F.; Bai, L. Parathyroid Hormone-Like Hormone Induces Epithelial-to-Mesenchymal Transition of Intestinal Epithelial Cells by Activating the Runt-Related Transcription Factor 2. Am. J. Pathol. 2018, 188, 1374–1388. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, Y.; Sun, B. The Molecular Mechanisms of Liver Fibrosis and Its Potential Therapy in Application. Int. J. Mol. Sci. 2022, 23, 12572. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.F.; Liu, C.P.; Li, L.X.; Xue, M.M.; Xie, F.; Guo, Y.; Bai, L. Activated effects of parathyroid hormone-related protein on human hepatic stellate cells. PLoS ONE 2013, 8, e76517. [Google Scholar] [CrossRef] [PubMed]
- Gardi, C.; Arezzini, B.; Fortino, V.; Comporti, M. Effect of free iron on collagen synthesis, cell proliferation and MMP-2 expression in rat hepatic stellate cells. Biochem. Pharm. 2002, 64, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Tang, J.; Diao, N.; Liao, Y.; Shi, J.; Xu, X.; Xie, F.; Bai, L. Parathyroid hormone-related protein activates HSCs via hedgehog signalling during liver fibrosis development. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1984–1994. [Google Scholar] [CrossRef]
- Torday, J.S.; Rehan, V.K. The evolutionary continuum from lung development to homeostasis and repair. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L608–L611. [Google Scholar] [CrossRef]
- Torday, J.S.; Rehan, V.K. Stretch-stimulated surfactant synthesis is coordinated by the paracrine actions of PTHrP and leptin. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 283, L130–L135. [Google Scholar] [CrossRef]
- Torday, J.; Rehan, V. Neutral lipid trafficking regulates alveolar type II cell surfactant phospholipid and surfactant protein expression. Exp. Lung Res. 2011, 37, 376–386. [Google Scholar] [CrossRef]
- Oruqaj, L.; Forst, S.; Schreckenberg, R.; Inserte, J.; Poncelas, M.; Bañeras, J.; Garcia-Dorado, D.; Rohrbach, S.; Schlüter, K.D. Effect of high fat diet on pulmonary expression of parathyroid hormone-related protein and its downstream targets. Heliyon 2016, 2, e00182. [Google Scholar] [CrossRef]
- Rehan, V.K.; Sakurai, R.; Wang, Y.; Santos, J.; Huynh, K.; Torday, J.S. Reversal of nicotine-induced alveolar lipofibroblast-to-myofibroblast transdifferentiation by stimulants of parathyroid hormone-related protein signaling. Lung 2007, 185, 151–159. [Google Scholar] [CrossRef]
- Sawada, Y.; Zhang, B.; Okajima, F.; Izumi, T.; Takeuchi, T. PTHrP increases pancreatic β-cell-specific functions in well-differentiated cells. Mol. Cell Endocrinol. 2001, 182, 265–275. [Google Scholar] [CrossRef]
- Sawada, Y.; Kameya, T.; Aizama, T.; Izumi, T.; Takeuchi, T. Proprotein-Processing Endoprotease Furin and its Substrate Parathyroid Hormone-Related Protein are Coexpressed in Insulinoma Cells. Endocr. Pathol. 2000, 11, 31–39. [Google Scholar] [CrossRef]
- Guthalu Kondegowda, N.; Joshi-Gokhale, S.; Harb, G.; Williams, K.; Zhang, X.Y.; Takane, K.K.; Zhang, P.; Scott, D.K.; Stewart, A.F.; Garcia-Ocaña, A.; et al. Parathyroid hormone-related protein enhances human ß-cell proliferation and function with associated induction of cyclin-dependent kinase 2 and cyclin E expression. Diabetes 2010, 59, 3131–3138. [Google Scholar] [CrossRef]
