
Angiotensin-Related Peptides and Their Role in Pain Regulation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
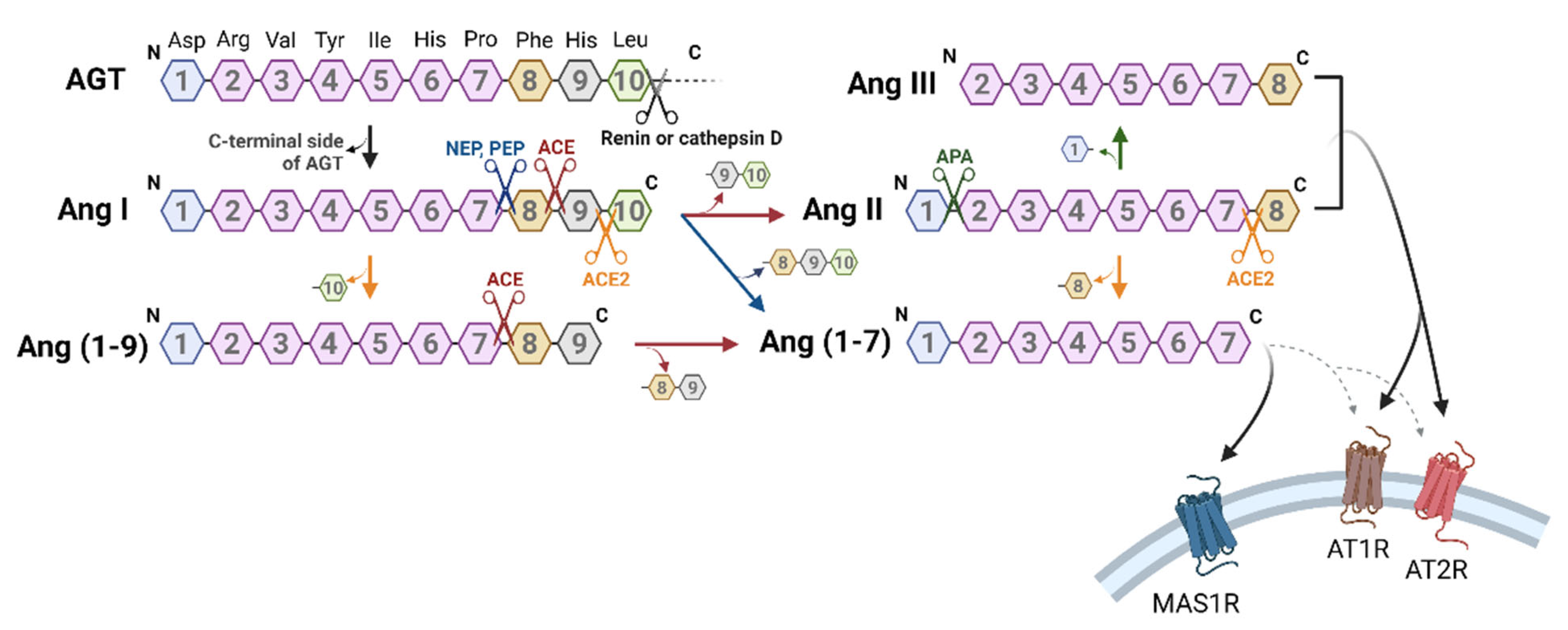
2. Biosynthetic Mechanism of Angiotensin-Related Peptides
3. Mechanisms of Pain Regulation in the Central Nervous System by Angiotensin-Related Peptides
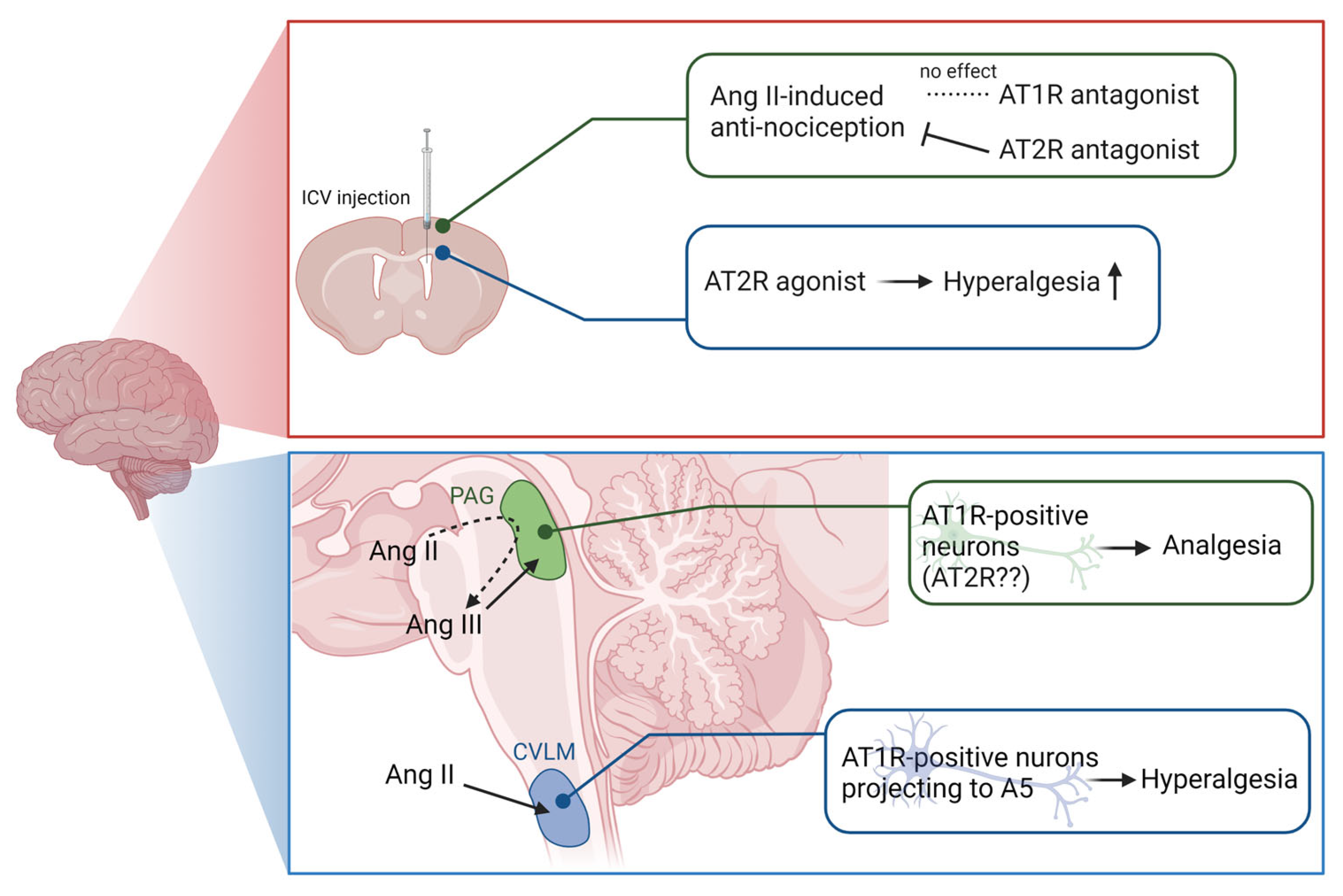
3.1. Brain
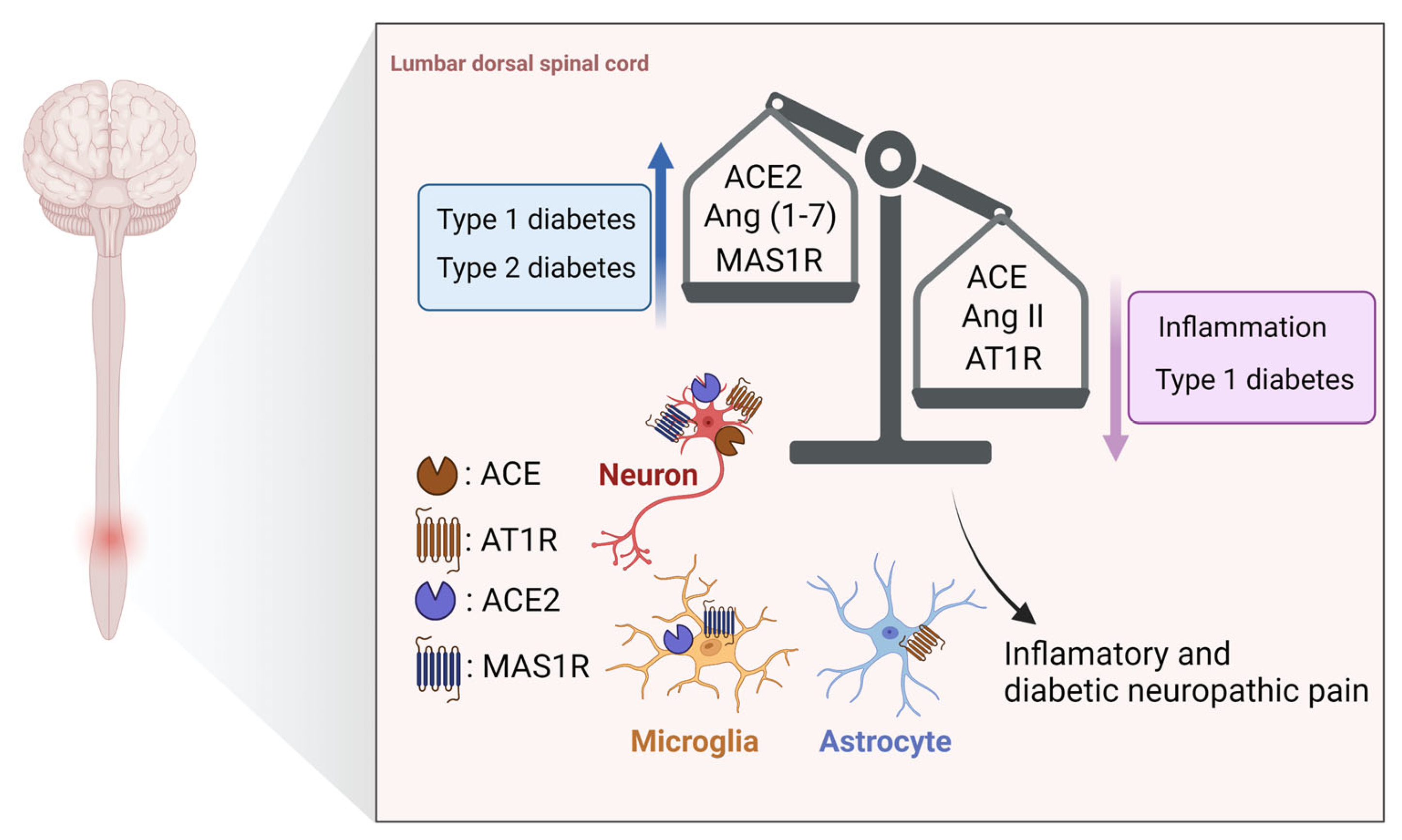
3.2. Spinal Cord
4. Mechanisms of Pain Regulation in Peripheral Tissue by Ang-Related Peptides
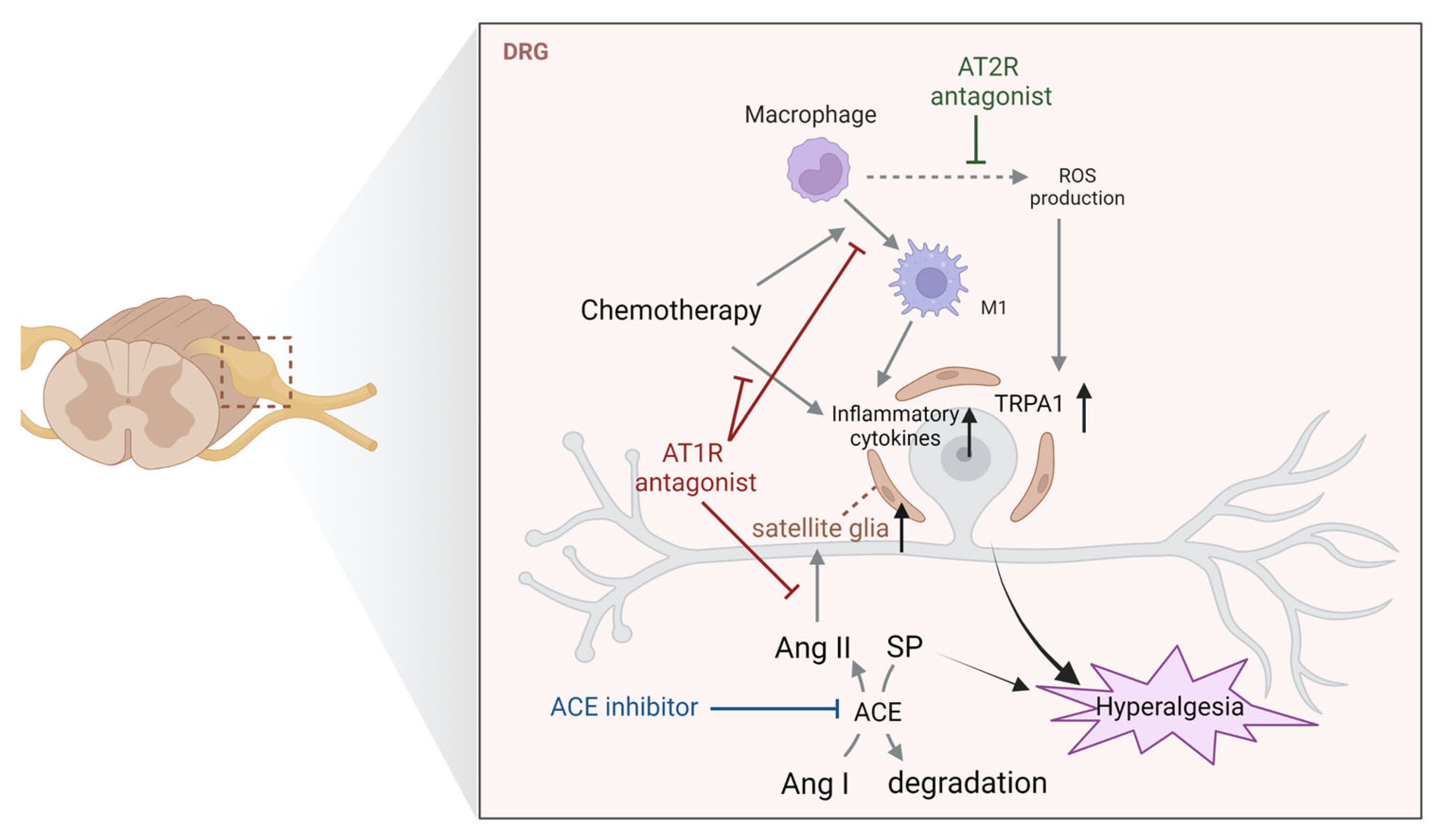
4.1. Dorsal Root Ganglia
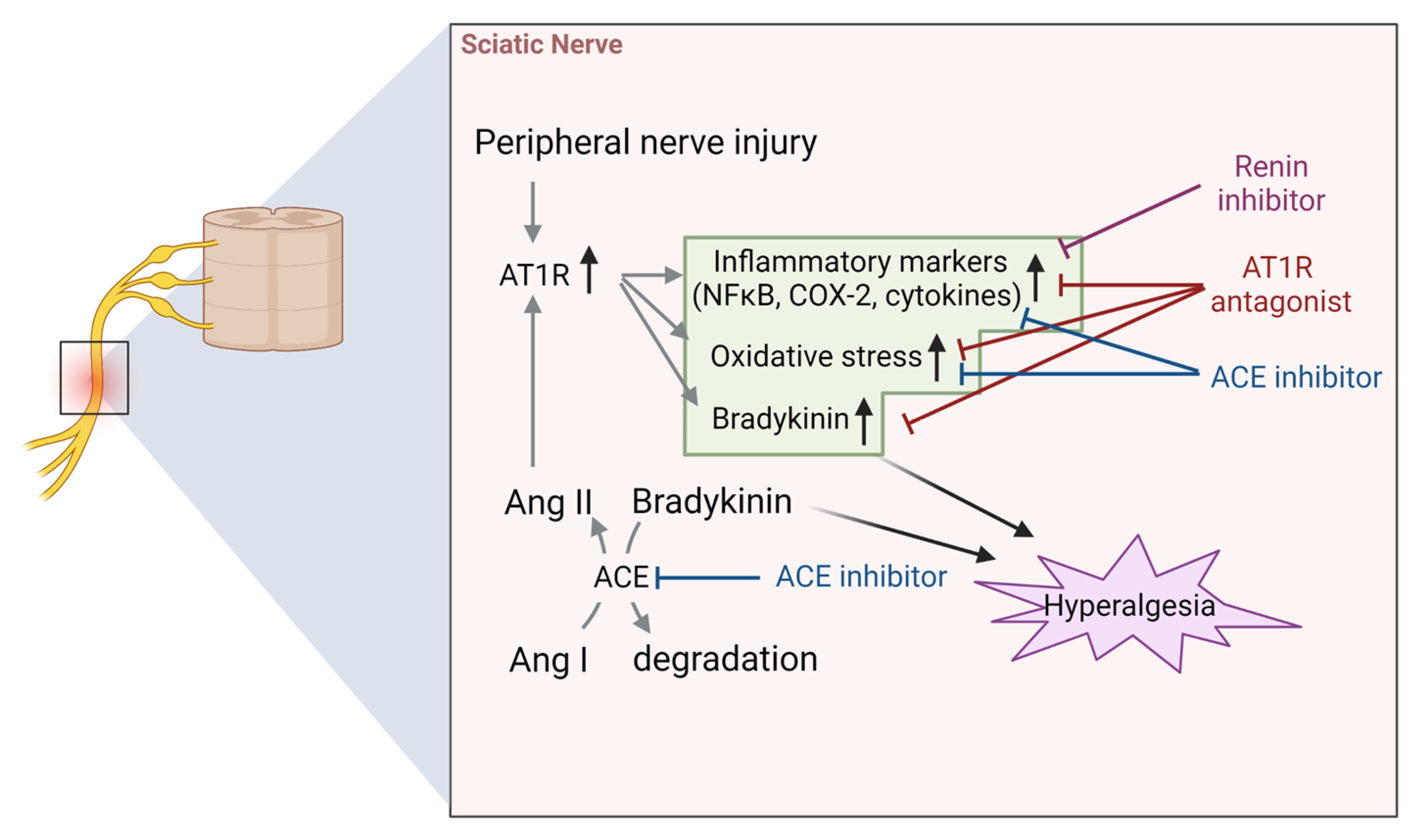
4.2. Sciatic Nerve
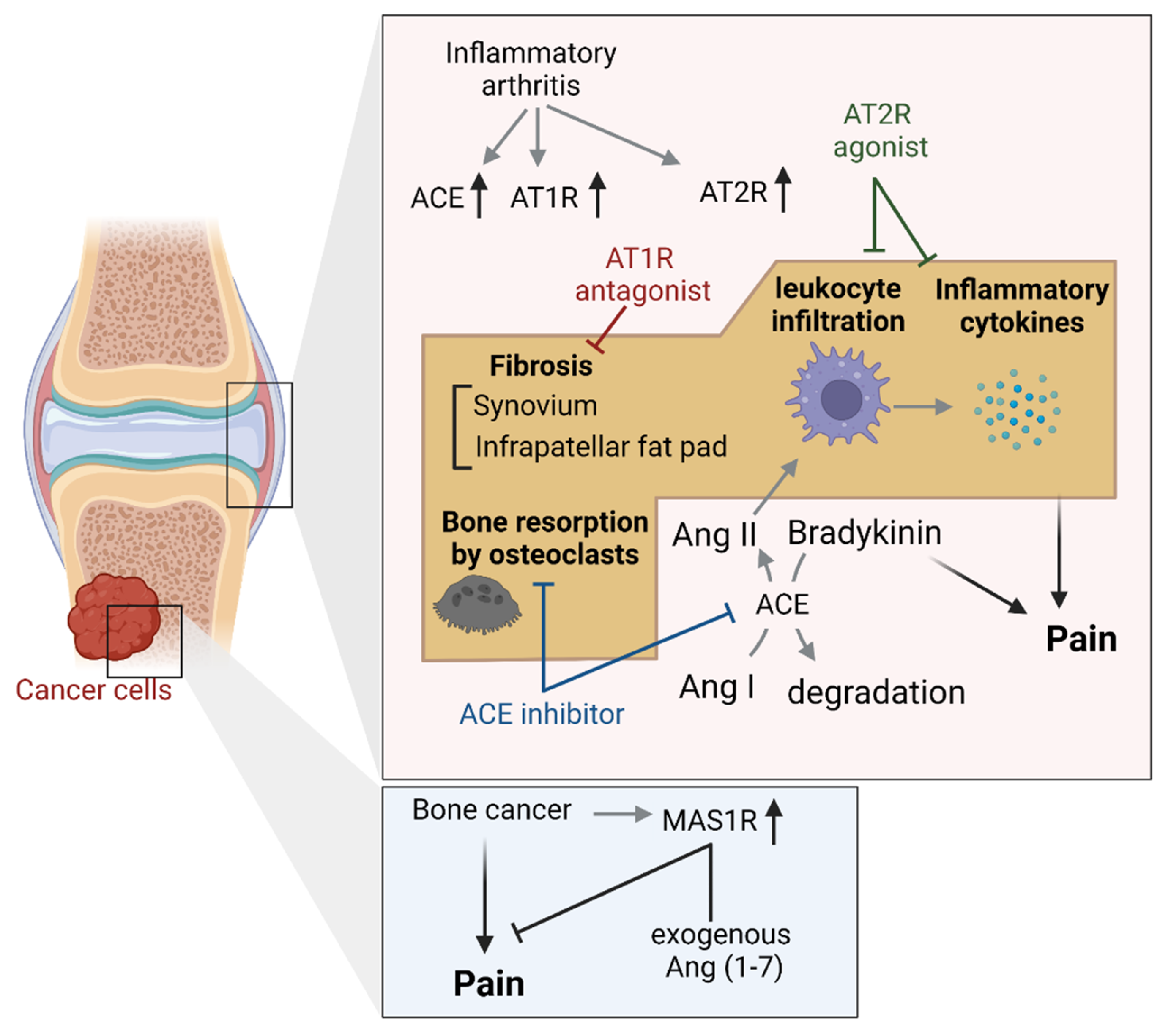
4.3. Joint and Bone Tissues
4.4. Other Findings in Peripheral Pain Models
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalupahana, N.S.; Moustaid-Moussa, N. The renin-angiotensin system: A link between obesity, inflammation and insulin resistance. Obes. Rev. 2012, 13, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Siragy, H.M.; Carey, R.M. Role of the intrarenal renin-angiotensin-aldosterone system in chronic kidney disease. Am. J. Nephrol. 2010, 31, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Simoes, E.S.A.C.; Miranda, A.S.; Rocha, N.P.; Teixeira, A.L. Renin angiotensin system in liver diseases: Friend or foe? World J. Gastroenterol. 2017, 23, 3396–3406. [Google Scholar] [CrossRef] [PubMed]
- Baltatu, O.C.; Campos, L.A.; Bader, M. Local renin-angiotensin system and the brain—A continuous quest for knowledge. Peptides 2011, 32, 1083–1086. [Google Scholar] [CrossRef]
- Frigolet, M.E.; Torres, N.; Tovar, A.R. The renin-angiotensin system in adipose tissue and its metabolic consequences during obesity. J. Nutr. Biochem. 2013, 24, 2003–2015. [Google Scholar] [CrossRef]
- Patil, J.; Schwab, A.; Nussberger, J.; Schaffner, T.; Saavedra, J.M.; Imboden, H. Intraneuronal angiotensinergic system in rat and human dorsal root ganglia. Regul. Pept. 2010, 162, 90–98. [Google Scholar] [CrossRef]
- Ogata, Y.; Nemoto, W.; Nakagawasai, O.; Yamagata, R.; Tadano, T.; Tan-No, K. Involvement of spinal angiotensin II system in streptozotocin-induced diabetic neuropathic pain in mice. Mol. Pharmacol. 2016, 90, 205–213. [Google Scholar] [CrossRef]
- Tsukamoto, I.; Akagi, M.; Inoue, S.; Yamagishi, K.; Mori, S.; Asada, S. Expressions of local renin-angiotensin system components in chondrocytes. Eur. J. Histochem. 2014, 58, 2387. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1–7)/MAS axis of the renin-angiotensin system: Focus on angiotensin-(1–7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef]
- Caroccia, B.; Vanderriele, P.E.; Seccia, T.M.; Piazza, M.; Lenzini, L.; Prisco, S.; Torresan, F.; Domenig, O.; Iacobone, M.; Poglitsch, M.; et al. Aldosterone and cortisol synthesis regulation by angiotensin-(1–7) and angiotensin-converting enzyme 2 in the human adrenal cortex. J. Hypertens. 2021, 39, 1577–1585. [Google Scholar] [CrossRef]
- Teixeira, L.B.; Parreiras, E.S.L.T.; Bruder-Nascimento, T.; Duarte, D.A.; Simoes, S.C.; Costa, R.M.; Rodriguez, D.Y.; Ferreira, P.A.B.; Silva, C.A.A.; Abrao, E.P.; et al. Ang-(1–7) is an endogenous beta-arrestin-biased agonist of the AT(1) receptor with protective action in cardiac hypertrophy. Sci. Rep. 2017, 7, 11903. [Google Scholar] [CrossRef]
- Goru, S.K.; Kadakol, A.; Malek, V.; Pandey, A.; Sharma, N.; Gaikwad, A.B. Diminazene aceturate prevents nephropathy by increasing glomerular ACE2 and AT2 receptor expression in a rat model of type1 diabetes. Br. J. Pharmacol. 2017, 174, 3118–3130. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Mogi, M.; Nakaoka, H.; Iwanami, J.; Min, L.J.; Kanno, H.; Tsukuda, K.; Chisaka, T.; Bai, H.Y.; Wang, X.L.; et al. Possible role of angiotensin-converting enzyme 2 and activation of angiotensin II type 2 receptor by angiotensin-(1–7) in improvement of vascular remodeling by angiotensin II type 1 receptor blockade. Hypertension 2014, 63, e53-59. [Google Scholar] [CrossRef] [PubMed]
