
Compositional Structure of the Genome: A Review


Abstract
Simple Summary
Abstract
1. Introduction
2. DNA Sequence Segmentation
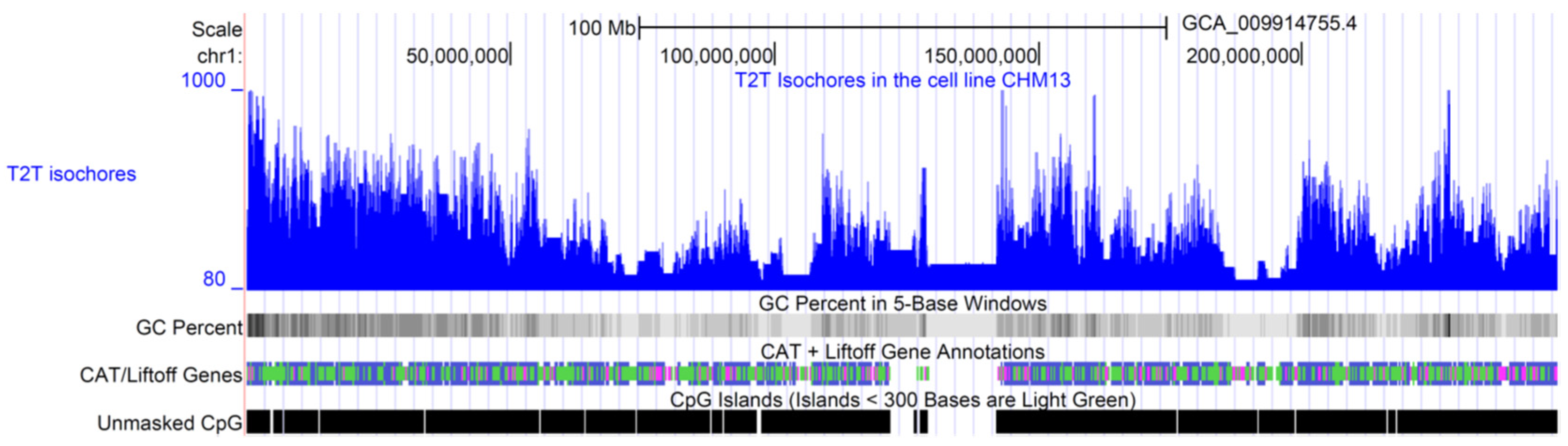
3. Prediction of Isochore Boundaries at the Sequence Level
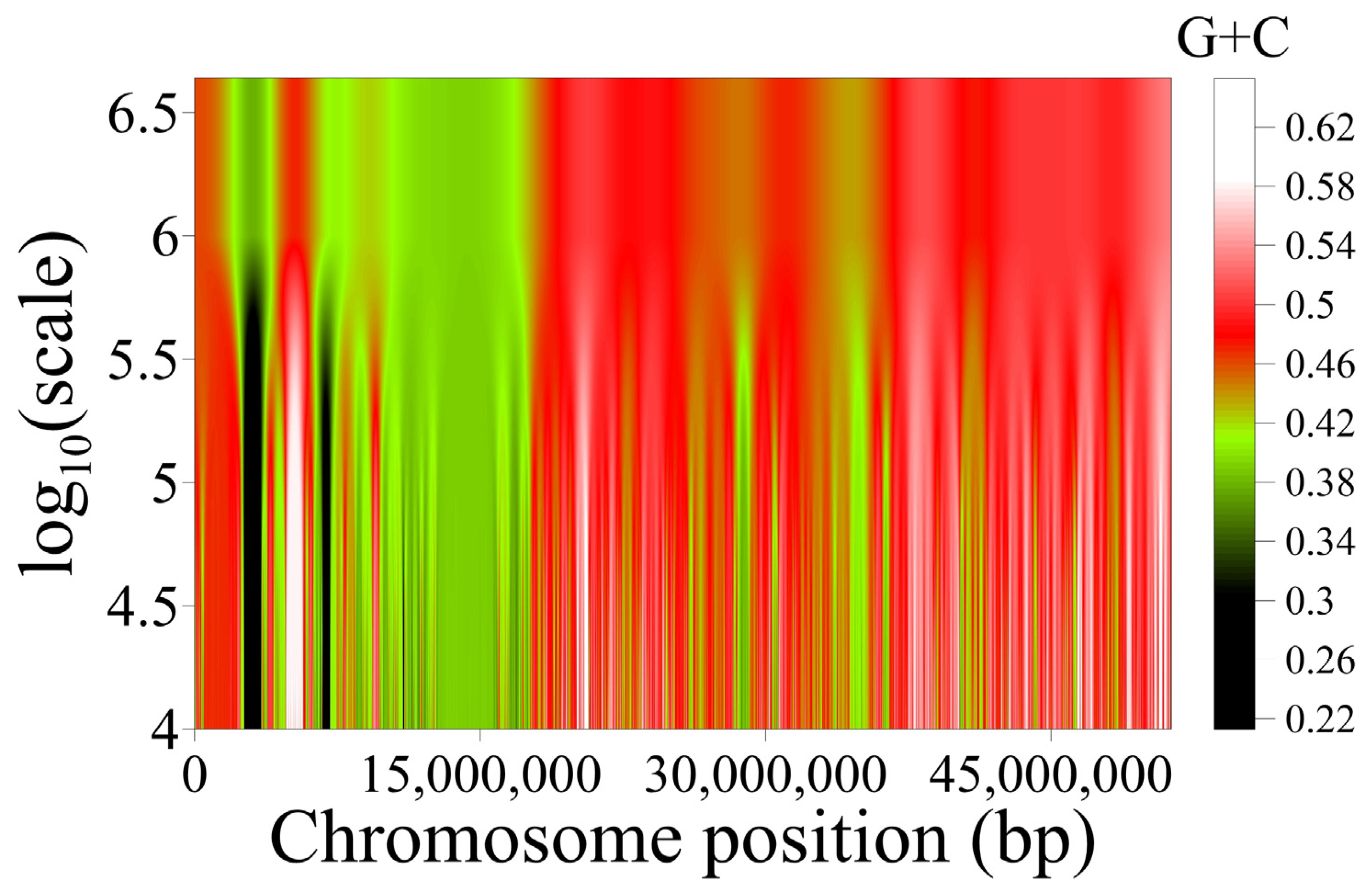
4. Long-Range Correlations and Compositional Superstructures in the Genome
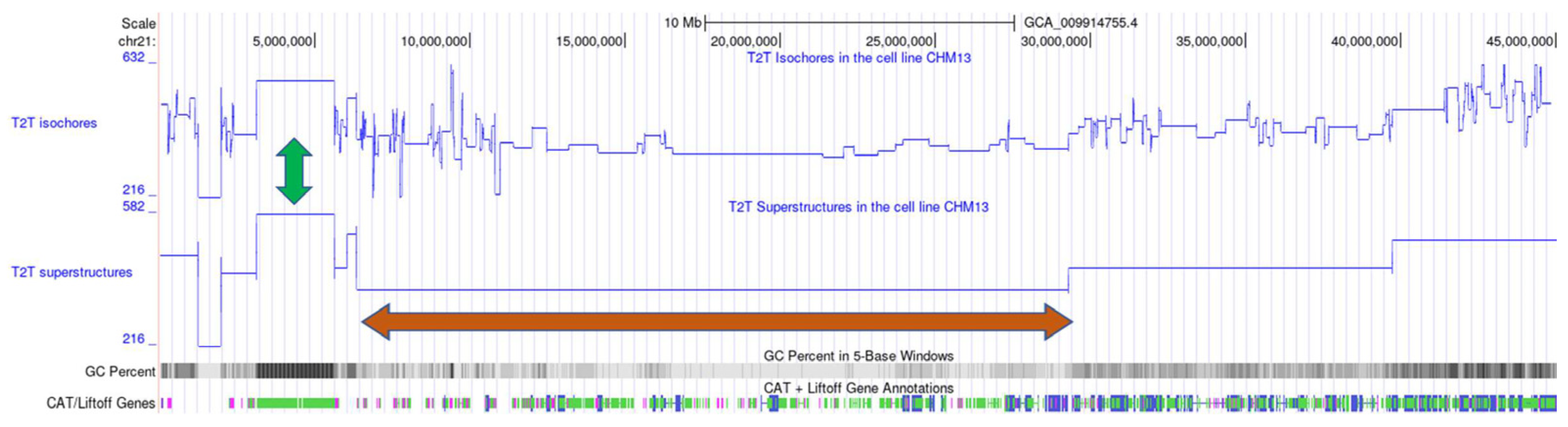
4.1. Detection of Genome Compositional Superstructures by Segmentation
4.2. Hierarchical Organization of Compositional Genome Structures
4.3. Functional Significance of Compositional Structures
5. Segment Compositional Signature (DJS)
6. Sequence Compositional Complexity (SCC)
- The SCC value is 0 if no segments are identified in the sequence, indicating that it is compositionally homogeneous, such as a random sequence.
- By using a statistical significance threshold over the segmentation step, SCC ensures that the difference between each pair of adjacent domains is not merely due to statistical fluctuations.
- SCC has a high sensitivity to sequence changes. A single nucleotide substitution, or a small indel, can often be sufficient to alter the number, length, or nucleotide frequencies of compositional domains and, consequently, affect the resulting SCC value.
- It increases/decreases with both the number of segments and the degree of compositional differences among them. In this way, SCC is analogous to the measure used by McShea and Brandon [74] for obtaining complexity estimates based on morphological characters: an organism is more complex if it has a greater number of parts and/or a higher differentiation among these parts.