- Williams, K.; Abanquah, D.; Joshi-Gokhale, S.; Otero, A.; Lin, H.; Guthalu, N.K.; Zhang, X.; Mozar, A.; Bisello, A.; Stewart, A.F.; et al. Systemic and acute administration of parathyroid hormone-related peptide(1–36) stimulates endogenous beta cell proliferation while preserving function in adult mice. Diabetologia 2011, 54, 2867–2877. [Google Scholar] [CrossRef]
- Bhatia, V.; Kim, S.O.; Aronson, J.F.; Chao, C.; Hellmich, M.R.; Falzon, M. Role of parathyroid hormone-related protein in the pro-inflammatory and pro-fibrogenic response associated with acute pancreatitis. Regul. Pept. 2012, 175, 49–60, Erratum in: Regul. Pept. 2014, 192–193, 59. [Google Scholar] [CrossRef]
- Sadr-Azodi, O.; Oskarsson, V.; Discacciati, A.; Videhult, P.; Askling, J.; Ekbom, A. Pancreatic Cancer Following Acute Pancreatitis: A Population-based Matched Cohort Study. Am. J. Gastroenterol. 2018, 113, 1711–1719. [Google Scholar] [CrossRef]
- Rastellini, C.; Han, S.; Bhatia, V.; Cao, Y.; Liu, K.; Gao, X.; Ko, T.C.; Greeley, G.H., Jr.; Falzon, M. Induction of chronic pancreatitis by pancreatic duct ligation activates BMP2, apelin, and PTHrP expression in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G554–G565. [Google Scholar] [CrossRef][Green Version]
- Gao, X.; Cao, Y.; Yang, W.; Duan, C.; Aronson, J.F.; Rastellini, C.; Chao, C.; Hellmich, M.R.; Ko, T.C. BMP2 inhibits TGF-β-induced pancreatic stellate cell activation and extracellular matrix formation. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G804–G813. [Google Scholar] [CrossRef]
- Han, S.; Englander, E.W.; Gomez, G.A.; Aronson, J.F.; Rastellini, C.; Garofalo, R.P.; Kolli, D.; Quertermous, T.; Kundu, R.; Greeley, G.H., Jr. Pancreatitis activates pancreatic apelin-APJ axis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G139–G150. [Google Scholar] [CrossRef]
- Morrell, N.W.; Archer, S.L.; Defelice, A.; Evans, S.; Fiszman, M.; Martin, T.; Saulnier, M.; Rabinovitch, M.; Schermuly, R.; Stewart, D.; et al. Anticipated classes of new medications and molecular targets for pulmonary arterial hypertension. Pulm. Circ. 2013, 3, 226–244. [Google Scholar] [CrossRef]
- Okoumassoun, L.E.; Russo, C.; Denizeau, F.; Averill-Bates, D.; Henderson, D.J.E. Parathyroid hormone-related protein (PTHrP) inhibits mitochondrial-dependent apoptosis through CK2. J. Cell. Physiol. 2007, 212, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Hanif, I.M.; Hanif, I.M.; Shazib, M.A.; Ahmad, K.A.; Pervaiz, S. Casein Kinase II: An attractive target for anti-cancer drug design. Int. J. Biochem. Cell Biol. 2010, 42, 1602–1605. [Google Scholar] [CrossRef] [PubMed]
- Ardura, J.A.; Rayego-Mateos, S.; Rámila, D.; Ruiz-Ortega, M.; Esbrit, P. Parathyroid Hormone–Related Protein Promotes Epithelial–Mesenchymal Transition. J. Am. Soc. Nephrol. 2010, 21, 237–248. [Google Scholar] [CrossRef]
- Hua, K.; Li, Y.; Zhou, H.; Hu, X.; Chen, Y.; He, R.; Luo, R.; Zhou, R.; Bi, D.; Jin, H. Haemophilus parasuis Infection Disrupts Adherens Junctions and Initializes EMT Dependent on Canonical Wnt/β-Catenin Signaling Pathway. Front. Cell. Infect. Microbiol. 2018, 8, 324. [Google Scholar] [CrossRef] [PubMed]
