
Surrounded by Kindred: Spermophilus major Hybridization with Other Spermophilus Species in Space and Time

 , ,
, ,  ,
,
Abstract
Simple Summary
Abstract
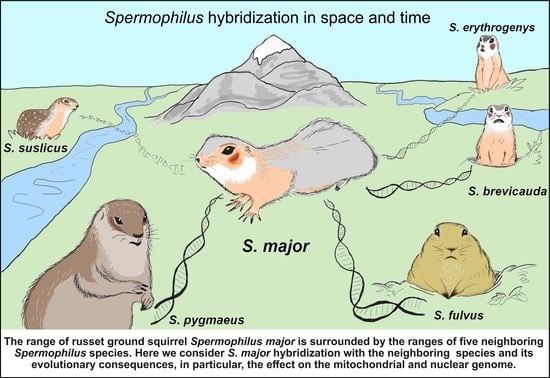
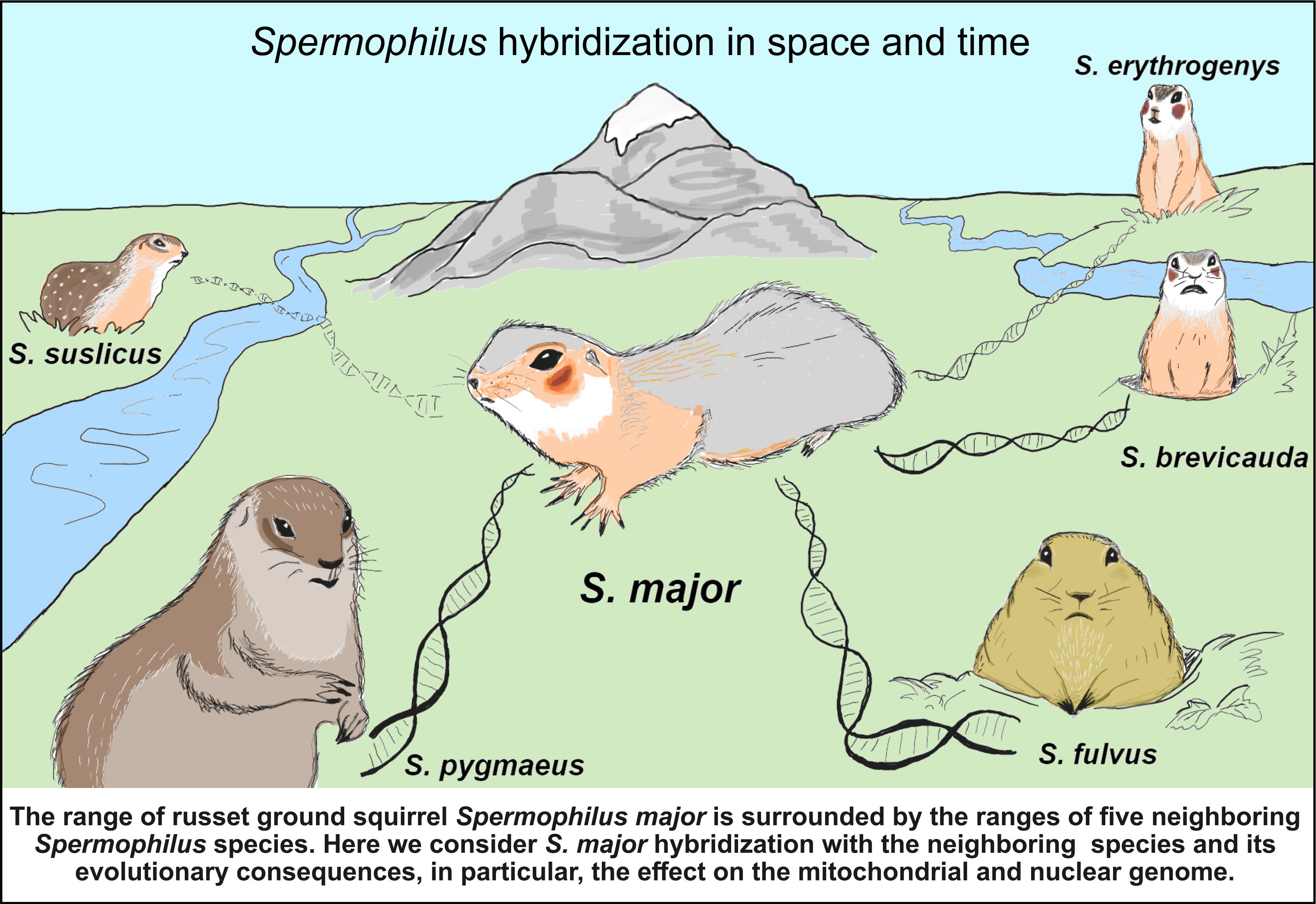
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. DNA Extraction, Amplification, Sequencing, and Restriction Analyzes
2.3. Molecular Data Analyzes
3. Results
3.1. Variability of Mitochondrial Molecular Markers
3.2. Variability of Nuclear Molecular Markers
3.2.1. SmcY Variability
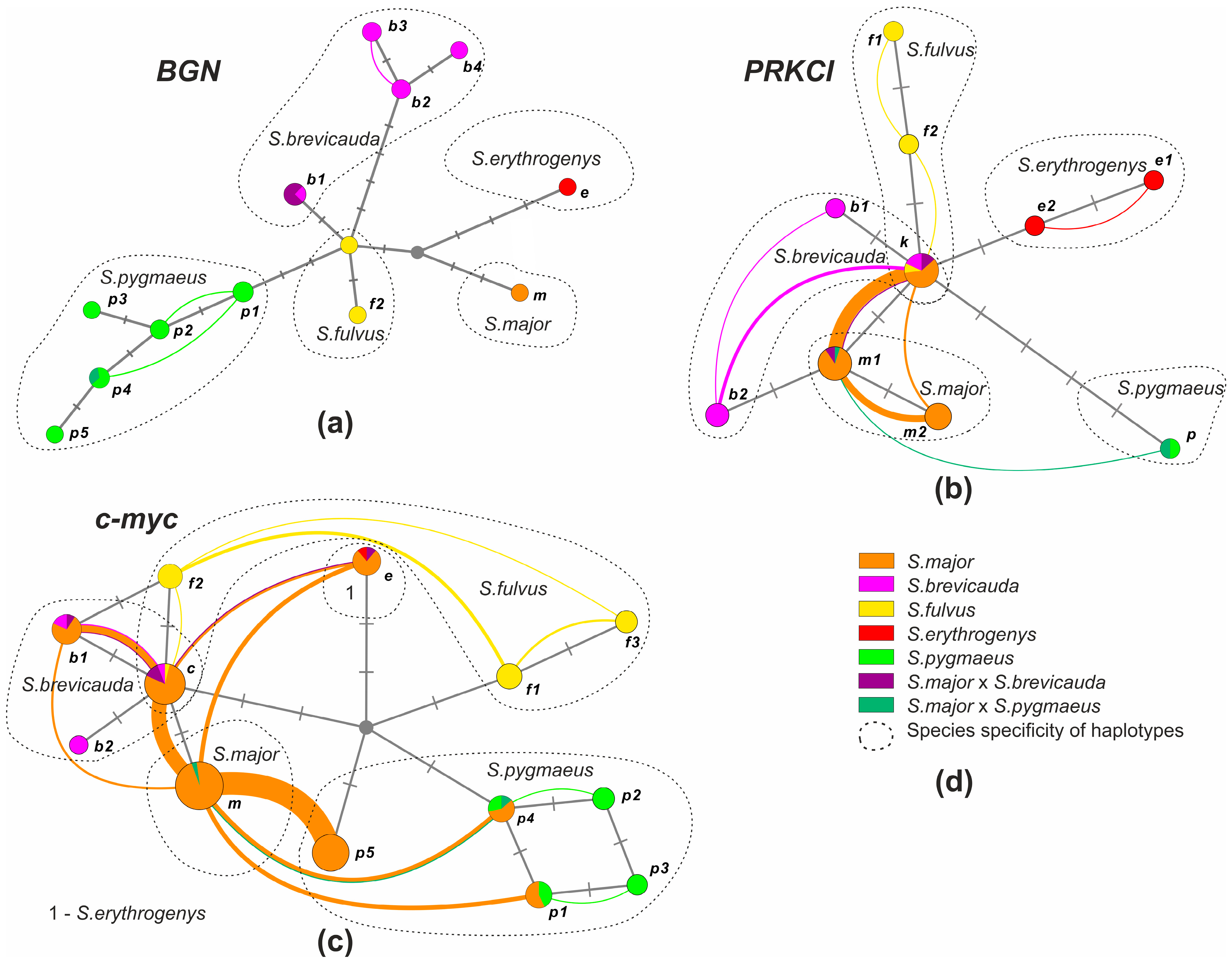
3.2.2. BGN Variability
3.2.3. PRKCI Variability
3.2.4. c-myc Variability
3.2.5. i6p53 Variability
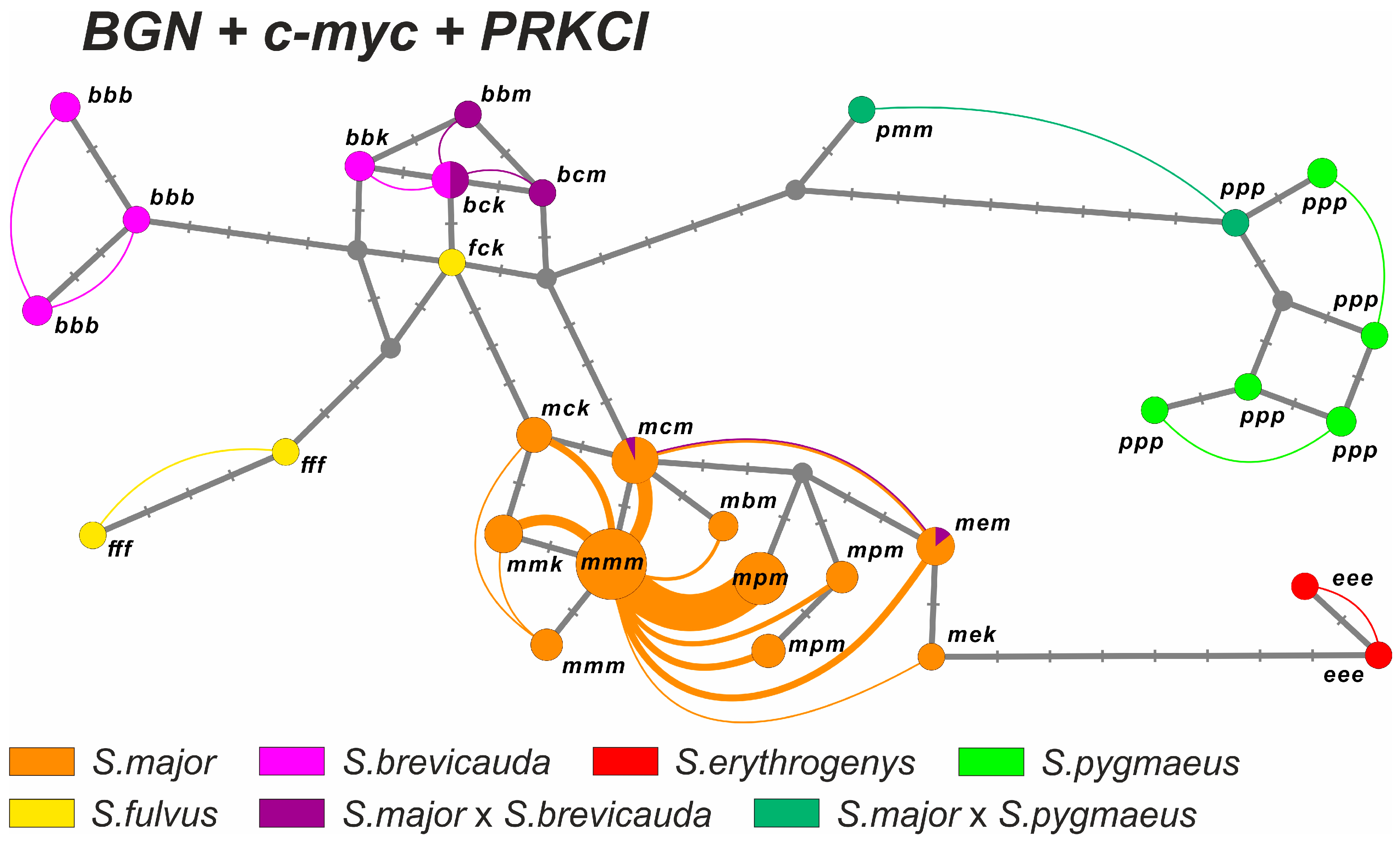
3.2.6. Consensus Sequence Analysis
3.3. Analysis of S. major Haplotypic Diversity
4. Discussion
4.1. Hybridization in Space
4.1.1. Cis-Ural Part of the S. major Range
4.1.2. Trans-Ural Part of the S. major Range
4.2. Hybridization over Time
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, G.; Davis, B.W.; Eizirik, E.; Murphy, W.J. Phylogenomic evidence for ancient hybridization in the genomes of living cats (Felidae). Genome Res. 2016, 26, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Palkopoulou, E.; Lipson, M.; Mallick, S.; Nielsen, S.; Rohland, N.; Baleka, S.; Karpinski, E.; Ivancevic, A.M.; Thu-Hien, T.; Kortschak, D.R.; et al. A comprehensive genomic history of extinct and living elephants. Proc. Natl. Acad. Sci. USA 2018, 115, E2566–E2574. [Google Scholar] [CrossRef] [PubMed]
- Zolotareva, K.I.; Belokon, M.M.; Belokon, Y.S.; Rutovskaya, M.V.; Hlyap, L.A.; Starykov, V.P.; Politov, D.V.; Lebedev, V.S.; Bannikova, A.A. Genetic diversity and structure of the hedgehogs Erinaceus europaeus and Erinaceus roumanicus: Evidence for ongoing hybridization in Eastern Europe. Biol. J. Linn. Soc. 2021, 132, 174–195. [Google Scholar] [CrossRef]
- Kerhoulas, N.J.; Gunderson, A.M.; Olson, L.E. Complex history of isolation and gene flow in hoary, Olympic, and endangered Vancouver Island marmots. J. Mammal. 2015, 96, 810–826. [Google Scholar] [CrossRef]
- Kovalev, S.Y.; Golovljova, I.V.; Mukhacheva, T.A. Natural hybridization between Ixodes ricinus and Ixodes persulcatus ticks evidenced by molecular genetics methods. Ticks Tick-Borne Dis. 2016, 7, 113–118. [Google Scholar] [CrossRef]
- Smith, K.L.; Hale, J.M.; Kearney, M.R.; Austin, J.J.; Melville, J. Molecular patterns of introgression in a classic hybrid zone between the Australian tree frogs, Litoria ewingii and L. áparaewingi: Evidence of a tension zone. Mol. Ecol. 2013, 22, 1869–1883. [Google Scholar] [CrossRef]
- Dufresnes, C.; Litvinchuk, S.N.; Rozenblut-Kościsty, B.; Rodrigues, N.; Perrin, N.; Crochet, P.A.; Jeffries, D.L. Hybridization and introgression between toads with different sex chromosome systems. Evol. Lett. 2020, 4, 444–456. [Google Scholar] [CrossRef]
- Tynkkynen, K.; Grapputo, A.; Kotiaho, J.S.; Rantala, M.J.; Väänänen, S.; Suhonen, J. Hybridization in Calopteryx damselflies: The role of males. Anim. Behav. 2008, 75, 1431–1439. [Google Scholar] [CrossRef]
- Abbott, R.; Albach, D.; Ansell, S.; Arntzen, J.W.; Baird, S.J.; Bierne, N.; Boughman, J.; Brelsford, A.; Buerkle, C.A.; Buggs, R.; et al. Hybridization and speciation. J. Evol. Biol. 2013, 26, 229–246. [Google Scholar] [CrossRef]