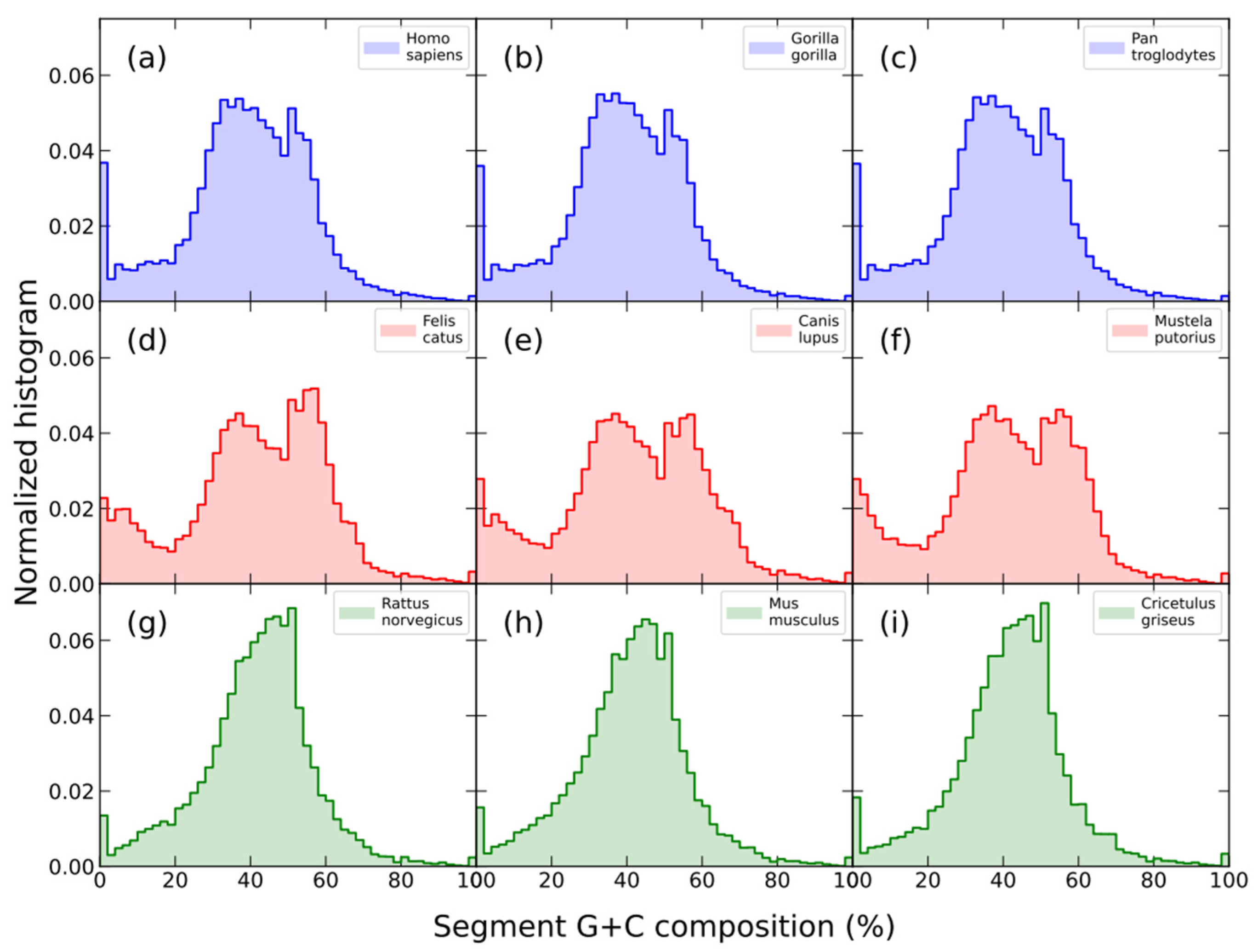
7. Phylogenetic Trends of Compositional Genome Structure
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, V.; Goren, A.; Bernstein, B. Charting Histone Modifications and the Functional Organization of Mammalian Genomes. Nat. Rev. Genet. 2010, 12, 7–18. [Google Scholar] [CrossRef]
- Bernardi, G. Structural and Evolutionary Genomics: Natural Selection in Genome Evolution; Elsevier: Amsterdam, The Netherlands, 2004; ISBN 9780444512550. [Google Scholar]
- Moya, A.; Oliver, J.L.; Verdú, M.; Delaye, L.; Arnau, V.; Bernaola-Galván, P.; de la Fuente, R.; Díaz, W.; Gómez-Martín, C.; González, F.; et al. Driven Progressive Evolution of Genome Sequence Complexity in Cyanobacteria. Sci. Rep. 2020, 10, 19073. [Google Scholar] [CrossRef] [PubMed]
- Elhaik, E.; Graur, D. A Comparative Study and a Phylogenetic Exploration of the Compositional Architectures of Mammalian Nuclear Genomes. PLoS Comput. Biol. 2014, 10, e1003925. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Macaya, G.; Bernardi, G. An Analysis of Eukaryotic Genomes by Density Gradient Centrifugation. J. Mol. Biol. 1976, 108, 219–235. [Google Scholar] [CrossRef]
- Bernardi, G.; Olofsson, B.; Filipski, J.; Zerial, M.; Salinas, J.; Cuny, G.; Meunier-Rotival, M.; Rodier, F. The Mosaic Genome of Warm-Blooded Vertebrates. Science 1985, 228, 953–958. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, G. Isochores and the Evolutionary Genomics of Vertebrates. Gene 2000, 241, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Clay, O.; Bernardi, G. The Isochores in Human Chromosomes 21 and 22. Biochem. Biophys. Res. 2001, 285, 855–856. [Google Scholar] [CrossRef]
- Bernardi, G. The Genomic Code: A Pervasive Encoding/Molding of Chromatin Structures and a Solution of the “Non-Coding DNA” Mystery. BioEssays 2019, 41, 1900106. [Google Scholar] [CrossRef]
- Lamolle, G.; Sabbia, V.; Musto, H.; Bernardi, G. The Short-Sequence Design of DNA and Its Involvement in the 3-D Structure of the Genome. Sci. Rep. 2018, 8, 17820. [Google Scholar] [CrossRef]
- Bernardi, G. The Formation of Chromatin Domains Involves a Primary Step Based on the 3-D Structure of DNA. Sci. Rep. 2018, 8, 17821. [Google Scholar] [CrossRef]
- Jabbari, K.; Bernardi, G. An Isochore Framework Underlies Chromatin Architecture. PLoS ONE 2017, 12, e0168023. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The Complete Sequence of a Human Genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kaneko, K. Long-Range Correlations and Partial 1/Fa Spectrum in a Noncoding DNA Sequence. Europhys. Lett. 1992, 17, 555–660. [Google Scholar] [CrossRef]
- Peng, C.C.-K.K.; Buldyrev, S.V.S.; Goldberger, A.L.; Havlin, S.; Sciortino, F.; Simons, M.; Stanley, H.E. Long-Range Correlations in Nucleotide Sequences. Nature 1992, 356, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Voss, R.F. Evolution of Long-Range Fractal Correlations and 1/Fnoise in DNA Base Sequences. Phys. Rev. Lett. 1992, 68, 3805–3808. [Google Scholar] [CrossRef]
- Bernaola-Galván, P.; Carpena, P.; Román-Roldán, R.; Oliver, J.L. Study of Statistical Correlations in DNA Sequences. Gene 2002, 300, 105–115. [Google Scholar] [CrossRef]
- Filipski, J.; Thiery, J.P.; Bernardi, G. An Analysis of the Bovine Genome by Cs2SO4-Ag Density Gradient Centrifugation. J. Mol. Biol. 1973, 80, 177–197. [Google Scholar] [CrossRef]