- Marques-Lopes, J.; Pinto, M.; Pinho, D.; Morato, M.; Patinha, D.; Albino-Teixeira, A.; Tavares, I. Microinjection of angiotensin II in the caudal ventrolateral medulla induces hyperalgesia. Neuroscience 2009, 158, 1301–1310. [Google Scholar] [CrossRef]
- Marques-Lopes, J.; Pinho, D.; Albino-Teixeira, A.; Tavares, I. The hyperalgesic effects induced by the injection of angiotensin II into the caudal ventrolateral medulla are mediated by the pontine A5 noradrenergic cell group. Brain Res. 2010, 1325, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Pelegrini-da-Silva, A.; Martins, A.R.; Prado, W.A. A new role for the renin-angiotensin system in the rat periaqueductal gray matter: Angiotensin receptor-mediated modulation of nociception. Neuroscience 2005, 132, 453–463. [Google Scholar] [CrossRef]
- Pelegrini-da-Silva, A.; Rosa, E.; Guethe, L.M.; Juliano, M.A.; Prado, W.A.; Martins, A.R. Angiotensin III modulates the nociceptive control mediated by the periaqueductal gray matter. Neuroscience 2009, 164, 1263–1273. [Google Scholar] [CrossRef]
- Restrepo, Y.M.; Noto, N.M.; Speth, R.C. CGP42112: The full AT2 receptor agonist and its role in the renin-angiotensin-aldosterone system: No longer misunderstood. Clin. Sci. 2022, 136, 1513–1533. [Google Scholar] [CrossRef]
- Georgieva, D.; Georgiev, V. The role of angiotensin II and of its receptor subtypes in the acetic acid-induced abdominal constriction test. Pharmacol. Biochem. Behav. 1999, 62, 229–232. [Google Scholar] [CrossRef]
- Bach, F.W.; Yaksh, T.L. Release into ventriculo-cisternal perfusate of beta-endorphin- and Met-enkephalin-immunoreactivity: Effects of electrical stimulation in the arcuate nucleus and periaqueductal gray of the rat. Brain Res. 1995, 690, 167–176. [Google Scholar] [CrossRef]
- Sakagawa, T.; Okuyama, S.; Kawashima, N.; Hozumi, S.; Nakagawasai, O.; Tadano, T.; Kisara, K.; Ichiki, T.; Inagami, T. Pain threshold, learning and formation of brain edema in mice lacking the angiotensin II type 2 receptor. Life Sci. 2000, 67, 2577–2585. [Google Scholar] [CrossRef] [PubMed]
- Pechlivanova, D.; Petrov, K.; Grozdanov, P.; Nenchovska, Z.; Tchekalarova, J.; Stoynev, A. Intracerebroventricular infusion of angiotensin AT2 receptor agonist novokinin aggravates some diabetes-mellitus-induced alterations in Wistar rats. Can. J. Physiol. Pharmacol. 2018, 96, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Ganten, D.; Hokfelt, T.; Bolme, P. Immunohistochemical evidence for the existence of angiotensin II-containing nerve terminals in the brain and spinal cord in the rat. Neurosci. Lett. 1976, 2, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, W.; Ogata, Y.; Nakagawasai, O.; Yaoita, F.; Tadano, T.; Tan-No, K. Angiotensin (1–7) prevents angiotensin II-induced nociceptive behaviour via inhibition of p38 MAPK phosphorylation mediated through spinal Mas receptors in mice. Eur. J. Pain 2014, 18, 1471–1479. [Google Scholar] [CrossRef]
- Nemoto, W.; Ogata, Y.; Nakagawasai, O.; Yaoita, F.; Tanado, T.; Tan-No, K. The intrathecal administration of losartan, an AT1 receptor antagonist, produces an antinociceptive effect through the inhibiton of p38 MAPK phosphorylation in the mouse formalin test. Neurosci. Lett. 2015, 585, 17–22. [Google Scholar] [CrossRef]