- Ardura, J.A.; Sanz, A.B.; Ortiz, A.; Esbrit, P. Parathyroid hormone–related protein protects renal tubuloepithelial cells from apoptosis by activating transcription factor Runx2. Kidney Int. 2013, 83, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Schneider, R.K. Parathyroid hormone-related protein and regulation of cell survival in the kidney. Kidney Int 2013, 83, 777–779. [Google Scholar] [CrossRef]
- Hochane, M.; Raison, D.; Coquard, C.; Béraud, C.; Bethry, A.; Danilin, S.; Massfelder, T.; Barthelmebs, M. Parathyroid hormone-related protein modulates inflammation in mouse mesangial cells and blunts apoptosis by enhancing COX-2 expression. Am. J. Physiol. Cell Physiol. 2018, 314, C242–C253. [Google Scholar] [CrossRef]
- Danilin, S.; Sourbier, C.; Thomas, L.; Rothhut, S.; Lindner, V.; Helwig, J.J.; Jacqmin, D.; Lang, H.; Massfelder, T. von Hippel-Lindau tumor suppressor gene-dependent mRNA stabilization of the survival factor parathyroid hormone-related protein in human renal cell carcinoma by the RNA-binding protein HuR. Carcinogenesis 2009, 30, 387–396. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Linker, R.A.; Deng, J.; Kaltschmidt, C. Cyclooxygenase-2 is a neuronal target gene of NF-kappaB. BMC Mol. Biol. 2002, 3, 16. [Google Scholar] [CrossRef]
- Cooper, M.E.; Vranes, D.; Youssef, S.; Stacker, S.A.; Cox, A.J.; Rizkalla, B.; Casley, D.J.; Bach, L.A.; Kelly, D.J.; Gilbert, R.E. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes 1999, 48, 2229–2239. [Google Scholar] [CrossRef]
- Burns, K.D. Angiotensin II and its receptors in the diabetic kidney. Am. J. Kidney Dis. 2000, 36, 449–467. [Google Scholar] [CrossRef]
- Izquierdo, A.; López-Luna, P.; Ortega, A.; Romero, M.; Guitiérrez-Tarrés, M.A.; Arribas, I.; Alvarez, M.J.; Esbrit, P.; Bosch, R.J. The parathyroid hormone-related protein system and diabetic nephropathy outcome in streptozotocin-induced diabetes. Kidney Int. 2006, 69, 2171–2177. [Google Scholar] [CrossRef]
- Esbrit, P.; Santos, S.; Ortega, A.; Fernández-Agulló, T.; Vélez, E.; Troya, S.; Garrido, P.; Peña, A.; Bover, J.; Bosch, R.J. Parathyroid hormone-related protein as a renal regulating factor. From vessels to glomeruli and tubular epithelium. Am. J. Nephrol. 2001, 21, 179–184. [Google Scholar] [CrossRef]
- Romero, M.; Ortega, A.; Izquierdo, A.; López-Luna, P.; Bosch, R.J. Parathyroid hormone-related protein induces hypertrophy in podocytes via TGF-beta(1) and p27(Kip1): Implications for diabetic nephropathy. Nephrol. Dial. Transpl. 2010, 25, 2447–2457. [Google Scholar] [CrossRef]
- Ortega, A.; Romero, M.; Izquierdo, A.; Troyano, N.; Arce, Y.; Ardura, J.A.; Arenas, M.I.; Bover, J.; Esbrit, P.; Bosch, R.J. Parathyroid hormone-related protein is a hypertrophy factor for human mesangial cells: Implications for diabetic nephropathy. J. Cell. Physiol. 2012, 227, 1980–1987. [Google Scholar] [CrossRef]
- Chen, H.M.; Dai, J.J.; Zhu, R.; Peng, F.F.; Wu, S.Z.; Yu, H.; Krepinsky, J.C.; Zhang, B.F. Parathyroid hormone-related protein induces fibronectin up-regulation in rat mesangial cells through reactive oxygen species/Src/EGFR signaling. Biosci. Rep. 2019, 39, BSR20182293. [Google Scholar] [CrossRef]