- Bloesch, Z.; Nauheimer, L.; Almeida, T.E.; Crayn, D.; Field, A.R. HybPhaser identifies hybrid evolution in Australian Thelypteridaceae. Mol. Phylogenetics Evol. 2022, 173, 107526. [Google Scholar] [CrossRef]
- Feder, J.L.; Nosil, P.; Wacholder, A.C.; Egan, S.P.; Berlocher, S.H.; Flaxman, S.M. Genome-wide congealing and rapid transitions across the speciation continuum during speciation with gene flow. J. Hered. 2014, 105, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Fontsere, C.; de Manuel, M.; Marques-Bonet, T.; Kuhlwilm, M. Admixture in mammals and how to understand its functional implications: On the abundance of gene flow in mammalian species, its impact on the genome, and roads into a functional understanding. Bioessays 2019, 41, 1900123. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, Z.; Aliabadian, M.; Ghorbani, F.; Moghaddam, F.Y.; Lissovsky, A.A.; Obst, M.; Olsson, U. Unidirectional introgression and evidence of hybrid superiority over parental populations in Eastern Iranian Plateau population of hares (Mammalia: Lepus Linnaeus, 1758). J. Mamm. Evol. 2020, 27, 723–743. [Google Scholar] [CrossRef]
- Johannesson, K.; Le Moan, A.; Perini, S.; André, C. A Darwinian laboratory of multiple contact zones. Trends Ecol. Evol. 2020, 35, 1021–1036. [Google Scholar] [CrossRef] [PubMed]
- Colella, J.P.; Lan, T.; Schuster, S.C.; Talbot, S.L.; Cook, J.A.; Lindqvist, C. Whole-genome analysis of Mustela erminea finds that pulsed hybridization impacts evolution at high latitudes. Commun. Biol. 2018, 1, 51. [Google Scholar] [CrossRef]
- Ackermann, R.R.; Arnold, M.L.; Baiz, M.D.; Cahill, J.A.; Cortés-Ortiz, L.; Evans, B.J.; Grant, B.R.; Grant, P.R.; Hallgrimsson, B.; Humphreys, R.A.; et al. Hybridization in human evolution: Insights from other organisms. Evol. Anthropol. Issues News Rev. 2019, 28, 189–209. [Google Scholar] [CrossRef]
- Gautier, M.; Moazami-Goudarzi, K.; Levéziel, H.; Parinello, H.; Grohs, C.; Rialle, S.; Kowalczyk, R.; Flori, L. Deciphering the wisent demographic and adaptive histories from individual whole-genome sequences. Mol. Biol. Evol. 2016, 33, 2801–2814. [Google Scholar] [CrossRef]
- Edwards, C.J.; Suchard, M.A.; Lemey, P.; Welch, J.J.; Barnes, I.; Fulton, T.L.; Barnett, R.; O’Connell, T.C.; Coxon, P.; Monaghan, N.; et al. Ancient hybridization and an Irish origin for the modern polar bear matriline. Curr. Biol. 2011, 21, 1251–1258. [Google Scholar] [CrossRef]
- Kovacs, K.M.; Lydersen, C.; Hammill, M.O.; White, B.N.; Wilson, P.J.; Malik, S. A harp seal × hooded seal hybrid. Mar. Mammal Sci. 1997, 13, 460–468. [Google Scholar] [CrossRef]
- Franco-Trecu, V.; Abud, C.; Feijoo, M.; Kloetzer, G.; Casacuberta, M.; Costa-Urrutia, P. Sex beyond species: The first genetically analyzed case of intergeneric fertile hybridization in pinnipeds. Evol. Dev. 2016, 18, 127–136. [Google Scholar] [CrossRef]
- Canestrelli, D.; Porretta, D.; Lowe, W.H.; Bisconti, R.; Carere, C.; Nascetti, G. The tangled evolutionary legacies of range expansion and hybridization. Trends Ecol. Evol. 2016, 31, 677–688. [Google Scholar] [CrossRef]
- Hewitt, G.M. Speciation, hybrid zones and phylogeography—Or seeing genes in space and time. Mol. Ecol. 2001, 10, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Garroway, C.J.; Bowman, J.; Cascaden, T.J.; Holloway, G.L.; Mahan, C.G.; Malcolm, J.R.; Steele, M.A.; Turner, G.; Wilson, P.J. Climate change induced hybridization in flying squirrels. Glob. Chang. Biol. 2010, 16, 113–121. [Google Scholar] [CrossRef]