- Oliver, J.L.; Carpena, P.; Hackenberg, M.; Bernaola-Galván, P. IsoFinder: Computational Prediction of Isochores in Genome Sequences. Nucleic Acids Res. 2004, 32, W287–W292. [Google Scholar] [CrossRef]
- Costantini, M.; Clay, O.; Auletta, F.; Bernardi, G. An Isochore Map of Human Chromosomes. Genome Res. 2006, 16, 536–541. [Google Scholar] [CrossRef]
- Carpena, P.; Bernaola-Galván, P.; Coronado, A.V.; Hackenberg, M.; Oliver, J.L. Identifying Characteristic Scales in the Human Genome. Phys. Rev. E 2007, 75, 032903. [Google Scholar] [CrossRef]
- Viswanathan, G.M.; Buldyrev, S.V.; Havlin, S.; Stanley, H.E. Quantification of DNA Patchiness Using Long-Range Correlation Measures. Biophys. J. 1997, 72, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.L.; Bernaola-Galván, P.; Hackenberg, M.; Carpena, P. Phylogenetic Distribution of Large-Scale Genome Patchiness. BMC Evol. Biol. 2008, 8, 107. [Google Scholar] [CrossRef] [PubMed]
- Carpena, P.; Oliver, J.L.; Hackenberg, M.; Coronado, A.V.; Barturen, G.; Bernaola-Galván, P. High-Level Organization of Isochores into Gigantic Superstructures in the Human Genome. Phys. Rev. E 2011, 83, 031908. [Google Scholar] [CrossRef]
- Román-Roldán, R.; Bernaola-Galván, P.; Oliver, J. Sequence Compositional Complexity of DNA through an Entropic Segmentation Method. Phys. Rev. Lett. 1998, 80, 1344–1347. [Google Scholar] [CrossRef]
- Bonnici, V.; Franco, G.; Manca, V. A Word Recurrence Based Algorithm to Extract Genomic Dictionaries. arXiv 2020, arXiv:2009.10449. [Google Scholar]
- Almeida, J.S.; Carriço, J.A.; Maretzek, A.; Noble, P.A.; Fletcher, M. Analysis of Genomic Sequences by Chaos Game Representation. Bioinformatics 2001, 17, 429–437. [Google Scholar] [CrossRef]
- Gell-Mann, M.; Lloyd, S. Information Measures, Effective Complexity, and Total Information. Complexity 1996, 2, 44–52. [Google Scholar] [CrossRef]
- de la Fuente, R.; Díaz-Villanueva, W.; Arnau, V.; Moya, A. Genomic Signature in Evolutionary Biology: A Review. Biology 2023, 12, 322. [Google Scholar] [CrossRef]
- Bernardi, G. The Human Genome: Organization and Evolutionary History. Annu. Rev. Genet. 1995, 29, 445–476. [Google Scholar] [CrossRef]
- Elhaik, E.; Graur, D.; Josić, K. Comparative Testing of DNA Segmentation Algorithms Using Benchmark Simulations. Mol. Biol. Evol. 2010, 27, 1015–1024. [Google Scholar] [CrossRef]
- Larhammar, D.; Chatzidimitriou-Dreismann, C. a Biological Origins of Long-Range Correlations and Compositional Variations in DNA. Nucleic Acids Res. 1993, 21, 5167–5170. [Google Scholar] [CrossRef] [PubMed]
- Stanley, H.E.; Buldyrev, S.V.; Goldberger, A.L.; Goldberger, Z.D.; Havlin, S.; Mantegna, R.N.; Ossadnik, S.M.; Peng, C.K.; Simons, M. Statistical Mechanics in Biology: How Ubiquitous Are Long-Range Correlations? Phys. A Stat. Mech. Its Appl. 1994, 205, 214–253. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Buldyrev, S.; Havlin, S. Mosaic Organization of DNA Nucleotides. Phys. Rev. E 1994, 49, 1685. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.L.; Román-Roldán, R.; Pérez, J.; Bernaola-Galván, P. SEGMENT: Identifying Compositional Domains in DNA Sequences. Bioinformatics 1999, 15, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Bernaola-Galván, P.; Román-Roldán, R.; Oliver, J.L. Compositional Segmentation and Long-Range Fractal Correlations in DNA Sequences. Phys. Rev. E 1996, 53, 5181–5189. [Google Scholar] [CrossRef]
