Simple Summary
Cell therapy is emerging as a promising modality to treat cancers such as hematological malignancies and solid tumors; hence there is a need to develop processes to manufacture and maintain functional and lasting cells. The use of cytokines, as well as transcription and growth factors, is critical to ensure effective cell therapeutics. This is especially important for allogeneic cell therapies that employ induced pluripotent stem cells (iPSC). The use of iPSC offers the potential to treat a large number of patients with consistent material without relying on limited donor cells and the delay associated with processing immediately before treatment. This paper demonstrates the importance and use of cytokines and growth factors in driving the iPSC-to-effector differentiation and expansion process, which leads to the generation of functional and persistent immune-effector cells such as natural killer cells or T cells.
Abstract
Cytokines and other growth factors are essential for cell expansion, health, function, and immune stimulation. Stem cells have the additional reliance on these factors to direct differentiation to the appropriate terminal cell type. Successful manufacturing of allogeneic cell therapies from induced pluripotent stem cells (iPSCs) requires close attention to the selection and control of cytokines and factors used throughout the manufacturing process, as well as after administration to the patient. This paper employs iPSC-derived natural killer cell/T cell therapeutics to illustrate the use of cytokines, growth factors, and transcription factors at different stages of the manufacturing process, ranging from the generation of iPSCs to controlling of iPSC differentiation into immune-effector cells through the support of cell therapy after patient administration.
Keywords:
cell therapy; allogeneic; pluripotent; stem cell; natural killer; T cell; cytokine; growth factor; differentiation; expansion 1. Background
Cell therapies are demonstrating promise as treatments for refractory and relapsed cancers with substantial advantages over traditional therapies. Some autologous CAR-Ts are even becoming a standard of care for treating certain leukemias, lymphomas, and multiple myeloma. Despite the benefit of autologous approaches to cell therapy, long manufacturing time and reliance on the patient as the donor are serious drawbacks. Allogeneic approaches share some of the same challenges as autologous processes [1], as well as unique challenges, such as the need to match human leukocyte antigen (HLA) between donor and patient. HLA mismatching can lead to alloreactivity due to donor immune cell recognition and, subsequently, graft versus host disease. However, the ability of allogeneic sources to provide a ready source of treatment cells is a substantial advantage to ailing patients who need cells in a timely manner [2] and should enable greater access to these advanced therapies. Unlike autologous therapy relying on a limited number of donor cells, allogeneic manufacturing processes can be scaled up, which further reduces the cost of goods.
The use of induced pluripotent stem cells (iPSC) that are derived from a single donor has the potential to provide the most consistent and characterizable cells for the lifetime of a given product. The ability to span the product lifecycle owes to the ability of iPSC to self-renew, thus providing a replenishable source of cells sufficient for the treatment of many patients. An unequivocal attribute of pluripotent stem cells (PSC) is the ability to perform multiple rounds of genetic engineering while preserving genomic stability and maintaining unlimited self-renewal. This enables the final PSC to be precision edited with a profile that allows for the final differentiated effector cell to be functionally enhanced. For immuno-oncology applications, the inclusion of chimeric antigen receptors (CAR), elimination of HLA class I/II on the cell surface, and homeostatic cytokine support provide iPSC cell therapies with the ability to recognize tumor antigens, evade aforementioned host-immune responses, and enhance effector cell persistence, respectively [3,4,5]. In this case, engineering of the iPSC is only performed once, thereby enabling the creation of a master cell bank and ensuring consistent and pure, gene-edited starting material for the subsequent differentiation and expansion process (Figure 1). For other allogeneic processes that are dependent on multiple donors and repeated genetic engineering of different batches of source cells, greater lot-to-lot, and genetic engineering variability may be observed.
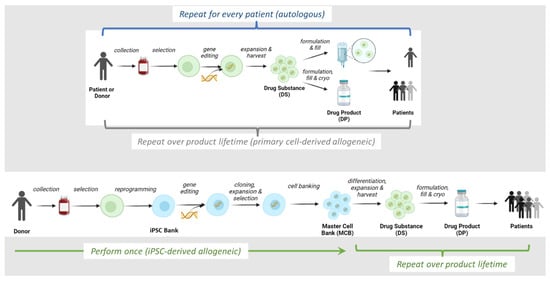
Figure 1.
Allogeneic and autologous cell therapy manufacturing schemes. Created using ©2023 BioRender.
To realize the potential of iPSC-based, allogeneic cell therapeutic approaches, the cells need to be guided through differentiation and expansion using precise exposure and signaling from cytokines and other growth factors to arrive at the desired functional cell type. There are several reports of the successful differentiation of iPSC to various immune-effector cell types, such as natural killer (NK) [6,7] and T cells [8,9]. The use of transcription factors and cytokines early in the process is also needed for reprogramming of source cells. In addition to iPSC, hematopoietic stem cells (HSC) or hematopoietic progenitor cells (HPC) have also been shown to generate immune cells. HSC/HPC-derived cell manufacturing processes also require specific cytokines and signaling ligands [10]. This review is built on the knowledge of cytokine application to cell differentiation, expanding to the potential use of iPSC as sourcing material for allogeneic cell therapy, and will discuss the role of cytokines and growth factors within an industrial-scale manufacturing process.
2. Overview of Allogeneic IPSC-Derived Effector Cell Manufacturing Process
In the production of iPSC-derived effector cell therapies (Figure 1), somatic cells are isolated, followed by reprogramming to iPSC, during which multiple transcription factors are required (Section 3). iPSC are then engineered with exogenous genes (e.g., CAR) to target cancer cells. Specific cytokine and media components have been optimized for the expansion of iPSC (Section 3). During the differentiation of iPSC to functional immune cells, HSC or HPC are used as an intermediate, splitting the entire manufacturing process into two distinct sections: iPSC to HPC (Section 4) and HPC to NK/T differentiation (Section 5). Various approaches will be discussed regarding the use of cytokines and growth factors in differentiation, maturation, activation, as well as cryopreservation (Section 6). In Section 7, we will discuss new methods for endogenously engineering cytokines. Lastly, Section 8 will cover critical cytokines (i.e., IL-2), which are used in conjunction with the manufactured cell products to supply clinical applications.
The use of cytokines in traditional cell therapy products is primarily focused on expanding functional effector cells after isolation from patients or donors. However, iPSC-derived cell therapy products require a diverse range of cytokines that act individually and synergistically during multiple stages of the manufacturing process, from iPSC to intermediate HSC/HPC and to final functional effector cells. Controlling the type and level of cytokines used becomes even more critical in developing a robust manufacturing process for iPSC-derived cell therapy, given the diverse range of cytokines required at multiple stages of the manufacturing process.