- Mallet, J.; Besansky, N.; Hahn, M.W. How reticulated are species? BioEssays 2016, 38, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Brandler, O.V.; Kapustina, S.Y.; Nikol’skii, A.A.; Kolesnikov, V.V.; Badmaev, B.B.; Adiya, Y. A study of hybridization between Marmota baibacina and M. sibirica in their secondary contact zone in Mongolian Altai. Front. Ecol. Evol. 2021, 9, 363. [Google Scholar] [CrossRef]
- Chavez, A.S.; Saltzberg, C.J.; Kenagy, G.J. Genetic and phenotypic variation across a hybrid zone between ecologically divergent tree squirrels (Tamiasciurus). Mol. Ecol. 2011, 20, 3350–3366. [Google Scholar] [CrossRef]
- Haines, M.L.; Luikart, G.; Amish, S.J.; Smith, S.; Latch, E.K. Evidence for adaptive introgression of exons across a hybrid swarm in deer. BMC Evol. Biol. 2019, 19, 199. [Google Scholar] [CrossRef]
- Thornton, W.A.; Creel, G.C.; Trimble, R.E. Hybridization in the fox genus Vulpes in west Texas. Southwest. Nat. 1971, 15, 473–484. [Google Scholar] [CrossRef]
- Teeter, K.C.; Thibodeau, L.M.; Gompert, Z.; Buerkle, C.A.; Nachman, M.W.; Tucker, P.K. The variable genomic architecture of isolation between hybridizing species of house mice. Evolution 2010, 64, 472–485. [Google Scholar] [CrossRef]
- Kryukov, A.P.; Chelomina, G.N.; Pavlenko, M.V. Hybrid Animal Zones: Classification and Study Methods. In The Present-Day Approaches to Studies of Variability; Kryukov, A.P., Ed.; FEB AS USSR: Vladivostok, Russia, 1989; pp. 25–31. [Google Scholar]
- Harrison, R.G.; Larson, E.L. Heterogeneous genome divergence, differential introgression, and the origin and structure of hybrid zones. Mol. Ecol. 2016, 25, 2454–2466. [Google Scholar] [CrossRef]
- Levänen, R.; Kunnasranta, M.; Pohjoismäki, J. Mitochondrial DNA introgression at the northern edge of the brown hare (Lepus europaeus) range. Ann. Zool. Fenn. 2018, 55, 15–24. [Google Scholar] [CrossRef]
- Melo-Ferreira, J.; Boursot, P.; Suchentrunk, F.; Ferrand, N.; Alves, P.C. Invasion from the cold past: Extensive introgression of mountain hare (Lepus timidus) mitochondrial DNA into three other hare species in northern Iberia. Mol. Ecol. 2005, 14, 2459–2464. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.P.; Farelo, L.; Vilela, J.; Vanderpool, D.; Alves, P.C.; Good, J.M.; Boursot, P.; Melo-Ferreira, J. Range expansion underlies historical introgressive hybridization in the Iberian hare. Sci. Rep. 2017, 7, 40788. [Google Scholar] [CrossRef] [PubMed]
- Eberlein, C.; Hénault, M.; Fijarczyk, A.; Charron, G.; Bouvier, M.; Kohn, L.M.; Anderson, J.B.; Landry, C.R. Hybridization is a recurrent evolutionary stimulus in wild yeast speciation. Nat. Commun. 2019, 10, 923. [Google Scholar] [CrossRef]
- Wright, E.A.; Roberts, E.K.; Platt, R.N.; Bayouth, J.V.; Conway, W.C.; Bradley, R.D. Mitochondrial capture and subsequent genetic divergence generates a novel haplogroup: Evidence from ancient and ongoing hybridization in mule and white-tailed deer. J. Mammal. 2022, 103, 723–736. [Google Scholar] [CrossRef]
- Abramson, N.I.; Rodchenkova, E.N.; Fokin, M.V.; Rakitin, S.B.; Gileva, E.A. Recent and ancient introgression of mitochondrial DNA between the red (Clethrionomys rutilus) and bank (Clethrionomys glareolus) voles (Rodentia, Cricetidae). Dokl. Biol. Sci. 2009, 425, 147–150. [Google Scholar] [CrossRef]