- Bernaola-Galván, P.; Oliver, J.L.; Hackenberg, M.; Coronado, A.V.; Ivanov, P.C.; Carpena, P. Segmentation of Time Series with Long-Range Fractal Correlations. Eur. Phys. J. B 2012, 85, 211. [Google Scholar] [CrossRef]
- Bernaola-Galván, P.; Grosse, I.; Carpena, P.; Oliver, J.L.; Román-Roldán, R. Finding Borders between Coding and Noncoding DNA Regions by an Entropic Segmentation Method. Phys. Rev. Lett. 2000, 85, 1342–1345. [Google Scholar] [CrossRef]
- Tenzen, T.; Yamagata, T.; Fukagawa, T.; Sugaya, K.; Ando, A.; Inoko, H.; Gojobori, T.; Fujiyama, A.; Okumura, K.; Ikemura, T. Precise Switching of DNA Replication Timing in the GC Content Transition Area in the Human Major Histocompatibility Complex. Mol. Cell. Biol. 1997, 17, 4043–4050. [Google Scholar] [CrossRef]
- Eisenbarth, I.; Vogel, G.; Krone, W.; Vogel, W.; Assum, G. An Isochore Transition in the NF1 Gene Region Coincides with a Switch in the Extent of Linkage Disequilibrium. Am. J. Hum. Genet. 2000, 67, 873–880. [Google Scholar] [CrossRef]
- Costantini, M.; Alvarez-Valin, F.; Costantini, S.; Cammarano, R.; Bernardi, G. Compositional Patterns in the Genomes of Unicellular Eukaryotes. BMC Genom. 2013, 14, 755. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, C.-T. Isochore Structures in the Genome of the Plant Arabidopsis Thaliana. J. Mol. Evol. 2004, 59, 227–238. [Google Scholar] [CrossRef]
- Fortes, G.G.; Bouza, C.; Martínez, P.; Sánchez, L. Diversity in Isochore Structure among Cold-Blooded Vertebrates Based on GC Content of Coding and Non-Coding Sequences. Genetica 2007, 129, 281–289. [Google Scholar] [CrossRef]
- Nekrutenko, A.; Li, W.; Nekrutenko, A.; Li, W. Assessment of Compositional Heterogeneity within and between Eukaryotic Genomes Assessment of Compositional Heterogeneity within and between Eukaryotic Genomes. Genome Res. 2000, 10, 1986–1995. [Google Scholar] [CrossRef]
- Eyre-Walker, A.; Hurst, L.D. The Evolution of Isochores. Nat. Rev. Genet. 2001, 2, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Costantini, M.; Musto, H. The Isochores as a Fundamental Level of Genome Structure and Organization: A General Overview. J. Mol. Evol. 2017, 84, 93–103. [Google Scholar] [CrossRef]
- Eyre-Walker, A. Evidence That Both G + C Rich and G + C Poor Isochores Are Replicated Early and Late in the Cell Cycle. Nucleic Acids Res 1992, 20, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Francino, M.; Ochman, H. Isochores Result from Mutation Not Selection. Nature 1999, 400, 30–31. [Google Scholar] [CrossRef] [PubMed]
- Fryxell, K.J.; Zuckerkandl, E. Cytosine Deamination Plays a Primary Role in the Evolution of Mammalian Isochores. Mol. Biol. Evol. 2000, 17, 1371–1383. [Google Scholar] [CrossRef]
- Piganeau, G.; Mouchiroud, D.; Duret, L.; Gautier, C. Expected Relationship between the Silent Substitution Rate and the GC Content: Implications for the Evolution of Isochores. J. Mol. Evol. 2002, 54, 129–133. [Google Scholar] [CrossRef]
- Kahl, G. Genomic Sequencing. In The Dictionary of Genomics, Transcriptomics and Proteomics; Markono Print Media Pte Ltd.: Singapore, 2015; p. 1. [Google Scholar]
- Oliver, J.L.; Bernaola-Galván, P.; Carpena, P.; Román-Roldán, R. Isochore Chromosome Maps of Eukaryotic Genomes. Gene 2001, 276, 47–56. [Google Scholar] [CrossRef]