- Nemoto, W.; Ogata, Y.; Nakagawasai, O.; Yaoita, F.; Tadano, T.; Tan-No, K. Involvement of p38 MAPK activation mediated through AT1 receptors on spinal astrocytes and neurons in angiotensin II- and III-induced nociceptive behavior in mice. Neuropharmacology 2015, 99, 221–231. [Google Scholar] [CrossRef]
- Nemoto, W.; Yamagata, R.; Nakagawasai, O.; Nakagawa, K.; Hung, W.Y.; Fujita, M.; Tadano, T.; Tan-No, K. Effect of spinal angiotensin-converting enzyme 2 activation on the formalin-induced nociceptive response in mice. Eur. J. Pharmacol. 2020, 872, 172950. [Google Scholar] [CrossRef]
- Nemoto, W.; Yamagata, R.; Ogata, Y.; Nakagawasai, O.; Tadano, T.; Tan-No, K. Inhibitory effect of angiotensin (1–7) on angiotensin III-induced nociceptive behaviour in mice. Neuropeptides 2017, 65, 71–76. [Google Scholar] [CrossRef]
- Aguilar, C.F.; Dhanaraj, V.; Guruprasad, K.; Dealwis, C.; Badasso, M.; Cooper, J.B.; Wood, S.P.; Blundell, T.L. Comparisons of the three-dimensional structures, specificities and glycosylation of renins, yeast proteinase A and cathepsin D. Adv. Exp. Med. Biol. 1995, 362, 155–166. [Google Scholar] [CrossRef]
- Naseem, R.H.; Hedegard, W.; Henry, T.D.; Lessard, J.; Sutter, K.; Katz, S.A. Plasma cathepsin D isoforms and their active metabolites increase after myocardial infarction and contribute to plasma renin activity. Basic Res. Cardiol. 2005, 100, 139–146. [Google Scholar] [CrossRef]
- Moon, C.; Lee, T.K.; Kim, H.; Ahn, M.; Lee, Y.; Kim, M.D.; Sim, K.B.; Shin, T. Immunohistochemical study of cathepsin D in the spinal cords of rats with clip compression injury. J. Vet. Med. Sci. 2008, 70, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Pavel, J.; Tang, H.; Brimijoin, S.; Moughamian, A.; Nishioku, T.; Benicky, J.; Saavedra, J.M. Expression and transport of Angiotensin II AT1 receptors in spinal cord, dorsal root ganglia and sciatic nerve of the rat. Brain Res. 2008, 1246, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, W.; Nakagawasai, O.; Yaoita, F.; Kanno, S.I.; Yomogida, S.; Ishikawa, M.; Tadano, T.; Tan-No, K. Angiotensin II produces nociceptive behavior through spinal AT1 receptor-mediated p38 mitogen-activated protein kinase activation in mice. Mol. Pain 2013, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, A.J.; Copits, B.A.; Mickle, A.D.; Karlsson, P.; Kadunganattil, S.; Haroutounian, S.; Tadinada, S.M.; de Kloet, A.D.; Valtcheva, M.V.; McIlvried, L.A.; et al. Angiotensin II Triggers Peripheral Macrophage-to-Sensory Neuron Redox Crosstalk to Elicit Pain. J. Neurosci. 2018, 38, 7032–7057. [Google Scholar] [CrossRef]
- Ogata, Y.; Nemoto, W.; Yamagata, R.; Nakagawasai, O.; Shimoyama, S.; Furukawa, T.; Ueno, S.; Tan-No, K. Anti-hypersensitive effect of angiotensin (1–7) on streptozotocin-induced diabetic neuropathic pain in mice. Eur. J. Pain 2019, 23, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, R.; Nemoto, W.; Nakagawasai, O.; Takahashi, K.; Tan-No, K. Downregulation of spinal angiotensin converting enzyme 2 is involved in neuropathic pain associated with type 2 diabetes mellitus in mice. Biochem. Pharmacol. 2020, 174, 113825. [Google Scholar] [CrossRef]
- Yamagata, R.; Nemoto, W.; Fujita, M.; Nakagawasai, O.; Tan-No, K. Angiotensin (1–7) Attenuates the Nociceptive Behavior Induced by Substance P and NMDA via Spinal MAS1. Biol. Pharm. Bull. 2021, 44, 742–746. [Google Scholar] [CrossRef]
- Seretny, M.; Currie, G.L.; Sena, E.S.; Ramnarine, S.; Grant, R.; MacLeod, M.R.; Colvin, L.A.; Fallon, M. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: A systematic review and meta-analysis. Pain 2014, 155, 2461–2470. [Google Scholar] [CrossRef]