- Thompson, C.W.; Stangl, F.B.; Bradley, R.D. Ancient hybridization and subsequent mitochondrial capture in ground squirrels (genus Ictidomys). Mus. Tex. Tech Univ. 2015, 113, 1–24. [Google Scholar]
- Fadakar, D.; Malekian, M.; Hemami, M.R.; Lerp, H.; Rezaei, H.R.; Bärmann, E.V. Repeated hybridization of two closely related gazelle species (Gazella bennettii and Gazella subgutturosa) in central Iran. Ecol. Evol. 2020, 10, 11372–11386. [Google Scholar] [CrossRef]
- Currat, M.; Ruedi, M.; Petit, R.J.; Excoffier, L. The hidden side of invasions: Massive introgression by local genes. Evolution 2008, 62, 1908–1920. [Google Scholar] [CrossRef]
- Gromov, I.M.; Bibikov, D.I.; Kalabukhov, N.I.; Meier, N.N. Fauna of the USSR: Mammals; Volume 3, Issue 2: Ground Squirrels (Marmotinae); Pavlovsky, E.N., Ed.; Nauka: Leningrad, Russia, 1965; p. 467. (In Russian) [Google Scholar]
- Helgen, K.M.; Cole, F.R.; Helgen, L.E.; Wilson, D.E. Generic revision in the Holarctic ground squirrel genus Spermophilus. J. Mammal. 2009, 90, 270–305. [Google Scholar] [CrossRef]
- Ermakov, O.A.; Titov, S.V.; Surin, V.L.; Formozov, N.A. Molecular genetic study of maternal and paternal liniages of hybridization of ground squirrels (Spermophilus: Rodentia, Sciuridae). Byull. Mosk. O-va Ispyt. Prir. Otd. Biol. 2006, 111, 30–35. (In Russian) [Google Scholar]
- Kryštufek, B.; Vohralík, V. Taxonomic Revision of the Palaearctic Rodents (Rodentia): Sciuridae: Xerinae 1 (Eutamias and Spermophilus); Linx: Praha, Czech Republic, 2012; Volume 43, pp. 17–111. [Google Scholar]
- Nikol’skii, A.A. On the problem of boundary between the ranges of Citellus maior and C. erythrogenys (Rodentia, Sciuridae) in Northern Kazakhstan. Zool. Zh. 1984, 63, 256–262. (In Russian) [Google Scholar]
- Sinitsa, M.V.; Pogodina, N.V.; Kryuchkova, L.Y. The skull of Spermophilus nogaici (Rodentia: Sciuridae: Xerinae) and the affinities of the earliest Old World ground squirrels. Zool. J. Linn. Soc. 2019, 186, 826–864. [Google Scholar] [CrossRef]
- Ognev, S.I. The Mammals of Russia (USSR) and Adjacent Countries; USSR Academy of Sciences Publishing: Moscow, Russia; Leningrad, Russia, 1947; Volume 5, pp. 65–85. (In Russian) [Google Scholar]
- Afanas’ev, A.V.; Sludskiy, A.A.; Korelov, M.N.; Bazhanov, V.S.; Strautman, E.I. Mammals of Kazakhstan; Afanas’ev, A.V., Ed.; Izdatel’stvo AN Kaz. SSR Publ.: Almaty, Russia, 1953; p. 536. (In Russian) [Google Scholar]
- Ermakov, O.A.; Surin, V.L.; Titov, S.V.; Tagiev, A.F.; Luk’yanenko, A.V.; Formozov, N.A. A Molecular Genetic Study of Hybridization in Four Species of Ground Squirrels (Spermophilus: Rodentia, Sciuridae). Russ. J. Genet. 2002, 7, 796–809. [Google Scholar] [CrossRef]
- Titov, S.V.; Kuz’min, A.A.; Simakov, M.D.; Kartavov, N.A. New data about hybridization of speckled (Spermophilus suslicus Güld.), and russet (Spermophilus major Pall.) ground squirrels in a wide zone of sympatry. Izv Penz. Gos. Pedagog. Univ. Im. i V.G. Belinskogo 2020, 1, 23–35. (In Russian) [Google Scholar] [CrossRef]
- Ermakov, O.A.; Titov, S.V. Dynamics of Spermophilus major (Rodentia, Sciuridae) range boundaries in the Volga river region. Zool. Zh. 2000, 79, 503–509. (In Russian) [Google Scholar]