3. Use of Transcription Factors to Generate Pluripotent Starting Material
Over the past two decades, meaningful advances have been made in cellular reprogramming that have facilitated the progression of pluripotent stem cells for use in cell therapies [11]. The discovery of induced pluripotent stem cells (iPSC) has provided a self-renewable source of allogeneic starting material which eliminates the ethical concerns linked to embryonic stem cells. The term was coined by Yamanaka after he was able to generate murine iPSC using four essential transcription factors, Oct3/4, Sox2, Klf4, and c-Myc [12]. Yamanaka was awarded the Nobel Prize in 2012 for this work, which he shared with John Gurdon for the discovery of reprogramming to generate pluripotent cells. Following this work, the successful reprogramming to human iPSC by Thomson included similar factors of Oct3/4, Sox2, Nanog, c-Myc, and Lin28 [13]. The derivation of iPSC from somatic cells provided a turning point for the field of regenerative medicine and cell therapies, but initial reprogramming strategies limited the potential due to the use of retroviral and lentiviral transgene deliveries. These methods provided low reprogramming efficiencies, permanent exogenous DNA integration, the potential for transgene reactivation, and the risk of retroviral replication. To overcome these challenges, several methods have been developed to improve cellular reprogramming based on viral and non-viral strategies [14]. One of the important criteria to achieve for iPSC therapies are ‘footprint-free’ reprogramming strategies that leave the iPSC with no exogenously integrated DNA [15], therefore, leaving a few methods ideal for reprogramming. These transgene delivery methods include viral approaches such as Sendai viruses, adeno-associated viruses (AAV), and adenovirus (AdV), all of which have been shown to reprogram somatic tissues; however, AdV and AAV have been shown to have low efficiency [16,17,18,19]. Non-viral reprogramming methods make it possible to derive iPSC with limited genomic integration risk and can include mRNA, miRNA, protein, chemical, and episomal reprogramming [15]. These strategies are attractive because they allow for an untouched genome during cellular reprogramming and the generation of cells that are indistinguishable from embryonic stem cells. The major drawback to many of these methods is the low efficiency of iPSC reprogramming and the limited ability to reprogram a variety of somatic cell types [20,21].
One aspect of cell reprogramming that is heavily debated is the choice of somatic starting material. The ideal source of somatic tissue should be easily accessible, susceptible to reprogramming methods, and able to expand under culture conditions [22,23,24]. The ideal somatic cell type has not been identified, with groups generating iPSC lines from neuronal progenitors, keratinocytes, hepatocytes, B cells, fibroblast, peripheral blood mononuclear cells, T cells, and erythrocyte progenitors [22,23,24]. Recent work suggests the potential for the employment of cancer cells as the source for iPSC generation [25]. Although the reprogramming method and somatic cell debate will likely persist, the ultimate goal is to produce iPSC lines that can be differentiated to provide a functionally reliable and robust cell therapy for patients on a global scale.
Since iPSC can serve as starting material for allogeneic cell therapy, large-scale generation of iPSC requires appropriate protocols and controls in place to ensure that master cell banks maintain pluripotency and the ability to differentiate downstream into the required effector cell. Conventional two-dimensional cultures have been shown to maintain iPSC expansion potential and quality. This can often come with added complexity, such as feeder cells, which creates difficulties in scaling up, purity considerations, and non-human pathogen risks [12]. On the other hand, chemically defined media such as E8, B8, and mTesR have been developed in the past decade, which eliminates the need for supporting cells and allows for iPSC banks to be produced at industrial-scale with minimal risk to product and patient safety [26,27,28]. More recently, three-dimensional culture methods using microcarriers, hydrogel-based encapsulation, or suspension bioreactors exhibit potential for larger scale and more efficient production of iPSC [29,30,31]. In either case, critical growth factors are required for the maintenance and further expansion of iPSC. TGF-β family, bFGF, and activin A play a role in stem-cell renewal, maintaining pluripotency, and mediating lineage commitment [32].
4. IPSC to HPC Differentiation Process
To generate lymphoid immune-effector cells, iPSC are differentiated to a hematopoietic stem-cell state, or hematopoietic progenitor cells (HPC). As peripheral blood mononuclear cells (PBMC), cord blood (CB), and bone marrow isolated cells can be used as multipotent progenitor cells, iPSC-derived HPC can be used as a source for a broad range of blood cells from lymphoid, myeloid, megakaryocytic or erythroid lineages. A typical research-scale pluripotent stem cell (PSC)-to-HPC process can be performed in 2-D monolayer adherent culture or 3-D embryonic body formation suspension culture [33]. Currently, it is thought that there are two distinct stages, mesodermal and meso-endothelial, during the differentiation to hematopoietic progenitor cells from pluripotent stem cells [34]. Although these differentiations can be done with stromal feeder cells and bovine serum, this can create bottlenecks in the clinical application of the resulting products; therefore, the transition to feeder-free and serum-free media with a combination of cytokine cocktails has been achieved [35,36,37]. These involve an orchestrated sequence of growth factor and cytokine stimulations that mimics embryonic development. iPSC can be patterned to generate the mesodermal lineage by stimulation with BMP4 and FGF2 early during differentiation and then later presented with VEGF to coordinate the progression of mesodermal to meso-endothelial cell transition [38]. Following the formation of the meso-endothelium, cells will start expressing hemogenic markers (CD34, FLK1, and Kit) at both the early and late stages of the meso-endothelium. These resulting multipotent progenitors are responsive to cytokines IL-3, IL-6, IGF-1/2, FLT3, and SCF, which results in cell expansion and the release of single hematopoietic progenitor cells that can maintain multipotency in culture [39].
Despite the application of key cytokines that drive the differentiation process from iPSC to HPC in 2-D or 3-D systems, scaling up of the differentiation process to generate large amounts of HPC remains challenging. The consistency of the final yield, characteristics, and purity of HPC, as well as further maintenance of multipotency, require more sophisticated controls within large-scale processes. Suspension cultures using stirred tanks or vertical wheel bioreactors can produce three-dimensional cell aggregates that are amenable to both iPSC and HPC expansion or differentiation, thereby providing a path to large-scale clinical or commercial cell manufacturing [40,41].
5. Differentiation and Activation Requirements for Production of Immune-Effector Cells
Industrial-scale manufacturing of allogeneic cell therapies requires a deep understanding of the biological mechanisms that commit HPC to mature functional effector cells. In this section, the role of cytokines toward differentiation, maturation, and activation of two of the most widely used types of immune cells (NK and T cells) will be reviewed. Differentiation to specific types of blood cells (e.g., T, NK, erythrocytes, macrophages) is guided by a series of cytokine and factor exposures (Figure 2) [6,7,42,43]. For example, a T cell can be derived from different progenitor stem cells through different pathways using distinct cytokines and exposure times [44]. This process must imitate the body’s cues for hematopoietic cell development while taking into account tissue, fluid, and mechanical engineering challenges to replicate it at an industrial manufacturing scale.
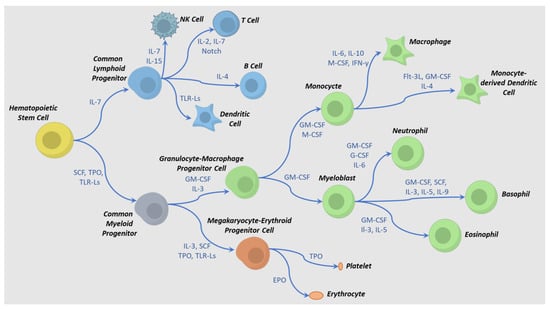
Figure 2.