- Frachet, S.; Danigo, A.; Labriffe, M.; Bessaguet, F.; Quinchard, B.; Deny, N.; Baffert, K.A.; Deluche, E.; Sturtz, F.; Demiot, C.; et al. Renin-Angiotensin-System Inhibitors for the Prevention of Chemotherapy-Induced Peripheral Neuropathy: OncoToxSRA, a Preliminary Cohort Study. J. Clin. Med. 2022, 11, 2939. [Google Scholar] [CrossRef]
- Kim, E.; Hwang, S.H.; Kim, H.K.; Abdi, S.; Kim, H.K. Losartan, an Angiotensin II Type 1 Receptor Antagonist, Alleviates Mechanical Hyperalgesia in a Rat Model of Chemotherapy-Induced Neuropathic Pain by Inhibiting Inflammatory Cytokines in the Dorsal Root Ganglia. Mol. Neurobiol. 2019, 56, 7408–7419. [Google Scholar] [CrossRef]
- Kalynovska, N.; Diallo, M.; Sotakova-Kasparova, D.; Palecek, J. Losartan attenuates neuroinflammation and neuropathic pain in paclitaxel-induced peripheral neuropathy. J. Cell. Mol. Med. 2020, 24, 7949–7958. [Google Scholar] [CrossRef] [PubMed]
- Pavel, J.; Oroszova, Z.; Hricova, L.; Lukacova, N. Effect of subpressor dose of angiotensin II on pain-related behavior in relation with neuronal injury and activation of satellite glial cells in the rat dorsal root ganglia. Cell. Mol. Neurobiol. 2013, 33, 681–688. [Google Scholar] [CrossRef]
- Shiers, S.; Ray, P.R.; Wangzhou, A.; Sankaranarayanan, I.; Tatsui, C.E.; Rhines, L.D.; Li, Y.; Uhelski, M.L.; Dougherty, P.M.; Price, T.J. ACE2 and SCARF expression in human dorsal root ganglion nociceptors: Implications for SARS-CoV-2 virus neurological effects. Pain 2020, 161, 2494–2501. [Google Scholar] [CrossRef] [PubMed]
- McFarland, A.J.; Yousuf, M.S.; Shiers, S.; Price, T.J. Neurobiology of SARS-CoV-2 interactions with the peripheral nervous system: Implications for COVID-19 and pain. Pain Rep. 2021, 6, e885. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qin, Y.; Liu, T.; Hao, D. Chronic nerve injury-induced Mas receptor expression in dorsal root ganglion neurons alleviates neuropathic pain. Exp. Ther. Med. 2015, 10, 2384–2388. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, A.J.; Mickle, A.D.; Golden, J.P.; Mack, M.R.; Halabi, C.M.; de Kloet, A.D.; Samineni, V.K.; Kim, B.S.; Krause, E.G.; Gereau, R.W., IV; et al. Macrophage angiotensin II type 2 receptor triggers neuropathic pain. Proc. Natl. Acad. Sci. USA 2018, 115, E8057–E8066. [Google Scholar] [CrossRef]
- Bessaguet, F.; Danigo, A.; Magy, L.; Sturtz, F.; Desmouliere, A.; Demiot, C. Candesartan prevents resiniferatoxin-induced sensory small-fiber neuropathy in mice by promoting angiotensin II-mediated AT2 receptor stimulation. Neuropharmacology 2017, 126, 142–150. [Google Scholar] [CrossRef]
- Bouchenaki, H.; Bernard, A.; Bessaguet, F.; Frachet, S.; Richard, L.; Sturtz, F.; Magy, L.; Bourthoumieu, S.; Demiot, C.; Danigo, A. Neuroprotective Effect of Ramipril Is Mediated by AT2 in a Mouse MODEL of Paclitaxel-Induced Peripheral Neuropathy. Pharmaceutics 2022, 14, 848. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Muralidharan, A.; Smith, M.T. Attenuation of the Infiltration of Angiotensin II Expressing CD3+ T-Cells and the Modulation of Nerve Growth Factor in Lumbar Dorsal Root Ganglia—A Possible Mechanism Underpinning Analgesia Produced by EMA300, An Angiotensin II Type 2 (AT(2)) Receptor Antagonist. Front. Mol. Neurosci. 2017, 10, 389. [Google Scholar] [CrossRef]
- Muralidharan, A.; Wyse, B.D.; Smith, M.T. Analgesic efficacy and mode of action of a selective small molecule angiotensin II type 2 receptor antagonist in a rat model of prostate cancer-induced bone pain. Pain Med. 2014, 15, 93–110. [Google Scholar] [CrossRef]