- Titov, S.V.; Ermakov, O.A.; Surin, V.L.; Formozov, N.A.; Kosatkin, M.V.; Shilova, S.A.; Shmyrov, A.A. Molecular genetic and bioacoustic diagnostics russet (Spermophilus major Pallas, 1778) and yellow (S. fulvus Lichtenstein, 1823) ground squirrel from mixed colony. Byull. Mosk. O-va Ispyt. Prir. Otd. Biol. 2005, 110, 72–77. (In Russian) [Google Scholar]
- Ermakov, O.A.; Surin, V.L.; Titov, S.V.; Zborovsky, S.S.; Formozov, N.A. A search for Y-chromosomal species-specific markers and their use for hybridization analysis in ground squirrels (Spermophilus: Rodentia, Sciuridae). Russ. J. Genet. 2006, 42, 429–438. [Google Scholar] [CrossRef]
- Zhang, Y. Distribution of Mammalian Species in China; China Forestry Publishing House: Beijing, China, 1997. [Google Scholar]
- Brandler, O.V.; Tukhbatullin, A.R.; Kapustina, S.Y.; Schepetov, D.M.; Titov, S.V.; Ermakov, O.A. Variability of Mitochondrial DNA Control Region and Phylogeography of Russet Ground Squirrel (Spermophilus major, Sciuridae, Rodentia). Russ. J. Genet. 2021, 57, 825–835. [Google Scholar] [CrossRef]
- Matrosova, V.A.; Ivanova, A.D.; Volodina, E.V.; Volodin, I.A.; Alexandrov, D.Y.; Sibiryakova, O.V.; Ermakov, O.A. Phylogenetic relationship and variation of alarm call traits of populations of red-cheeked ground squirrels (Spermophilus erythrogenys sensu lato) suggest taxonomic delineation. Integr. Zool. 2019, 14, 341–353. [Google Scholar] [CrossRef]
- Vasil’eva, M.V. The systematic position of the Palaearctic ground squirrels of the genus Citellus Oken, 1816. In Proceedings of the I Annual Scientific Reporting Conference of the Faculty of Biology and Soil of MSU, MSU, Moscow, Russia, 9–12 March 1964; pp. 125–127. (In Russian). [Google Scholar]
- Nikol’skii, A.A.; Starikov, V.P. Variation of the Alarm Call in Ground Squirrels Spermophilus major and S. erythrogenys (Rodentia, Sciuridae) in the Contact Zone in the Kurgan Oblast. Russ. J. Zool. 1997, 1, 340–351. [Google Scholar]
- Spiridonova, L.N.; Chelomina, G.N.; Starikov, V.P.; Korablev, V.P.; Zvirka, M.V.; Lyapunova, E.A. RAPD-PCR Analysis of Ground Squirrels from the Tobol-Ishim Interfluve: Evidence for Interspecific Hybridization between Ground Squirrel Species Spermophilus major and S. erythrogenys. Russ. J. Genet. 2005, 41, 991–1001. [Google Scholar] [CrossRef]
- Gromov, I.M.; Erbaeva, M.A. The Mammals of Russia and Adjacent Territories; Scarlato, O.A., Ed.; Nauka: St. Petersburg, Russia, 1995; Volume Lagomorphs and rodents; pp. 131–132. (In Russian) [Google Scholar]
- Wilson, D.E.; Reeder, D.M. (Eds.) Mammal Species of the World. A Taxonomic and Geographic, 3rd ed.; Johns Hopkins University Press: Baltimore, MA, USA, 2005; p. 2142. [Google Scholar]
- Spiridonova, L.N.; Chelomina, G.N.; Tsuda, K.; Yonekava, H.; Starikov, V.P. Genetic evidence of extensive introgression of short-tailed ground squirrel genes in a hybridization zone of Spermophilus major and S. erythrogenys, inferred from sequencing of the mtDNA cytochrome b gene. Russ. J. Genet. 2006, 42, 802–809. [Google Scholar] [CrossRef]
- Ermakov, O.A.; Simonov, E.P.; Surin, V.L.; Titov, S.V.; Brandler, O.V.; Ivanova, N.V. Implications of hybridization, NUMTs, and overlooked diversity for DNA barcoding of Eurasian ground squirrels. PLoS ONE 2015, 10, e0117201. [Google Scholar] [CrossRef] [PubMed]
- Aljanabi, S.M.; Martinez, I. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucl. Acids Res. 1997, 25, 4692–4693. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Fritch, E.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Lab. Press: New York, NY, USA, 1989; p. 398. [Google Scholar]