Cytokine-directed differentiation pathways for hematopoietic cell lineages. Examples of cytokine combinations that affect intermediate and terminal differentiation are shown. Adapted from references [45,46]. Abbreviations: TLR-Ls = toll-like receptor ligands, M-CSF = macrophage colony stimulatory factor, SCF = stem cell factor, EPO = erythropoietin, IL = interleukin, TPO = thrombopoietin, G-CSF = granulocyte colony-stimulating factor, GM-CSF = granulocyte-macrophage colony-stimulating factor, IFN-γ = interferon gamma, Flt-3L = FMS-related tyrosine kinase 3 receptor ligand.
Terminal differentiation of an iPSC is modeled after somatic pathways of embryonic development in the case of NK cells or thymic development in the case of T cells (Figure 2). Both lymphocytes require stimulation via ligands that prompt downstream signaling through notch and integrin pathways and initiate early lymphoid commitment. For T cells, a few interleukins and growth factors, such as IL-7, SCF, TPO, and FLT3L, are involved in early commitment, activation, and subsequent contraction and formation of memory cells [47,48,49,50]. When activated by an antigen, T cells produce and respond to IL-2, IL-4, and IL-7 by greatly expanding their numbers. The presence of IL-2 is especially critical since regulatory T cells have the ability to downregulate IL-2 expression, having an anti-proliferative effect and eventual apoptosis. Both the presence of IL-7 and IL-15 after this contraction phase can direct a small fraction of the T cells to a persistent memory phenotype. For NK cells, some of the same cytokines are involved in development and functioning, although others, such as IL-12 and IL-18, are also critical for NK function [51]. Several cytokines (e.g., IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21) play a role in the development of NK cells in the bone marrow and lymph nodes. Different combinations of these cytokines result in various NK phenotypes [52,53,54]; therefore, controlling the levels of these cytokines is critical during the manufacturing process.
After lymphoid progenitor commitment to mature functional cells, the immune cells play a critical role in the acute innate immune response of invading organisms, immunogens, viral-infected cells, and tumor cells [45,46]. These cells survey the microenvironment and control tumor initiation by recognizing tumor cells. Upon engagement, NK and T cells produce large amounts of cytokines and chemokines, which can recruit other immune cells such as dendritic cells, monocytes, macrophages, T cells, and B cells. A tumor microenvironment can significantly impact the recruitment of subpopulations of immune cells and influence the distinct effector role in a tumor-specific context. Researchers have found several cytokines, including IL-12, IL-18, and IL-15, which are secreted by dendritic cells and macrophages and play an important role in NK activation [55,56,57]. Within the IL-12 family, IL-27 possesses pro- and anti-inflammatory effects within the immune system. In recent studies, IL-27 has been found to serve as a regulator in NK priming, activation, and cytolytic function [58]. These impacts on NK cells can provide a synergetic effect with other related cytokines, such as IL-18 and IL-12 [58]. Within the IL-27 family, IL-4 has also been shown to serve as a mediator in the crosstalk between NK and macrophages. In the IL-4-rich environment, a different sub-set of CD11-low-expressing NK cells has been discovered. Crosstalk between IL-4-induced macrophages and NK cells has been shown to enhance the level of IL-15 [59].
A growing body of evidence suggests that different NK subsets, presenting specific phenotypic and functional attributes, can recognize targets through activating, inhibitory, and cytokine receptors [60,61]. A memory-like NK subset has emerged as a potential immune effector for cancer therapy. The commitment of naïve NK cells to a memory-like phenotype, identified by the expression of CD25, CD69, and down-regulation of CD62L, can be induced by simultaneous stimulation of IL-12, IL-15, and IL-18 [62]. These cytokine-induced memory-type NK cells possess enhanced in vivo expansion capability, IFN-γ production, increased cell persistence, and in vitro/in vivo cytolytic effect [63], [NCT01898793].
Similar to NK responsiveness to these cytokines, T cells express identical receptors that can synergistically elevate cytokine production, in vitro and in vivo expansion and potent cytolytic activities. T cell activation and maturation rely on a transition from naïve T to either activated T or memory T cells [64]. Multiple pathways have been found, including antigen-dependent (i.e., TCR signaling) and cytokine-triggered activation. IL-2, IL-4, IL-6, IL-7, IL-12, IL-15, IL-18, and TNF-α are part of the major cytokines that drive the proliferation of multiple T cell subsets [65,66]. IL-7 and IL-15 have been found to have a substantial effect on the proliferation of effector memory CD4+ T cell and central memory T cell but not naïve T cells, while TNF-α, IL-6, and IL-10 enhanced IL-7/IL-15-induced proliferation [67].
Memory CD8+ T cells have been shown to be activated by cytokines such as IL-12 and IL-18 in an individual or combination manner [68,69]. The activation of CD8+ T cells typically results in IFN-γ production and the upregulation of CD69 expression. A synergetic effect of cytokines on INF-γ production by memory T cells has been shown in which IL-12 supports IL-18-induced activation by lowering its threshold [68]. A comprehensive analysis of multiple cytokines, such as IL-15 or IL-2, has also shown a synergetic effect with IL-12 or IL-18 in T cell activation. [70]. IL-15, unlike IL-12 and IL-18, has been shown as both an activation or inhibition mediator to CD8+ T cells. IL-15 is able to stimulate the upregulation of NKG2D expression of CD8+ T cells, resulting in enhanced cytolytic function of these T cells. Beside cytokine level and cytokine-cytokine synergetic effects, cytokine receptors also play an important role in T cell activation and the transition from naïve T cell into memory T cell [64,71]. For example, the response of each T cell subset has been shown to correlate with the expression level of cytokine receptors, such as IL-2 and IL-15 receptor beta [67]. The cytokine/cytokine receptor-induced signaling triggers a downstream pathway for T cell activation, differentiation, proliferation, and survival [71]. These signaling pathways include STAT3/5, PI3K/AKT, and MAPK pathways [72,73].
Placing these findings into the context of allogeneic cell therapy, in vitro iPSC-derived NK and T cells can be influenced by similar cytokines to mimic the commitment to memory and effector subsets at various scales of production [50,74,75,76]. Although feeder cells were used with the support of key cytokines in iPSC-derived T cell generation, safety, manufacturing of feeder cells, and the use of the feeder cells within iPSC-derived differentiation process at a larger scale remain a challenge. A potential feeder-free large-scale production of iPSC-derived T cells for off-the-shell allogeneic immunotherapy has shown feasibility with the use of cytokine combinations at various stages of differentiation [48]. Despite these advances, the development of manufacturing processes capable of producing iPSC-derived immune cells at a commercial scale is in a nascent state. As this field evolves, one of the key challenges revolves around the need for surface-bound ligands that direct immune-effector cell differentiation and activation, thereby complicating the transition of these unit operations into more scalable bioreactor configurations, such as rocking or stirred tank bioreactors.