- Liao, Z.; Chakrabarty, A.; Mu, Y.; Bhattacherjee, A.; Goestch, M.; Leclair, C.M.; Smith, P.G. A Local Inflammatory Renin-Angiotensin System Drives Sensory Axon Sprouting in Provoked Vestibulodynia. J. Pain 2017, 18, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.S.C.; Dworkin, R.H.; McCarthy, T.D.; Anand, P.; Bountra, C.; McCloud, P.I.; Hill, J.; Cutter, G.; Kitson, G.; Desem, N.; et al. EMA401, an orally administered highly selective angiotensin II type 2 receptor antagonist, as a novel treatment for postherpetic neuralgia: A randomised, double-blind, placebo-controlled phase 2 clinical trial. Lancet 2014, 383, 1637–1647. [Google Scholar] [CrossRef]
- Rice, A.S.C.; Dworkin, R.H.; Finnerup, N.B.; Attal, N.; Anand, P.; Freeman, R.; Piaia, A.; Callegari, F.; Doerr, C.; Mondal, S.; et al. Efficacy and safety of EMA401 in peripheral neuropathic pain: Results of 2 randomised, double-blind, phase 2 studies in patients with postherpetic neuralgia and painful diabetic neuropathy. Pain 2021, 162, 2578–2589. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.G.; Choi, S.R.; Kang, D.W.; Kim, J.; Park, J.B.; Kim, H.W. Inhibition of angiotensin converting enzyme induces mechanical allodynia through increasing substance P expression in mice. Neurochem. Int. 2021, 146, 105020. [Google Scholar] [CrossRef] [PubMed]
- Skidgel, R.A.; Erdos, E.G. Cleavage of peptide bonds by angiotensin I converting enzyme. Agents Actions Suppl. 1987, 22, 289–296. [Google Scholar] [CrossRef]
- Matsas, R.; Kenny, A.J.; Turner, A.J. The metabolism of neuropeptides. The hydrolysis of peptides, including enkephalins, tachykinins and their analogues, by endopeptidase-24.11. Biochem. J. 1984, 223, 433–440. [Google Scholar] [CrossRef]
- Blumberg, S.; Teichberg, V.I.; Charli, J.L.; Hersh, L.B.; McKelvy, J.F. Cleavage of substance P to an N-terminal tetrapeptide and a C-terminal heptapeptide by a post-proline Cleaving enzyme from bovine brain. Brain Res. 1980, 192, 477–486. [Google Scholar] [CrossRef]
- Heymann, E.; Mentlein, R. Liver dipeptidyl aminopeptidase IV hydrolyzes substance P. FEBS Lett. 1978, 91, 360–364. [Google Scholar] [CrossRef]
- Azaryan, A.V.; Galoyan, A.A. Substrate specificity of cerebral cathepsin D and high-Mr aspartic endopeptidase. J. Neurosci. Res. 1988, 19, 268–271. [Google Scholar] [CrossRef]
- Kageyama, T. Rabbit procathepsin E and cathepsin E: Nucleotide sequence of cDNA, hydrolytic specificity for biologically active peptides and gene expression during development. Eur. J. Biochem. 1993, 216, 717–728. [Google Scholar] [CrossRef]
- Harford-Wright, E.; Thornton, E.; Vink, R. Angiotensin-converting enzyme (ACE) inhibitors exacerbate histological damage and motor deficits after experimental traumatic brain injury. Neurosci. Lett. 2010, 481, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Pavel, J.; Saavedra, J.M.; Brimijoin, S. Type-1 angiotensin receptors are expressed and transported in motor and sensory axons of rat sciatic nerves. Neuropeptides 2009, 43, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Jaggi, A.S.; Singh, N. Exploring the potential of telmisartan in chronic constriction injury-induced neuropathic pain in rats. Eur. J. Pharmacol. 2011, 667, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Kukkar, A.; Singh, N.; Jaggi, A.S. Neuropathic pain-attenuating potential of aliskiren in chronic constriction injury model in rats. J. Renin Angiotensin Aldosterone Syst. 2013, 14, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.P.; Di Pietro, F.; Alshelh, Z.; Peck, C.C.; Murray, G.M.; Vickers, E.R.; Henderson, L.A. Brainstem Pain-Control Circuitry Connectivity in Chronic Neuropathic Pain. J. Neurosci. 2018, 38, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, P.; Weiss, S.; Jansen, A.; Wienen, W.; Stangier, J.; Rascher, W.; Culman, J.; Unger, T. AT1 receptor antagonist telmisartan administered peripherally inhibits central responses to angiotensin II in conscious rats. J. Pharmacol. Exp. Ther. 2001, 298, 62–70. [Google Scholar]