- Oliver, J.L.; Carpena, P.; Román-Roldán, R.; Mata-Balaguer, T.; Mejías-Romero, A.; Hackenberg, M.; Bernaola-Galván, P. Isochore Chromosome Maps of the Human Genome. Gene 2002, 300, 117–127. [Google Scholar] [CrossRef]
- Wen, S.-Y.; Zhang, C.-T. Identification of Isochore Boundaries in the Human Genome Using the Technique of Wavelet Multiresolution Analysis. Biochem. Biophys. Res. Commun. 2003, 311, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Fearnhead, P.; Vasilieou, D. Bayesian Analysis of Isochores. J. Am. Stat. Assoc. 2009, 104, 132–141. [Google Scholar] [CrossRef]
- Kent, J. Genome Browser Software. Available online: https://users.soe.ucsc.edu/~kent/ (accessed on 16 December 2016).
- Nassar, L.R.; Barber, G.P.; Benet-Pagès, A.; Casper, J.; Clawson, H.; Diekhans, M.; Fischer, C.; Gonzalez, J.N.; Hinrichs, A.S.; Lee, B.T.; et al. The UCSC Genome Browser Database: 2023 Update. Nucleic Acids Res. 2022, 51, D1188–D1195. [Google Scholar] [CrossRef]
- Li, W.; Kaneko, K. DNA Correlations. Nature 1992, 360, 635–636. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.; Cremer, C. Chromosome Territories, Nuclear Architecture and Gene Regulation in Mammalian Cells. Nat. Rev. Genet. 2001, 2, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.; Cremer, M. Chromosome Territories. Cold Spring Harb. Perspect. Biol. 2010, 2, a003889. [Google Scholar] [CrossRef]
- Li, W.; Stolovitzky, G.; Bernaola-Galva, P.; Oliver, L. Compositional Heterogeneity within, and Uniformity between, DNA Sequences of Yeast Chromosomes. Genome 1998, 8, 916–928. [Google Scholar] [CrossRef]
- Wolf, Y.I.; Katsnelson, M.I.; Koonin, E.V. Physical Foundations of Biological Complexity. Proc. Natl. Acad. Sci. USA 2018, 115, E8678–E8687. [Google Scholar] [CrossRef]
- Hackenberg, M.; Bernaola-Galván, P.; Carpena, P.; Oliver, J.L. The Biased Distribution of Alus in Human Isochores Might Be Driven by Recombination. J. Mol. Evol. 2005, 60, 365–377. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.J.; Gene, T.; Consortium, O.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; et al. Gene Ontology: Tool for the Unification of Biology. Nature 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Deschavanne, P.J.; Giron, A.; Vilain, J.; Fagot, G.; Fertil, B. Genomic Signature: Characterization and Classification of Species Assessed by Chaos Game Representation of Sequences. Mol. Biol. Evol. 1999, 16, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Bonnici, V.; Manca, V. Informational Laws of Genome Structures. Sci. Rep. 2016, 6, 28840. [Google Scholar] [CrossRef]
- McHardy, A.C.; Martín, H.G.; Tsirigos, A.; Hugenholtz, P.; Rigoutsos, I. Accurate Phylogenetic Classification of Variable-Length DNA Fragments. Nat. Methods 2007, 4, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Dufraigne, C.; Fertil, B.; Lespinats, S.; Giron, A.; Deschavanne, P. Detection and Characterization of Horizontal Transfers in Prokaryotes Using Genomic Signature. Nucleic Acids Res. 2005, 33, e6. [Google Scholar] [CrossRef]
- Mrazek, J. Phylogenetic Signals in DNA Composition: Limitations and Prospects. Mol. Biol. Evol. 2009, 26, 1163–1169. [Google Scholar] [CrossRef]
- Bernaola-Galván, P.; Oliver, J.L.; Gómez-Martín, C.; Carpena, P. Segment Compositional Signature. 2023; in preparation. [Google Scholar]