6. Cryopreservation
Ensuring that cells survive the cryopreservation process is essential, and the primary focus is the development of a cryo-formulation that maintains cell functionality through the cryopreservation process [77,78]. Although cytokines themselves are not typically included in stabilizing formulations for cryopreservation, prior exposure in the expansion process appear to be important for post-cryo cell health and functionality. Recovery of iPSC seems improved if they are cryopreserved during the log growth phase. Cells going beyond this and entering the stationary phase tend to have poorer survival. Some have observed that cryopreservation 2–4 days after passaging yields good recovery [79]. Chemically defined media containing factors such as FGF-2 and TGF-β1 contribute to the health of stem cells and have been shown to improve survival through cryopreservation [80,81].
In allogeneic immunotherapies, an ‘off-the-shelf’ supply chain requires an approach that reliably preserves the functionality and potency of cryopreserved therapeutic cells, which can be infused immediately after thaw. Effector T cell therapies appear to be amenable to cryopreservation and freeze-thaw cycles; however, there is evidence of CAR-T cells showing signs of cell damage and apoptosis after cryopreservation [82]. When cytokines such as IL-7 and IL-15 are added to a previously cryopreserved preparation, they have been shown to increase the number and functionality of T cells [83]. NK cells are particularly sensitive to lower recovery and loss of functionality after cryopreservation. Compared to fresh NK cells, cryopreserved NK cells have been found to be much less motile and cytotoxic [84]. After cryopreservation, expanded NK cells demonstrate lower cytotoxicity with reduced expression of receptors like TRAIL and NKG2D. Exposure of NK cells to IL-2 post-thaw during in vitro resting reduces this effect [85]. Taken together, the addition of cytokines during T and NK cell manufacturing suggests a positive benefit to increased survival and anti-tumor activity of cryopreserved drug product material.
7. Engineering Cytokine Support into Allogeneic Cell Therapies
Obstacles in CAR-T therapy remain with regard to target specificity and difficulty in T cell activation due to the tumor microenvironment. Supplementation of exogenous cytokines can improve cell health and persistence. For example, stem cell factor (SCF) serves as a regulator of the cell cycle and plays a key role in hematopoietic stem/progenitor cell survival and proliferation [86]. Cytokines and growth factors also serve as communicators between cells for the regulation and balancing of an immune response. Supplementation of exogenous IL-2 in cell culture is able to trigger T cell proliferation and activation [87]. Support from exogenous cytokines such as IL-2 and IL-7 has been used in vivo and in clinical trials for T cell activation and expansion.
Multiple strategies have been developed to improve the efficacy of cytokines in vivo and in clinical settings (Table 1). One of the approaches is to modify the biological structure of the cytokine itself [88,89]. It has recently been shown that by replacing the endogenous secretory motif of IL-24 (melanoma differentiation associated gene-7) with insulin secretory motif and amino acid substitutions, a new ’Superkine’ IL-24S can be created with higher secretion, enhanced stability, and increased anti-tumor activity in multiple cancer xenograft models [88].

Table 1.
List of cytokine/cell engineering.
Another approach to ensure effector cells are exposed to stimulating cytokines is to engineer cells to produce the cytokines themselves in a homeostatic fashion. Gene editing the effector cell or its precursor to secrete the desired cytokine ensures that the cytokine is present in the cell’s immediate environment. Gene editing to affect this type of autocrine support has been reported with IL-7, IL-12, and IL-15 [98]. One of the common methods to implement the expression of transgenic cytokine is by co-expression of cytokine-expressing gene and CAR in the same construct. By using retrovirus transduction, newly integrated cytokine-expressing genes can be controlled under the same promoter as CAR expression. For example, the co-expression of CAR and IL-12 enhanced CAR-T proliferation and IFN-gamma secretion in vitro, as well as anti-tumor efficacy toward ovarian cancer in vivo [90]. A new phenotype of NK-like CAR-T demonstrated the capability to target non-antigen-presenting tumors with the CAR/IL-12 co-expression system in CAR-T [91]. IL-7 is known as the key cytokine for T cell expansion and maturation and has been widely used in vivo and in clinical applications [92].
Researchers have also shown engineered tethered versions of cytokines to be effective [4]. Further cell engineering to include the expression of the cytokine receptor will ensure that the cell will effectively respond to the presence of the desired cytokine, either exogenously supplemented or secreted by the effector cells themselves. Constitutive expression of IL-7 receptor (C7R) was shown to actively stimulate IL-7 signaling in CAR-T cells without the existence of extracellular IL-7, which improved T cell proliferation, survival, and antitumor activity [93].
Another novel approach to facilitate the autocrine cytokine support and signaling is to create a cytokine/cytokine-receptor pair. Engineering CAR-T cells to express the orthogonal pairs of IL-2/IL-2 receptor (IL-2/IL-2R) was demonstrated to increase the specificity of T cell activation and reduced toxicity due to native IL-2 [94]. Similar approaches have been applied to IL-15/IL15R and IL-7/IL-7R expressing systems in cancer therapy [4,99,100]. In short summary, engineering the structure of cytokines or their expression in engineered immune cells enforces cytokine support in anti-tumor activities, as shown with in vivo mouse models and in clinical applications.
8. Use of Exogenous Cytokines in Cell Therapy Trials
The previous sections described the manufacturing of iPSC-derived cell therapeutics and specific points in the manufacturing process where cytokines are used to improve cell health, persistence, and function. The impact of cytokines, however, goes beyond the filled cell product. In some cases, successful delivery of a cell therapy benefits from additional exposure at the time and after administration to maintain the same effects on cell health and function as desired in the manufacturing environment. Therefore, the goal of delivering an effective therapeutic does not end at the cell product stage but rather with successful administration to the patient. As a result, manufacturing and delivery are inextricably linked.
Cytokines themselves traditionally have played a key role in cancer immunotherapy, with some being used as monotherapies (e.g., IL-2, IL-15) or in combinations with monoclonal antibodies (e.g., rituximab) or chemotherapeutics [101]. Many cytokines and factors, supplied exogenously, have been used or planned to be used in clinical trials with cellular-based therapies (Table 2).
Table 2.
Examples of clinical trials using exogenous cytokines and factors in conjunction with cell therapies.
While cytokines such as G-CSF (filgrastim), IFN-γ, GM-CSF (sargramostim), and stem cell factor (SCF) were in previous clinical trials with autologous stem-cell treatments, more recent trials seem to rely on IL-2 (aldesleukin). IL-2 was the first recombinantly produced cytokine and remains in use as an immunostimulatory with anti-cancer activity, and it is involved in the activation and growth of T and NK cells [102].
Most of the trials using a cytokine have IL-2 as an arm. The use of IL-2 is much more frequent and still used or planned for use in more recent clinical trials for both autologous and allogeneic cell therapies. IL-2 is typically administered prior to cell treatment for single-dose therapies, especially for T cells. IL-2 is used to stimulate T cell production for enhancing anti-cancer immunity. In the case of NK cells that may have multiple dosing, IL-2 can be used repeatedly throughout the treatment regimen between and after dosing.