- Gohlke, P.; Scholkens, B.; Henning, R.; Urbach, H.; Unger, T. Inhibition of converting enzyme in brain tissue and cerebrospinal fluid of rats following chronic oral treatment with the converting enzyme inhibitors ramipril and Hoe 288. J. Cardiovasc. Pharmacol. 1989, 14 (Suppl. S4), S32–S36. [Google Scholar] [CrossRef]
- Yamada, K.; Uchida, S.; Takahashi, S.; Takayama, M.; Nagata, Y.; Suzuki, N.; Shirakura, S.; Kanda, T. Effect of a centrally active angiotensin-converting enzyme inhibitor, perindopril, on cognitive performance in a mouse model of Alzheimer’s disease. Brain Res. 2010, 1352, 176–186. [Google Scholar] [CrossRef]
- Hegazy, N.; Rezq, S.; Fahmy, A. Mechanisms Involved in Superiority of Angiotensin Receptor Blockade over ACE Inhibition in Attenuating Neuropathic Pain Induced in Rats. Neurotherapeutics 2020, 17, 1031–1047. [Google Scholar] [CrossRef]
- Brusco, I.; Silva, C.R.; Trevisan, G.; de Campos Velho Gewehr, C.; Rigo, F.K.; La Rocca Tamiozzo, L.; Rossato, M.F.; Tonello, R.; Dalmolin, G.D.; de Almeida Cabrini, D.; et al. Potentiation of Paclitaxel-Induced Pain Syndrome in Mice by Angiotensin I Converting Enzyme Inhibition and Involvement of Kinins. Mol. Neurobiol. 2017, 54, 7824–7837. [Google Scholar] [CrossRef] [PubMed]
- Zanata, G.C.; Pinto, L.G.; da Silva, N.R.; Lopes, A.H.P.; de Oliveira, F.F.B.; Schivo, I.R.S.; Cunha, F.Q.; McNaughton, P.; Cunha, T.M.; Silva, R.L. Blockade of bradykinin receptors or angiotensin II type 2 receptor prevents paclitaxel-associated acute pain syndrome in mice. Eur. J. Pain 2021, 25, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Emanueli, C.; Grady, E.F.; Madeddu, P.; Figini, M.; Bunnett, N.W.; Parisi, D.; Regoli, D.; Geppetti, P. Acute ACE inhibition causes plasma extravasation in mice that is mediated by bradykinin and substance P. Hypertension 1998, 31, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Sulpizio, A.C.; Pullen, M.A.; Edwards, R.M.; Brooks, D.P. The effect of acute angiotensin-converting enzyme and neutral endopeptidase 24.11 inhibition on plasma extravasation in the rat. J. Pharmacol. Exp. Ther. 2004, 309, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Bujalska, M.; Makulska-Nowak, H. Bradykinin receptor antagonists and cyclooxygenase inhibitors in vincristine- and streptozotocin-induced hyperalgesia. Pharmacol. Rep. 2009, 61, 631–640. [Google Scholar] [CrossRef]
- Costa, R.; Motta, E.M.; Dutra, R.C.; Manjavachi, M.N.; Bento, A.F.; Malinsky, F.R.; Pesquero, J.B.; Calixto, J.B. Anti-nociceptive effect of kinin B(1) and B(2) receptor antagonists on peripheral neuropathy induced by paclitaxel in mice. Br. J. Pharmacol. 2011, 164, 681–693. [Google Scholar] [CrossRef]
- Campbell, D.J.; Duncan, A.M.; Kladis, A. Angiotensin-converting enzyme inhibition modifies angiotensin but not kinin peptide levels in human atrial tissue. Hypertension 1999, 34, 171–175. [Google Scholar] [CrossRef]
- Wang, Y.; Kou, J.; Zhang, H.; Wang, C.; Li, H.; Ren, Y.; Zhang, Y. The renin-angiotensin system in the synovium promotes periarticular osteopenia in a rat model of collagen-induced arthritis. Int. Immunopharmacol. 2018, 65, 550–558. [Google Scholar] [CrossRef]
- Wu, Y.; Li, M.; Zeng, J.; Feng, Z.; Yang, J.; Shen, B.; Zeng, Y. Differential Expression of Renin-Angiotensin System-related Components in Patients with Rheumatoid Arthritis and Osteoarthritis. Am. J. Med. Sci. 2020, 359, 17–26. [Google Scholar] [CrossRef]