- Lyons, L.A.; Laughlin, T.F.; Copeland, N.G.; Jenkins, N.A.; Womak, J.E.; O’Brien, S.J. Comparative anchor tagged sequences (CATS) for integrative mapping of mammalian genomes. Nat. Genet. 1997, 15, 47–56. [Google Scholar] [CrossRef]
- Matthee, C.A.; Burzlaff, J.D.; Taylor, J.F.; Davis, S.K. Mining the mammalian genome for artiodactyl systematics. Syst. Biol. 2001, 50, 367–390. [Google Scholar] [CrossRef] [PubMed]
- Matthee, C.A.; van Vuuren, B.J.; Bell, D.; Robinson, T.J. A molecular supermatrix of the rabbits and hares (Leporidae) allows for the identification of five intercontinental exchanges during the Miocene. Syst. Biol. 2004, 53, 433–447. [Google Scholar] [CrossRef]
- Steppan, S.J.; Storz, B.L.; Hoffmann, R.S. Nuclear DNA phylogeny of the squirrels (Mammalia: Rodentia) and the evolution of arboreality from c-myc and RAG1. Mol. Phylogenetics Evol. 2004, 30, 703–719. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Res. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Spöri, Y.; Flot, J.F. HaplowebMaker and CoMa: Two web tools to delimit species using haplowebs and conspecificity matrices. Methods Ecol. Evol. 2020, 11, 1434–1438. [Google Scholar] [CrossRef]
- Doyle, J.J. The irrelevance of allele tree topologies for species delimitation, and a non-topological alternative. Syst. Bot. 1995, 20, 574–588. [Google Scholar] [CrossRef]
- Petzold, A.; Hassanin, A. A comparative approach for species delimitation based on multiple methods of multi-locus DNA sequence analysis: A case study of the genus Giraffa (Mammalia, Cetartiodactyla). PLoS ONE 2020, 15, e0217956. [Google Scholar] [CrossRef] [PubMed]
- Hinckley, A.; Hawkins, M.T.; Maldonado, J.E.; Leonard, J.A. Evolutionary history and patterns of divergence in three tropical east Asian squirrels across the Isthmus of Kra. J. Biogeogr. 2023, 50, 1090–1102. [Google Scholar] [CrossRef]
- He, S.; Planes, S.; Sinclair-Taylor, T.H.; Berumen, M.L. Diagnostic nuclear markers for hybrid Nemos in Kimbe Bay, PNG-Amphiprion chrysopterus x Amphiprion sandaracinos hybrids. Mar. Biodivers. 2019, 49, 1261–1269. [Google Scholar] [CrossRef]
- Dellicour, S.; Flot, J.F. Delimiting species-poor data sets using single molecular markers: A study of barcode gaps, haplowebs and GMYC. Syst. Biol. 2015, 64, 900–908. [Google Scholar] [CrossRef]
- Baker, R.J.; Bradley, R.D. Speciation in mammals and the genetic species concept. J. Mammal. 2006, 87, 643–662. [Google Scholar] [CrossRef]
- Tukhbatullin, A.R.; Brandler, O.V. Current condition of populations and spatial-environmental features of habitats of russet ground squirrel Spermophilus major. RUDN J. Ecol. Life Saf. 2021, 29, 7–22. [Google Scholar] [CrossRef]
- Burton, R.S. The role of mitonuclear incompatibilities in allopatric speciation. Cell. Mol. Life Sci. 2022, 79, 1–18. [Google Scholar] [CrossRef]
- Toews, D.P.; Brelsford, A. The biogeography of mitochondrial and nuclear discordance in animals. Mol. Ecol. 2012, 21, 3907–3930. [Google Scholar] [CrossRef] [PubMed]
- Grant, P.R.; Grant, B.R. Triad hybridization via a conduit species. Proc. Natl. Acad. Sci. USA 2020, 117, 7888–7896. [Google Scholar] [CrossRef] [PubMed]
- Illarionov, A.G. Turgay Spillway. To the History of Formation and Development of the River Network of the Aral-Irtysh Region; UdSU: Izhevsk, Russia, 2013. (In Russian) [Google Scholar]