9. Conclusions
The use of cytokines is inextricably linked to the development and function of allogeneic cell therapeutics. For maximum benefit, these factors need to be controlled to optimize cell differentiation, activation, and expansion throughout the manufacturing of these emerging therapies. Continued improvements can be expected as more knowledge is gained into the complex responses of cell therapeutics.
Author Contributions
Conceptualization, C.-Y.K., J.A.M., C.J.B. and B.F.M.; writing—original draft preparation, C.-Y.K., J.A.M., C.J.B., B.M. and B.F.M.; writing—review and editing, C.-Y.K., J.A.M., C.J.B. and B.F.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Gregory Russotti, Kenneth Dow, and Elizabeth Krutoholow for the internal review of this manuscript.
Conflicts of Interest
The authors declare that Century Therapeutics is developing allogeneic cell products.
References
- Burke, C.J.; Zylberberg, C. Sources of Variability in Manufacturing of Cell Therapeutics. Regen. Eng. Transl. Med. 2019, 5, 332–340. [Google Scholar] [CrossRef]
- Borges, L.; Levitsky, H. The Challenge of Immune Rejection for Allogeneic Cell Therapies. Bioprocess Online 2022. Available online: https://www.cellandgene.com/doc/the-challenge-of-immune-rejection-for-allogeneic-cell-therapies-0001 (accessed on 23 December 2022).
- Koga, K.; Wang, B.; Kaneko, S. Current Status and Future Perspectives of HLA-Edited Induced Pluripotent Stem Cells. Inflamm Regen 2020, 40, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.A.; et al. Tethered IL-15 Augments Antitumor Activity and Promotes a Stem-Cell Memory Subset in Tumor-Specific T Cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7788–E7797. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, I.; Ho, W.J.; Marple, A.; Ravich, J.W.; Tam, A.; Rahnama, R.; Fearnow, A.; Rietberg, C.; Yanik, S.; Solomou, E.E.; et al. Engineering CAR-NK Cells to Secrete IL-15 Sustains Their Anti-AML Functionality but Is Associated with Systemic Toxicities. J. Immunother. Cancer 2021, 9, e003894. [Google Scholar] [CrossRef]
- Mesquitta, W.T.; Wandsnider, M.; Kang, H.J.; Thomson, J.; Moskvin, O.; Suknuntha, K.; Slukvin, I.I. UM171 Expands Distinct Types of Myeloid and NK Progenitors from Human Pluripotent Stem Cells. Sci. Rep. 2019, 9, 6622. [Google Scholar] [CrossRef]
- Ni, Z.; Knorr, D.A.; Kaufman, D.S. Hematopoietic and Nature Killer Cell Development from Human Pluripotent Stem Cells. Methods Mol. Biol. 2013, 1029, 33–41. [Google Scholar] [CrossRef]
- Wang, Z.; McWilliams-Koeppen, H.P.; Reza, H.; Ostberg, J.R.; Chen, W.; Wang, X.; Huynh, C.; Vyas, V.; Chang, W.C.; Starr, R.; et al. 3D-Organoid Culture Supports Differentiation of Human CAR+ IPSCs into Highly Functional CAR T Cells. Cell Stem Cell 2022, 29, 515–527. [Google Scholar] [CrossRef]
- Jing, R.; Scarfo, I.; Najia, M.A.; Lummertz da Rocha, E.; Han, A.; Sanborn, M.; Bingham, T.; Kubaczka, C.; Jha, D.K.; Falchetti, M.; et al. EZH1 Repression Generates Mature IPSC-Derived CAR T Cells with Enhanced Antitumor Activity. Cell Stem Cell 2022, 29, 1181–1196.e6. [Google Scholar] [CrossRef]
- Li, Y.R.; Zhou, Y.; Kim, Y.J.; Zhu, Y.; Ma, F.; Yu, J.; Wang, Y.C.; Chen, X.; Li, Z.; Zeng, S.; et al. Development of Allogeneic HSC-Engineered INKT Cells for off-the-Shelf Cancer Immunotherapy. Cell Rep. Med. 2021, 2, 100449. [Google Scholar] [CrossRef]
- Liu, G.; David, B.T.; Trawczynski, M.; Fessler, R.G. Advances in Pluripotent Stem Cells: History, Mechanisms, Technologies, and Applications. Stem. Cell Rev. Rep. 2020, 16, 3–32. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Scesa, G.; Adami, R.; Bottai, D. IPSC Preparation and Epigenetic Memory: Does the Tissue Origin Matter? Cells 2021, 10, 1470. [Google Scholar] [CrossRef] [PubMed]
- Malik, N.; Rao, M.S. A Review of the Methods for Human IPSC Derivation. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 997. [Google Scholar]
- Seki, T.; Yuasa, S.; Oda, M.; Egashira, T.; Yae, K.; Kusumoto, D.; Nakata, H.; Tohyama, S.; Hashimoto, H.; Kodaira, M.; et al. Generation of Induced Pluripotent Stem Cells from Human Terminally Differentiated Circulating t Cells. Cell Stem Cell 2010, 7, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Yuasa, S.; Fukuda, K. Derivation of Induced Pluripotent Stem Cells from Human Peripheral Circulating T Cells. Curr. Protoc. Stem Cell Biol. 2011, 18, 4A.3.1–4A.3.9. [Google Scholar] [CrossRef]
- Haridhasapavalan, K.K.; Borgohain, M.P.; Dey, C.; Saha, B.; Narayan, G.; Kumar, S.; Thummer, R.P. An Insight into Non-Integrative Gene Delivery Approaches to Generate Transgene-Free Induced Pluripotent Stem Cells. Gene 2019, 686, 146–159. [Google Scholar] [CrossRef]
- Hirsch, M.L.; Wolf, S.J.; Samulski, R.J. Delivering Transgenic DNA Exceeding the Carrying Capacity of AAV Vectors. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2016; Volume 1382. [Google Scholar]
- Li, J.; Song, W.; Pan, G.; Zhou, J. Advances in Understanding the Cell Types and Approaches Used for Generating Induced Pluripotent Stem Cells. J. Hematol. Oncol. 2014, 7, 50. [Google Scholar] [CrossRef]
- Rao, M.S.; Malik, N. Assessing IPSC Reprogramming Methods for Their Suitability in Translational Medicine. J. Cell Biochem. 2012, 113, 3061–3068. [Google Scholar] [CrossRef]
- al Abbar, A.; Ngai, S.C.; Nograles, N.; Alhaji, S.Y.; Abdullah, S. Induced Pluripotent Stem Cells: Reprogramming Platforms and Applications in Cell Replacement Therapy. Biores. Open Access 2020, 9, 121–136. [Google Scholar] [CrossRef]