- An, J.S.; Tsuji, K.; Onuma, H.; Araya, N.; Isono, M.; Hoshino, T.; Inomata, K.; Hino, J.; Miyazato, M.; Hosoda, H.; et al. Inhibition of fibrotic changes in infrapatellar fat pad alleviates persistent pain and articular cartilage degeneration in monoiodoacetic acid-induced rat arthritis model. Osteoarthr. Cartil. 2021, 29, 380–388. [Google Scholar] [CrossRef]
- Abbasi, B.; Pezeshki-Rad, M.; Akhavan, R.; Sahebari, M. Association between clinical and sonographic synovitis in patients with painful knee osteoarthritis. Int. J. Rheum. Dis. 2017, 20, 561–566. [Google Scholar] [CrossRef]
- AlQudah, M.; Hale, T.M.; Czubryt, M.P. Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol. 2020, 91–92, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Sriwatananukulkit, O.; Desclaux, S.; Tawonsawatruk, T.; Srikuea, R.; Himakhun, W.; Likitnukul, S.; Hemstapat, R. Effectiveness of losartan on infrapatellar fat pad/synovial fibrosis and pain behavior in the monoiodoacetate-induced rat model of osteoarthritis pain. Biomed. Pharmacother. 2023, 158, 114121. [Google Scholar] [CrossRef] [PubMed]
- De Sa, G.A.; Dos Santos, A.; Nogueira, J.M.; Dos Santos, D.M.; Amaral, F.A.; Jorge, E.C.; Caliari, M.V.; Queiroz-Junior, C.M.; Ferreira, A.J. Angiotensin II triggers knee joint lesions in experimental osteoarthritis. Bone 2021, 145, 115842. [Google Scholar] [CrossRef] [PubMed]
- Terenzi, R.; Manetti, M.; Rosa, I.; Romano, E.; Galluccio, F.; Guiducci, S.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Angiotensin II type 2 receptor (AT2R) as a novel modulator of inflammation in rheumatoid arthritis synovium. Sci. Rep. 2017, 7, 13293. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, S.; Zhu, J.; Yuan, J.; Wu, J.; Zhou, A.; Wu, Y.; Zhao, W.; Huang, Q.; Chang, Y.; et al. Angiotensin II type 2 receptor correlates with therapeutic effects of losartan in rats with adjuvant-induced arthritis. J. Cell. Mol. Med. 2013, 17, 1577–1587. [Google Scholar] [CrossRef]
- Sehnert, B.; Valero-Esquitino, V.; Schett, G.; Unger, T.; Steckelings, U.M.; Voll, R.E. Angiotensin AT(2) Receptor Stimulation Alleviates Collagen-Induced Arthritis by Upregulation of Regulatory T Cell Numbers. Front. Immunol. 2022, 13, 921488. [Google Scholar] [CrossRef]
- Sun, K.; Sun, X.; Sun, J.; Jiang, Y.; Lin, F.; Kong, F.; Li, F.; Zhu, J.; Huan, L.; Zheng, B.; et al. Tissue Renin-Angiotensin System (tRAS) Induce Intervertebral Disc Degeneration by Activating Oxidative Stress and Inflammatory Reaction. Oxid. Med. Cell. Longev. 2021, 2021, 3225439. [Google Scholar] [CrossRef]
- Tawfik, V.L.; Quarta, M.; Paine, P.; Forman, T.E.; Pajarinen, J.; Takemura, Y.; Goodman, S.B.; Rando, T.A.; Clark, J.D. Angiotensin receptor blockade mimics the effect of exercise on recovery after orthopaedic trauma by decreasing pain and improving muscle regeneration. J. Physiol. 2020, 598, 317–329. [Google Scholar] [CrossRef]
- Silva, C.R.; Oliveira, S.M.; Hoffmeister, C.; Funck, V.; Guerra, G.P.; Trevisan, G.; Tonello, R.; Rossato, M.F.; Pesquero, J.B.; Bader, M.; et al. The role of kinin B1 receptor and the effect of angiotensin I-converting enzyme inhibition on acute gout attacks in rodents. Ann. Rheum. Dis. 2016, 75, 260–268. [Google Scholar] [CrossRef]
- Forte, B.L.; Slosky, L.M.; Zhang, H.; Arnold, M.R.; Staatz, W.D.; Hay, M.; Largent-Milnes, T.M.; Vanderah, T.W. Angiotensin-(1–7)/Mas receptor as an antinociceptive agent in cancer-induced bone pain. Pain 2016, 157, 2709–2721. [Google Scholar] [CrossRef] [PubMed]
- Dursteler-MacFarland, K.M.; Kowalewski, R.; Bloch, N.; Wiesbeck, G.A.; Kraenzlin, M.E.; Stohler, R. Patients on injectable diacetylmorphine maintenance have low bone mass. Drug Alcohol Rev. 2011, 30, 577–582. [Google Scholar] [CrossRef]