- Velichko, A.A. (Ed.) Paleoclimates and Paleoenvironments of Extra-Tropical Regions of the Northern Hemisphere. Late Pleistocene-Holocene; GEOS: Moscow, Russia, 2009; p. 120. ISBN 978-5-89118-436-7. (In Russian) [Google Scholar]
- Svitoch, A.A. Paleogeography of the Greater Caspian sea. Vestn. Mosk. Unviersiteta Seriya Geogr. 2015, 4, 69–80. (In Russian) [Google Scholar]
- Yanina, T.A. The Ponto-Caspian region: Environmental consequences of climate change during the Late Pleistocene. Quat. Int. 2014, 345, 88–99. [Google Scholar] [CrossRef]
- Chemagina, D.; Strukova, T.; Pogodina, N.; Kuzmina, E.; Gudova, D. Ground squirrels of the genus Spermophilus from the Pleistocene and Holocene localities of the Middle and South Urals and Trans-Urals region: The dental features. Hist. Biol. 2019, 33, 40–53. [Google Scholar] [CrossRef]
- Rakotoarivelo, A.R.; O’Donoghue, P.; Bruford, M.W.; Moodley, Y. An ancient hybridization event reconciles mito-nuclear discordance among spiral-horned antelopes. J. Mammal. 2019, 100, 1144–1155. [Google Scholar] [CrossRef]
- Kosintsev, P.; Danukalova, G.; Osipova, E.; Yakovlev, A.; Alimbekova, L.; Popova-Lvova, M. Palaeoenvironment of the Late Pleistocene–Holocene interval in the Tanalyk river valley of the Southern Trans-Ural region (Russia). Quat. Int. 2013, 284, 74–84. [Google Scholar] [CrossRef]
- Yakovlev, A.G. Small mammals of the Late Neopleistocene and Holocene of the South Urals region. Geol. Collect. 2009, 8, 54–59. (In Russian) [Google Scholar]
- Kosintsev, P.A.; Bachura, O.P. Late Pleistocene and Holocene mammal fauna of the Southern Urals. Quat. Int. 2012, 284, 161–170. [Google Scholar] [CrossRef]
- Green, R.E.; Krause, J.; Briggs, A.W.; Maricic, T.; Stenzel, U.; Kircher, M.; Patterson, N.; Li, H.; Zhai, W.; Fritz, M.H.Y.; et al. A draft sequence of the Neandertal genome. Science 2010, 328, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Sarver, B.A.J.; Herrera, N.D.; Sneddon, D.; Hunter, S.S.; Settles, M.L.; Kronenberg, Z.; Demboski, J.R.; Good, J.M.; Sullivan, J. Diversification, Introgression, and Rampant Cytonuclear Discordance in Rocky Mountains Chipmunks (Sciuridae: Tamias). Syst. Biol. 2021, 70, 908–921. [Google Scholar] [CrossRef]
- Ge, D.; Wen, Z.; Feijó, A.; Lissovsky, A.; Zhang, W.; Cheng, J.; Yan, C.; She, H.; Zhang, D.; Cheng, Y.; et al. Genomic Consequences of and Demographic Response to Pervasive Hybridization Over Time in Climate-Sensitive Pikas. Mol. Biol. Evol. 2023, 40, msac274. [Google Scholar] [CrossRef] [PubMed]
- Brandler, O.V.; Tukhbatullin, A.R.; Nikol’skii, A.A. Comparative Analysis of the Alarm Call in Different Age and Sexual Groups of the Russet Ground Squirrel (Spermophilus major Pallas 1778). Russ. J. Dev. Biol. 2019, 50, 173–179. [Google Scholar] [CrossRef]
- Nikol’skii, A.A. Acoustic Signals of Mammals in the Evolutionary Process; Nauka: Moscow, Russia, 1984; p. 199. (In Russian) [Google Scholar]
- Kocher, T.D.; Thomas, W.K.; Meyer, A.; Edwards, S.V.; Pääbo, S.; Villablanca, F.X.; Wilson, A.C. Dynamics of mitochondrial DNA evolution in animals: Amplification and sequencing with conserved primers. Proc. Natl. Acad. Sci. USA 1989, 86, 6196–6200. [Google Scholar] [CrossRef]