- Ray, A.; Joshi, J.M.; Sundaravadivelu, P.K.; Raina, K.; Lenka, N.; Kaveeshwar, V.; Thummer, R.P. An Overview on Promising Somatic Cell Sources Utilized for the Efficient Generation of Induced Pluripotent Stem Cells. Stem Cell Rev. Rep. 2021, 17, 1954–1974. [Google Scholar] [CrossRef]
- Raab, S.; Klingenstein, M.; Liebau, S.; Linta, L. A Comparative View on Human Somatic Cell Sources for IPSC Generation. Stem Cells Int. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Shamsian, A.; Sahebnasagh, R.; Norouzy, A.; Hussein, S.H.; Ghahremani, M.H.; Azizi, Z. Cancer Cells as a New Source of Induced Pluripotent Stem Cells. Stem Cell Res. Ther. 2022, 13, 459. [Google Scholar] [CrossRef]
- Chen, G.; Gulbranson, D.R.; Hou, Z.; Bolin, J.M.; Ruotti, V.; Probasco, M.D.; Smuga-Otto, K.; Howden, S.E.; Diol, N.R.; Propson, N.E.; et al. Chemically Defined Conditions for Human IPSC Derivation and Culture. Nat. Methods 2011, 8, 424–429. [Google Scholar] [CrossRef]
- Yasuda, S.Y.; Ikeda, T.; Shahsavarani, H.; Yoshida, N.; Nayer, B.; Hino, M.; Vartak-Sharma, N.; Suemori, H.; Hasegawa, K. Chemically Defined and Growth-Factor-Free Culture System for the Expansion and Derivation of Human Pluripotent Stem Cells. Nat. Biomed. Eng. 2018, 2, 173–182. [Google Scholar] [CrossRef]
- Kuo, H.H.; Gao, X.; DeKeyser, J.M.; Fetterman, K.A.; Pinheiro, E.A.; Weddle, C.J.; Fonoudi, H.; Orman, M.V.; Romero-Tejeda, M.; Jouni, M.; et al. Negligible-Cost and Weekend-Free Chemically Defined Human IPSC Culture. Stem Cell Rep. 2020, 14, 256–270. [Google Scholar] [CrossRef]
- Borys, B.S.; So, T.; Colter, J.; Dang, T.; Roberts, E.L.; Revay, T.; Larijani, L.; Krawetz, R.; Lewis, I.; Argiropoulos, B.; et al. Optimized Serial Expansion of Human Induced Pluripotent Stem Cells Using Low-Density Inoculation to Generate Clinically Relevant Quantities in Vertical-Wheel Bioreactors. Stem Cells Transl. Med. 2020, 9, 1036–1052. [Google Scholar] [CrossRef]
- Elanzew, A.; Sommer, A.; Pusch-Klein, A.; Brüstle, O.; Haupt, S. A Reproducible and Versatile System for the Dynamic Expansion of Human Pluripotent Stem Cells in Suspension. Biotechnol. J. 2015, 10, 1589–1599. [Google Scholar] [CrossRef]
- Nogueira, D.E.S.; Rodrigues, C.A.V.; Carvalho, M.S.; Miranda, C.C.; Hashimura, Y.; Jung, S.; Lee, B.; Cabral, J.M.S. Strategies for the Expansion of Human Induced Pluripotent Stem Cells as Aggregates in Single-Use Vertical-WheelTM Bioreactors. J. Biol. Eng. 2019, 13, 1–14. [Google Scholar] [CrossRef]
- Gordeeva, O. TGFβ Family Signaling Pathways in Pluripotent and Teratocarcinoma Stem Cells’ Fate Decisions: Balancing between Self-Renewal, Differentiation, and Cancer. Cells 2019, 8, 1500. [Google Scholar] [CrossRef]
- Tursky, M.L.; Loi, T.H.; Artuz, C.M.; Alateeq, S.; Wolvetang, E.J.; Tao, H.; Ma, D.D.; Molloy, T.J. Direct Comparison of Four Hematopoietic Differentiation Methods from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2020, 15, 735–748. [Google Scholar] [CrossRef]
- Fang, F.; Xie, S.; Chen, M.; Li, Y.; Yue, J.; Ma, J.; Shu, X.; He, Y.; Xiao, W.; Tian, Z. Advances in NK Cell Production. Cell Mol. Immunol. 2022, 19, 460–481. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; D’Souza, S.; Uenishi, G.; Park, M.; Lee, J.; Slukvin, I. Generation of T Cells from Human and Nonhuman Primate Pluripotent Stem Cells. Bio. Protoc. 2020, 10, e3675. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Yu, Q.; Long, Y.; Luo, Q.; Li, H.; Han, Y.; Xu, Y.; Fu, S.; Zeng, X.; Wei, C.; et al. Enhanced HSC-like Cell Generation from Mouse Pluripotent Stem Cells in a 3D Induction System Cocultured with Stromal Cells. Stem Cell Res. Ther. 2021, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.F.; Kuo, H.C.; Chien, C.L.; Shun, C.T.; Yao, Y.L.; Ip, P.L.; Chuang, C.Y.; Wang, C.C.; Yang, Y.S.; Ho, H.N. Derivation, Characterization and Differentiation of Human Embryonic Stem Cells: Comparing Serum-Containing versus Serum-Free Media and Evidence of Germ Cell Differentiation. Hum. Reprod. 2007, 22, 567–577. [Google Scholar] [CrossRef]
- Bruveris, F.F.; Ng, E.S.; Stanley, E.G.; Elefanty, A.G. VEGF, FGF2, and BMP4 Regulate Transitions of Mesoderm to Endothelium and Blood Cells in a Human Model of Yolk Sac Hematopoiesis. Exp. Hematol. 2021, 103, 30–39.e2. [Google Scholar] [CrossRef]
- Mills, J.A.; Paluru, P.; Weiss, M.J.; Gadue, P.; French, D.L. Hematopoietic Differentiation of Pluripotent Stem Cells in Culture. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2014; Volume 1185. [Google Scholar] [CrossRef]
- Ackermann, M.; Rafiei Hashtchin, A.; Manstein, F.; Carvalho Oliveira, M.; Kempf, H.; Zweigerdt, R.; Lachmann, N. Continuous Human IPSC-Macrophage Mass Production by Suspension Culture in Stirred Tank Bioreactors. Nat. Protoc. 2022, 17, 513–519. [Google Scholar] [CrossRef]
- Pandey, P.R.; Tomney, A.; Woon, M.T.; Uth, N.; Shafighi, F.; Ngabo, I.; Vallabhaneni, H.; Levinson, Y.; Abraham, E.; Ben-Nun, I.F. End-to-End Platform for Human Pluripotent Stem Cell Manufacturing. Int. J. Mol. Sci. 2020, 21, 89. [Google Scholar] [CrossRef]
- Rieger, M.A.; Hoppe, P.S.; Smejkal, B.M.; Eitelhuber, A.C.; Schroeder, T. Hematopoietic Cytokines Can Instruct Lineage Choice. Science 2009, 325, 217–218. [Google Scholar] [CrossRef]