- Grey, A.; Rix-Trott, K.; Horne, A.; Gamble, G.; Bolland, M.; Reid, I.R. Decreased bone density in men on methadone maintenance therapy. Addiction 2011, 106, 349–354. [Google Scholar] [CrossRef] [PubMed]
- King, T.; Vardanyan, A.; Majuta, L.; Melemedjian, O.; Nagle, R.; Cress, A.E.; Vanderah, T.W.; Lai, J.; Porreca, F. Morphine treatment accelerates sarcoma-induced bone pain, bone loss, and spontaneous fracture in a murine model of bone cancer. Pain 2007, 132, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Kshirsagar, S.; Chang, L.; Schwartz, R.; Law, P.Y.; Yee, D.; Hebbel, R.P. Morphine stimulates angiogenesis by activating proangiogenic and survival-promoting signaling and promotes breast tumor growth. Cancer Res. 2002, 62, 4491–4498. [Google Scholar] [PubMed]
- Lennon, F.E.; Mirzapoiazova, T.; Mambetsariev, B.; Poroyko, V.A.; Salgia, R.; Moss, J.; Singleton, P.A. The Mu opioid receptor promotes opioid and growth factor-induced proliferation, migration and Epithelial Mesenchymal Transition (EMT) in human lung cancer. PLoS ONE 2014, 9, e91577. [Google Scholar] [CrossRef]
- Zylla, D.; Gourley, B.L.; Vang, D.; Jackson, S.; Boatman, S.; Lindgren, B.; Kuskowski, M.A.; Le, C.; Gupta, K.; Gupta, P. Opioid requirement, opioid receptor expression, and clinical outcomes in patients with advanced prostate cancer. Cancer 2013, 119, 4103–4110. [Google Scholar] [CrossRef]
- Costa, A.C.; Becker, L.K.; Moraes, E.R.; Romero, T.R.; Guzzo, L.; Santos, R.A.; Duarte, I.D. Angiotensin-(1–7) induces peripheral antinociception through mas receptor activation in an opioid-independent pathway. Pharmacology 2012, 89, 137–144. [Google Scholar] [CrossRef]
- Costa, A.; Galdino, G.; Romero, T.; Silva, G.; Cortes, S.; Santos, R.; Duarte, I. Ang-(1–7) activates the NO/cGMP and ATP-sensitive K+ channels pathway to induce peripheral antinociception in rats. Nitric Oxide 2014, 37, 11–16. [Google Scholar] [CrossRef]
- Chakrabarty, A.; Liao, Z.; Smith, P.G. Angiotensin II receptor type 2 activation is required for cutaneous sensory hyperinnervation and hypersensitivity in a rat hind paw model of inflammatory pain. J. Pain 2013, 14, 1053–1065. [Google Scholar] [CrossRef]
- Chakrabarty, A.; Liao, Z.; Mu, Y.; Smith, P.G. Inflammatory Renin-Angiotensin System Disruption Attenuates Sensory Hyperinnervation and Mechanical Hypersensitivity in a Rat Model of Provoked Vestibulodynia. J. Pain 2018, 19, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Nozu, T.; Miyagishi, S.; Nozu, R.; Ishioh, M.; Takakusaki, K.; Okumura, T. EMA401, an angiotensin II type 2 receptor antagonist blocks visceral hypersensitivity and colonic hyperpermeability in rat model of irritable bowel syndrome. J. Pharmacol. Sci. 2021, 146, 121–124. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nemoto, W.; Yamagata, R.; Nakagawasai, O.; Tan-No, K. Angiotensin-Related Peptides and Their Role in Pain Regulation. Biology 2023, 12, 755. https://doi.org/10.3390/biology12050755
Nemoto W, Yamagata R, Nakagawasai O, Tan-No K. Angiotensin-Related Peptides and Their Role in Pain Regulation. Biology. 2023; 12(5):755. https://doi.org/10.3390/biology12050755
Chicago/Turabian StyleNemoto, Wataru, Ryota Yamagata, Osamu Nakagawasai, and Koichi Tan-No. 2023. "Angiotensin-Related Peptides and Their Role in Pain Regulation" Biology 12, no. 5: 755. https://doi.org/10.3390/biology12050755
APA StyleNemoto, W., Yamagata, R., Nakagawasai, O., & Tan-No, K. (2023). Angiotensin-Related Peptides and Their Role in Pain Regulation. Biology, 12(5), 755. https://doi.org/10.3390/biology12050755