- Póvoa, G.; Diniz, L.M. Growth Hormone System: Skin interactions. Bras. Derm. 2011, 86, 1159–1165. [Google Scholar] [CrossRef]
- Shin, J.H.; Ji, C.; Casinghino, S.; McCarthy, T.L.; Centrella, M. Parathyroid hormone-related protein enhances insulin-like growth factor-I expression by fetal rat dermal fibroblasts. J. Biol. Chem. 1997, 272, 23498–23502. [Google Scholar] [CrossRef]
- Blomme, E.A.; Sugimoto, Y.; Lin, Y.C.; Capen, C.C.; Rosol, T.J. Parathyroid hormone-related protein is a positive regulator of keratinocyte growth factor expression by normal dermal fibroblasts. Mol. Cell. Endocrinol. 1999, 152, 189–197. [Google Scholar] [CrossRef]
- Miao, D.; Su, H.; He, B.; Gao, J.; Xia, Q.; Zhu, M.; Gu, Z.; Goltzman, D.; Karaplis, A.C. Severe growth retardation and early lethality in mice lacking the nuclear localization sequence and C-terminus of PTH-related protein. Proc. Natl. Acad. Sci. USA 2008, 105, 20309–20314. [Google Scholar] [CrossRef]
- Harvat, B.L.; Wang, A.; Seth, P.; Jetten, A.M. Up-regulation of p27Kip1, p21WAF1/Cip1 and p16Ink4a is associated with, but not sufficient for, induction of squamous differentiation. J. Cell Sci. 1998, 111, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Chen, G.; Lu, N.; Zhang, Y.; Jin, S.; Karaplis, A.; Goltzman, D.; Miao, D. Deficiency of the parathyroid hormone-related peptide nuclear localization and carboxyl terminal sequences leads to premature skin ageing partially mediated by the downregulation of p27. Exp. Derm. 2015, 24, 847–852. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Promoter | Exons Included at the 5′ End | Ever-Present Exons | Exons Included at the 3′ End Protein Isoform (Amino Acids) |
|---|---|---|---|
| P1 | I-II-III | V-VI | VII (139)-VIII (173)-IX (141) |
| I-III | |||
| I | |||
| P2 | III | V-VI | VII (139)-VIII (173)-IX (141) |
| P3 | IV | V-VI | VII (139)-VIII (173)-IX (141) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Librizzi, M.; Naselli, F.; Abruscato, G.; Luparello, C.; Caradonna, F. Parathyroid Hormone Related Protein (PTHrP)-Associated Molecular Signatures in Tissue Differentiation and Non-Tumoral Diseases. Biology 2023, 12, 950. https://doi.org/10.3390/biology12070950
Librizzi M, Naselli F, Abruscato G, Luparello C, Caradonna F. Parathyroid Hormone Related Protein (PTHrP)-Associated Molecular Signatures in Tissue Differentiation and Non-Tumoral Diseases. Biology. 2023; 12(7):950. https://doi.org/10.3390/biology12070950
Chicago/Turabian StyleLibrizzi, Mariangela, Flores Naselli, Giulia Abruscato, Claudio Luparello, and Fabio Caradonna. 2023. "Parathyroid Hormone Related Protein (PTHrP)-Associated Molecular Signatures in Tissue Differentiation and Non-Tumoral Diseases" Biology 12, no. 7: 950. https://doi.org/10.3390/biology12070950
APA StyleLibrizzi, M., Naselli, F., Abruscato, G., Luparello, C., & Caradonna, F. (2023). Parathyroid Hormone Related Protein (PTHrP)-Associated Molecular Signatures in Tissue Differentiation and Non-Tumoral Diseases. Biology, 12(7), 950. https://doi.org/10.3390/biology12070950