- Carlyle, J.R.; Zúñiga-Pflücker, J.C. Lineage Commitment and Differentiation of T and Natural Killer Lymphocytes in the Fetal Mouse. Immunol. Rev. 1998, 165, 63–74. [Google Scholar] [CrossRef]
- Smith, M.J.; Webber, B.R.; Mohtashami, M.; Stefanski, H.E.; Zúñiga-Pflücker, J.C.; Blazar, B.R. In Vitro T-Cell Generation from Adult, Embryonic, and Induced Pluripotent Stem Cells: Many Roads to One Destination. Stem Cells 2015, 33, 3174–3180. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Iriguchi, S.; Yasui, Y.; Kawai, Y.; Arima, S.; Kunitomo, M.; Sato, T.; Ueda, T.; Minagawa, A.; Mishima, Y.; Yanagawa, N.; et al. A Clinically Applicable and Scalable Method to Regenerate T-Cells from IPSCs for off-the-Shelf T-Cell Immunotherapy. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Vizcardo, R.; Masuda, K.; Yamada, D.; Ikawa, T.; Shimizu, K.; Fujii, S.I.; Koseki, H.; Kawamoto, H. Regeneration of Human Tumor Antigen-Specific T Cells from IPSCs Derived from Mature CD8+ T Cells. Cell Stem Cell 2013, 12, 31–36. [Google Scholar] [CrossRef]
- Nishimura, T.; Kaneko, S.; Kawana-Tachikawa, A.; Tajima, Y.; Goto, H.; Zhu, D.; Nakayama-Hosoya, K.; Iriguchi, S.; Uemura, Y.; Shimizu, T.; et al. Generation of Rejuvenated Antigen-Specific T Cells by Reprogramming to Pluripotency and Redifferentiation. Cell Stem Cell 2013, 12, 114–126. [Google Scholar] [CrossRef]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef]
- Euchner, J.; Sprissler, J.; Cathomen, T.; Fürst, D.; Schrezenmeier, H.; Debatin, K.M.; Schwarz, K.; Felgentreff, K. Natural Killer Cells Generated From Human Induced Pluripotent Stem Cells Mature to CD56brightCD16+NKp80+/- In-Vitro and Express KIR2DL2/DL3 and KIR3DL1. Front. Immunol. 2021, 12, 640672. [Google Scholar] [CrossRef]
- Grzywacz, B.; Kataria, N.; Kataria, N.; Blazar, B.R.; Miller, J.S.; Verneris, M.R. Natural Killer-Cell Differentiation by Myeloid Progenitors. Blood 2011, 117, 3548–3558. [Google Scholar] [CrossRef]
- Luevano, M.; Madrigal, A.; Saudemont, A. Generation of Natural Killer Cells from Hematopoietic Stem Cells in Vitro for Immunotherapy. Cell Mol. Immunol. 2012, 9, 310–320. [Google Scholar] [CrossRef]
- Pixley, F.J.; Stanley, E.R. Cytokines and Cytokine Receptors Regulating Cell Survival, Proliferation, and Differentiation in Hematopoiesis. In Handbook of Cell Signaling, 2nd ed.; Academic Press: Cambridge, MA, USA, 2010; Volume 3. [Google Scholar]
- Fiedler, K.; Brunner, C. Mechanisms Controlling Hematopoiesis. In Hematology—Science and Practice; IntechOpen: London, UK, 2012. [Google Scholar]
- Zhang, M.; Wen, B.; Anton, O.M.; Yao, Z.; Dubois, S.; Ju, W.; Sato, N.; DiLillo, D.J.; Bamford, R.N.; Ravetch, J.V.; et al. IL-15 Enhanced Antibody-Dependent Cellular Cytotoxicity Mediated by NK Cells and Macrophages. Proc. Natl. Acad. Sci. USA 2018, 115, E10924. [Google Scholar] [CrossRef]
- Parihar, R.; Dierksheide, J.; Hu, Y.; Carson, W.E. IL-12 Enhances the Natural Killer Cell Cytokine Response to Ab-Coated Tumor Cells. J. Clin. Investig. 2002, 110, 983–992. [Google Scholar] [CrossRef]
- Lusty, E.; Poznanski, S.M.; Kwofie, K.; Mandur, T.S.; Lee, D.A.; Ashkar, A.A. IL-18/IL-15/IL-12 Synergy Induces Elevated and Prolonged IFN-γ Production by Ex Vivo Expanded NK Cells Which Is Not Due to Enhanced STAT4 Activation. Mol. Immunol. 2017, 88, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Zwirner, N.W.; Ziblat, A. Regulation of NK Cell Activation and Effector Functions by the IL-12 Family of Cytokines: The Case of IL-27. Front. Immunol. 2017, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Kiniwa, T.; Enomoto, Y.; Terazawa, N.; Omia, A.; Miyataa, N.; Ishiwata, K.; Miyajima, A. NK Cells Activated by Interleukin-4 in Cooperation with Interleukin-15 Exhibit Distinctive Characteristics. Proc. Natl. Acad. Sci. USA 2016, 113, 10139–10144. [Google Scholar] [CrossRef] [PubMed]
- Gang, M.; Marin, N.D.; Wong, P.; Neal, C.C.; Marsala, L.; Foster, M.; Schappe, T.; Meng, W.; Tran, J.; Schaettler, M.; et al. CAR-Modified Memory-like NK Cells Exhibit Potent Responses to NK-Resistant Lymphomas. Blood 2020, 136, 2308–2318. [Google Scholar] [CrossRef]
- Mahapatra, S.; Mace, E.M.; Minard, C.G.; Forbes, L.R.; Vargas-Hernandez, A.; Duryea, T.K.; Makedonas, G.; Banerjee, P.P.; Shearer, W.T.; Orange, J.S. High-Resolution Phenotyping Identifies NK Cell Subsets That Distinguish Healthy Children from Adults. PLoS ONE 2017, 12, e0181134. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef]
- Freeman, B.E.; Raué, H.-P.; Hill, A.B.; Slifka, M.K. Cytokine-Mediated Activation of NK Cells during Viral Infection. J. Virol. 2015, 89, 7922–7931. [Google Scholar] [CrossRef]
- Schluns, K.S.; Lefrançois, L. Cytokine Control of Memory T-Cell Development and Survival. Nat. Rev. Immunol. 2003, 3, 269–279. [Google Scholar] [CrossRef]
- Zhang, Y.; Guan, X.Y.; Jiang, P. Cytokine and Chemokine Signals of T-Cell Exclusion in Tumors. Front. Immunol. 2020, 11, 594609. [Google Scholar] [CrossRef]
- Bevington, S.L.; Cauchy, P.; Withers, D.R.; Lane, P.J.L.; Cockerill, P.N. T Cell Receptor and Cytokine Signaling Can Function at Different Stages to Establish and Maintain Transcriptional Memory and Enable T Helper Cell Differentiation. Front. Immunol. 2017, 8, 204. [Google Scholar] [CrossRef]
- Geginat, J.; Sallusto, F.; Lanzavecchia, A. Cytokine-Driven Proliferation and Differentiation of Human Naive, Central Memory, and Effector Memory CD4+ T Cells. J. Exp. Med. 2001, 194, 1711–1720. [Google Scholar] [CrossRef]