- Steppan, S.J.; Akhverdyan, M.R.; Lyapunova, E.A.; Fraser, D.G.; Vorontsov, N.N.; Hoffmann, R.S.; Braun, M.J. Molecular phylogeny of the marmots (Rodentia: Sciuridae): Tests of evolutionary and biogeographic hypotheses. Syst. Biol. 1999, 48, 715–734. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.G.; Bogdanowicz, S.M.; Hoffmann, R.S.; Yensen, E.; Sherman, P.W. Phylogeny and evolutionary history of the ground squirrels (Rodentia: Marmotinae). J. Mamm. Evol. 2003, 10, 249–276. [Google Scholar] [CrossRef]
- Ekimova, I.; Korshunova, T.; Schepetov, D.; Neretina, T.; Sanamyan, N.; Martynov, A. Integrative systematics of northern and Arctic nudibranchs of the genus Dendronotus (Mollusca, Gastropoda), with descriptions of three new species. Zool. J. Linn. Soc. 2015, 173, 841–886. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | n | H | S/ƞ | h ± SD | π ± SD | k | Tajima’s D (p-Value) | Fs (p-Value) |
|---|---|---|---|---|---|---|---|---|
| A | 241 | 72 | 204/229 | 0.979 ± 0.002 | 0.0179 ± 0.009 | 18.335 | −1.4016 (0.01) | −9.6147 (0.07) |
| B | 223 | 66 | 80/85 | 0.976 ± 0.003 | 0.0078 ± 0.004 | 7.914 | −1.162 (0.14) | −24.43 (<0.01) |
| No. | Species/Sub-Species | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | S. major | 0.012 | 0.021 | – | 0.036 | 0.039 | 0.049 | 0.081 | |
| 2 | S. b. heptneri | 0.003 | 0.017 | – | 0.033 | 0.039 | 0.046 | 0.084 | |
| 3 | S. b. iliensis | 0.007 | 0.007 | – | 0.031 | 0.045 | 0.050 | 0.089 | |
| 4 | S. b. brevicauda | 0.024 | 0.024 | 0.026 | – | – | – | – | |
| 5 | S. b. intermedius | 0.031 | 0.031 | 0.034 | 0.027 | 0.047 | 0.056 | 0.094 | |
| 6 | S. fulvus | 0.033 | 0.032 | 0.035 | 0.032 | 0.044 | 0.049 | 0.091 | |
| 7 | S. erythrogenys | 0.054 | 0.054 | 0.055 | 0.050 | 0.054 | 0.051 | 0.088 | |
| 8 | S. pygmaeus | 0.106 | 0.106 | 0.107 | 0.106 | 0.109 | 0.102 | 0.113 |
| Species/Alleles | CR | SmcY 1 | BGN 2 | c-myc | PRKCI | i6p53 | Consensus |
|---|---|---|---|---|---|---|---|
| S. brevicauda | 0 | – | 1.29 | 6.3 3 | 3.6 3 | 0 | 3 3 |
| S. fulvus | 5.4 | 0 | 0 | 0 | |||
| S. erythrogenys | 0 | 0 | 0 | 1.7 | 0 | 0 | 0.4 |
| S. pygmaeus | 2.5 | 1.2 | 0.26 | 8 | 0.2 (1) 4 | 0.2 | 2.2 (2.4) 4 |
| Total | 7.9 | 1.2 | 1.55 | 16 | 3.8 (4.6) 4 | 0.2 | 5.6 (5.8) 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tukhbatullin, A.; Ermakov, O.; Kapustina, S.; Starikov, V.; Tambovtseva, V.; Titov, S.; Brandler, O. Surrounded by Kindred: Spermophilus major Hybridization with Other Spermophilus Species in Space and Time. Biology 2023, 12, 880. https://doi.org/10.3390/biology12060880
Tukhbatullin A, Ermakov O, Kapustina S, Starikov V, Tambovtseva V, Titov S, Brandler O. Surrounded by Kindred: Spermophilus major Hybridization with Other Spermophilus Species in Space and Time. Biology. 2023; 12(6):880. https://doi.org/10.3390/biology12060880
Chicago/Turabian StyleTukhbatullin, Andrey, Oleg Ermakov, Svetlana Kapustina, Vladimir Starikov, Valentina Tambovtseva, Sergey Titov, and Oleg Brandler. 2023. "Surrounded by Kindred: Spermophilus major Hybridization with Other Spermophilus Species in Space and Time" Biology 12, no. 6: 880. https://doi.org/10.3390/biology12060880
APA StyleTukhbatullin, A., Ermakov, O., Kapustina, S., Starikov, V., Tambovtseva, V., Titov, S., & Brandler, O. (2023). Surrounded by Kindred: Spermophilus major Hybridization with Other Spermophilus Species in Space and Time. Biology, 12(6), 880. https://doi.org/10.3390/biology12060880