- Grosse, I.; Bernaola-Galván, P.; Carpena, P.; Román-Roldán, R.; Oliver, J.; Stanley, H.E. Analysis of Symbolic Sequences Using the Jensen-Shannon Divergence. Phys. Rev. E 2002, 65, 16. [Google Scholar] [CrossRef]
- Lin, J. Divergence Measures Based on the Shannon Entropy. IEEE Trans. Inf. Theory 1991, 37, 145–151. [Google Scholar] [CrossRef]
- Beyer, W.A.; Stein, M.L.; Smith, T.F.; Ulam, S.M. A Molecular Sequence Metric and Evolutionary Trees. Math. Biosci. 1974, 19, 9–25. [Google Scholar] [CrossRef]
- McShea, D.W.; Brandon, R.N. Biology’s First Law: The Tendency for Diversity and Complexity to Increase in Evolutionary Systems; University of Chicago Press: Chicago, IL, USA, 2010; ISBN 9780226562278. [Google Scholar]
- Sergeev, V.N.; Gerasimenko, L.M.; Zavarzin, G.A. Proterozoic History and Present State of Cyanobacteria. Microbiology 2002, 71, 623–637. [Google Scholar] [CrossRef]
- Schirrmeister, B.E.; de Vos, J.M.; Antonelli, A.; Bagheri, H.C. Evolution of Multicellularity Coincided with Increased Diversification of Cyanobacteria and the Great Oxidation Event. Proc. Natl. Acad. Sci. USA 2013, 110, 1791–1796. [Google Scholar] [CrossRef] [PubMed]
- Bekker, A.; Holland, H.D.; Wang, P.-L.; Rumble, D.; Stein, H.J.; Hannah, J.L.; Coetzee, L.L.; Beukes, N.J. Dating the Rise of Atmospheric Oxygen. Nature 2004, 427, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Hedges, S.; Blair, J.E.; Venturi, M.L.; Shoe, J.L. A Molecular Timescale of Eukaryote Evolution and the Rise of Complex Multicellular Life. BMC Evol. Biol. 2004, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.-W.; Asri, M.; Ebler, J.; Doerr, D.; Haukness, M.; Hickey, G.; Lu, S.; Lucas, J.K.; Monlong, J.; Abel, H.J.; et al. A Draft Human Pangenome Reference. Nature 2023, 617, 312–324. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Length | GC% | ||||||
|---|---|---|---|---|---|---|---|
| Chromosome | N | Min | Median | Max | Min | Median | Max |
| chr1 | 1113 | 30,004 | 102,983 | 5,403,580 | 32.80 | 43.02 | 67.96 |
| chr2 | 897 | 30,004 | 128,804 | 4,304,270 | 31.57 | 41.20 | 66.39 |
| chr3 | 656 | 30,004 | 127,485 | 5,001,190 | 21.54 | 41.23 | 62.17 |
| chr4 | 427 | 30,004 | 234,223 | 5,642,550 | 23.80 | 39.15 | 72.64 |
| chr5 | 561 | 30,004 | 164,800 | 7,206,270 | 30.21 | 40.70 | 62.46 |
| chr6 | 510 | 30,004 | 163,262 | 3,500,380 | 32.19 | 40.85 | 58.39 |
| chr7 | 596 | 30,004 | 119,830 | 3,412,220 | 33.19 | 42.44 | 68.05 |
| chr8 | 449 | 30,004 | 147,916 | 4,875,420 | 33.27 | 41.36 | 63.98 |
| chr9 | 501 | 30,004 | 107,885 | 22,256,800 | 31.74 | 42.57 | 65.87 |
| chr10 | 562 | 30,004 | 121,504 | 3,195,210 | 32.63 | 42.09 | 72.51 |
| chr11 | 551 | 30,004 | 109,143 | 3,008,680 | 33.64 | 42.81 | 62.56 |
| chr12 | 505 | 30,005 | 115,060 | 3,649,550 | 32.99 | 42.49 | 63.91 |
| chr13 | 317 | 30,004 | 128,955 | 10,449,500 | 21.22 | 40.27 | 60.57 |
| chr14 | 426 | 30,004 | 101,284 | 3,881,480 | 21.89 | 42.01 | 63.58 |
| chr15 | 461 | 30,004 | 104,795 | 7,482,370 | 21.37 | 42.62 | 62.03 |
| chr16 | 457 | 30,004 | 82,508 | 12,645,100 | 33.24 | 44.86 | 66.31 |
| chr17 | 517 | 30,004 | 82,785 | 4,713,850 | 33.08 | 45.61 | 62.57 |
| chr18 | 237 | 30,004 | 180,918 | 3,584,850 | 34.03 | 40.14 | 56.08 |
| chr19 | 313 | 30,004 | 101,006 | 2,676,290 | 35.20 | 48.09 | 65.30 |
| chr20 | 314 | 30,004 | 101,682 | 2,232,290 | 32.89 | 44.17 | 65.15 |
| chr21 | 178 | 30,004 | 95,755 | 4,852,870 | 21.63 | 42.14 | 63.20 |
| chr22 | 342 | 30,004 | 68,700 | 1,690,830 | 21.25 | 46.26 | 64.72 |
| chrX | 366 | 30,004 | 166,946 | 14,835,700 | 22.41 | 40.66 | 62.24 |
| N | Minimum | Median | Maximum | |
|---|---|---|---|---|
| Length (bp) | 11,256 | 30,005.00 | 116,447.00 | 22,256,800.00 |
| GC% | 11,256 | 21.22 | 42.24 | 72.64 |
| Length | GC% | ||||||
|---|---|---|---|---|---|---|---|
| Chromosome | N | Min | Median | Max | Min | Median | Max |
| chr1 | 4 | 328,708.00 | 23,816,031.50 | 200,426,557.00 | 40.02 | 45.53 | 58.20 |
| chr2 | 4 | 3,360,333.00 | 51,107,636.50 | 137,121,146.00 | 38.23 | 43.43 | 50.92 |
| chr3 | 3 | 36,078,355.00 | 73,698,202.00 | 91,329,391.00 | 35.28 | 39.59 | 41.28 |
| chr4 | 4 | 489,000.00 | 4,883,872.00 | 183,318,201.00 | 37.50 | 44.89 | 55.27 |
| chr5 | 6 | 1,719,665.00 | 10,580,851.50 | 128,874,195.00 | 38.12 | 44.70 | 53.09 |
| chr6 | 5 | 4,367,830.00 | 18,410,867.00 | 104,707,592.00 | 37.67 | 41.22 | 46.35 |
| chr7 | 4 | 2,840,530.00 | 7,380,668.00 | 142,965,562.00 | 39.85 | 46.63 | 54.87 |
| chr8 | 5 | 532,709.00 | 18,506,340.00 | 74,252,182.00 | 38.17 | 41.71 | 55.45 |
| chr9 | 5 | 745,901.00 | 7,358,207.00 | 105,799,637.00 | 37.88 | 48.44 | 56.06 |
| chr10 | 6 | 31,699.00 | 790,594.50 | 122,866,079.00 | 41.00 | 45.76 | 72.51 |
| chr11 | 3 | 230,004.00 | 3,096,068.00 | 131,801,697.00 | 41.22 | 42.54 | 55.51 |
| chr12 | 5 | 295,312.00 | 8,431,710.00 | 98,848,568.00 | 39.05 | 45.18 | 52.94 |
| chr13 | 7 | 167,828.00 | 3,576,422.00 | 99,522,852.00 | 21.87 | 42.77 | 57.94 |
| chr14 | 6 | 935,799.00 | 5,282,438.00 | 78,687,833.00 | 39.51 | 45.26 | 55.98 |
| chr15 | 1 | 99,753,195.00 | 99,753,195.00 | 99,753,195.00 | 42.12 | 42.12 | 42.12 |
| chr16 | 8 | 2,323,621.00 | 6,794,467.50 | 37,570,616.00 | 36.35 | 46.71 | 57.98 |
| chr17 | 3 | 10,136,275.00 | 22,311,188.00 | 51,829,434.00 | 41.51 | 45.56 | 52.19 |
| chr18 | 6 | 299,493.00 | 2,825,872.00 | 64,251,224.00 | 35.92 | 42.88 | 53.05 |
| chr19 | 7 | 33,056.00 | 4,915,345.00 | 30,358,228.00 | 35.71 | 47.91 | 56.35 |
| chr20 | 4 | 2,912,961.00 | 16,593,634.00 | 30,110,026.00 | 40.53 | 45.21 | 55.82 |
| chr21 | 9 | 307,155.00 | 1,240,965.00 | 22,967,603.00 | 21.63 | 43.15 | 58.18 |
| chr22 | 2 | 17,629,880.00 | 25,662,463.00 | 33,695,046.00 | 41.31 | 44.77 | 48.23 |
| chrX | 6 | 272,109.00 | 1,306,918.50 | 148,323,701.00 | 39.21 | 46.71 | 55.04 |
| N | Minimum | Median | Maximum | |
|---|---|---|---|---|
| Length (bp) | 113 | 31,699.00 | 6,111,300.50 | 200,427,000.00 |
| GC% | 113 | 21.63 | 45.10 | 72.51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernaola-Galván, P.; Carpena, P.; Gómez-Martín, C.; Oliver, J.L. Compositional Structure of the Genome: A Review. Biology 2023, 12, 849. https://doi.org/10.3390/biology12060849
Bernaola-Galván P, Carpena P, Gómez-Martín C, Oliver JL. Compositional Structure of the Genome: A Review. Biology. 2023; 12(6):849. https://doi.org/10.3390/biology12060849
Chicago/Turabian StyleBernaola-Galván, Pedro, Pedro Carpena, Cristina Gómez-Martín, and Jose L. Oliver. 2023. "Compositional Structure of the Genome: A Review" Biology 12, no. 6: 849. https://doi.org/10.3390/biology12060849
APA StyleBernaola-Galván, P., Carpena, P., Gómez-Martín, C., & Oliver, J. L. (2023). Compositional Structure of the Genome: A Review. Biology, 12(6), 849. https://doi.org/10.3390/biology12060849