- Raué, H.-P.; Brien, J.D.; Hammarlund, E.; Slifka, M.K. Activation of Virus-Specific CD8+ T Cells by Lipopolysaccharide-Induced IL-12 and IL-18. J. Immunol. 2004, 173, 6873–6881. [Google Scholar] [CrossRef]
- Smeltz, R.B. Profound Enhancement of the IL-12/IL-18 Pathway of IFN-γ Secretion in Human CD8+ Memory T Cell Subsets via IL-15. J. Immunol. 2007, 178, 4786–4792. [Google Scholar] [CrossRef]
- Freeman, B.E.; Hammarlund, E.; Raué, H.P.; Slifka, M.K. Regulation of Innate CD8+ T-Cell Activation Mediated by Cytokines. Proc. Natl. Acad. Sci. USA 2012, 109, 9971–9976. [Google Scholar] [CrossRef]
- Huang, W.; August, A. The Signaling Symphony: T Cell Receptor Tunes Cytokine-Mediated T Cell Differentiation. J. Leukoc. Biol. 2015, 97, 477–485. [Google Scholar] [CrossRef]
- Rochman, Y.; Spolski, R.; Leonard, W.J. New Insights into the Regulation of T Cells by Γc Family Cytokines. Nat. Rev. Immunol. 2009, 9, 480–490. [Google Scholar] [CrossRef]
- Dwyer, C.J.; Knochelmann, H.M.; Smith, A.S.; Wyatt, M.M.; Rivera, G.O.R.; Arhontoulis, D.C.; Bartee, E.; Li, Z.; Rubinstein, M.P.; Paulos, C.M. Fueling Cancer Immunothery with Common Gamma Chain Cytokines. Front. Immunol. 2019, 10, 263. [Google Scholar] [CrossRef]
- Maeda, T.; Nagano, S.; Ichise, H.; Kataoka, K.; Yamada, D.; Ogawa, S.; Koseki, H.; Kitawaki, T.; Kadowaki, N.; Takaori-Kondo, A.; et al. Regeneration of CD8αβ T Cells from T-Cell-Derived IPSC Imparts Potent Tumor Antigen-Specific Cytotoxicity. Cancer Res. 2016, 76, 6839–6850. [Google Scholar] [CrossRef]
- Cichocki, F.; Bjordahl, R.; Gaidarova, S.; Mahmood, S.; Abujarour, R.; Wang, H.; Tuininga, K.; Felices, M.; Davis, Z.B.; Bendzick, L.; et al. IPSC-Derived NK Cells Maintain High Cytotoxicity and Enhance in Vivo Tumor Control in Concert with T Cells and Anti–PD-1 Therapy. Sci. Transl. Med. 2020, 12, 5618. [Google Scholar] [CrossRef] [PubMed]
- Knorr, D.A.; Ni, Z.; Hermanson, D.; Hexum, M.K.; Bendzick, L.; Cooper, L.J.N.; Lee, D.A.; Kaufman, D.S. Clinical-Scale Derivation of Natural Killer Cells From Human Pluripotent Stem Cells for Cancer Therapy. Stem Cells Transl. Med. 2013, 2, 274–283. [Google Scholar] [CrossRef]
- LI, R.; JOHNSON, R.; YU, G.; MCKENNA, D.H.; HUBEL, A. Preservation of Cell-Based Immunotherapies for Clinical Trials. Cytotherapy 2019, 21, 943–957. [Google Scholar] [CrossRef]
- Cottle, C.; Porter, A.P.; Lipat, A.; Turner-Lyles, C.; Nguyen, J.; Moll, G.; Chinnadurai, R. Impact of Cryopreservation and Freeze-Thawing on Therapeutic Properties of Mesenchymal Stromal/Stem Cells and Other Common Cellular Therapeutics. Curr. Stem Cell Rep. 2022, 8, 72–92. [Google Scholar] [CrossRef] [PubMed]
- Uhrig, M.; Ezquer, F.; Ezquer, M. Improving Cell Recovery: Freezing and Thawing Optimization of Induced Pluripotent Stem Cells. Cells 2022, 11, 799. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Yang, D.; Li, Q.; Tian, W.; Guo, W. The Establishment of a Chemically Defined Serum-Free Culture System for Human Dental Pulp Stem Cells. Stem Cell Res. Ther. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Liu, W.; Chen, G. Cryopreservation of Human Pluripotent Stem Cells in Defined Medium. Curr. Protoc. Stem Cell Biol. 2014, 2014, 1C.17.1–1C.17.13. [Google Scholar] [CrossRef] [PubMed]
- Panch, S.R.; Srivastava, S.K.; Elavia, N.; McManus, A.; Liu, S.; Jin, P.; Highfill, S.L.; Li, X.; Dagur, P.; Kochenderfer, J.N.; et al. Effect of Cryopreservation on Autologous Chimeric Antigen Receptor T Cell Characteristics. Mol. Ther. 2019, 27, 1275–1285. [Google Scholar] [CrossRef]
- Jennes, W.; Kestens, L.; Nixon, D.F.; Shacklett, B.L. Enhanced ELISPOT Detection of Antigen-Specific T Cell Responses from Cryopreserved Specimens with Addition of Both IL-7 and IL-15—The Amplispot Assay. J. Immunol. Methods 2002, 270, 99–108. [Google Scholar] [CrossRef]
- Mark, C.; Czerwinski, T.; Roessner, S.; Mainka, A.; Hörsch, F.; Heublein, L.; Winterl, A.; Sanokowski, S.; Richter, S.; Bauer, N.; et al. Cryopreservation Impairs 3-D Migration and Cytotoxicity of Natural Killer Cells. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Berg, M.; Lundqvist, A.; McCoy, P.; Samsel, L.; Fan, Y.; Tawab, A.; Childs, R. Clinical-Grade Ex Vivo-Expanded Human Natural Killer Cells up-Regulate Activating Receptors and Death Receptor Ligands and Have Enhanced Cytolytic Activity against Tumor Cells. Cytotherapy 2009, 11, 341–355. [Google Scholar] [CrossRef]
- Hassan, H.T.; Zander, A. Stem Cell Factor as a Survival and Growth Factor in Human Normal and Malignant Hematopoiesis. Acta Haematol. 1996, 95, 257–262. [Google Scholar] [CrossRef]
- Nelson, B.H. IL-2, Regulatory T Cells, and Tolerance. J. Immunol. 2004, 172, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.K.; Bhoopathi, P.; Maji, S.; Kumar, A.; Guo, C.; Mannangatti, P.; Li, J.; Wang, X.Y.; Sarkar, D.; Emdad, L.; et al. Enhanced Cancer Therapy Using an Engineered Designer Cytokine Alone and in Combination With an Immune Checkpoint Inhibitor. Front. Oncol. 2022, 12, 812560. [Google Scholar] [CrossRef]
- Kim, M.Y.; Jayasinghe, R.; Devenport, J.M.; Ritchey, J.K.; Rettig, M.P.; O’Neal, J.; Staser, K.W.; Kennerly, K.M.; Carter, A.J.; Gao, F.; et al. A Long-Acting Interleukin-7, RhIL-7-HyFc, Enhances CAR T Cell Expansion, Persistence, and Anti-Tumor Activity. Nat. Commun. 2022, 13, 3296. [Google Scholar] [CrossRef]
- Bell, M.; Gottschalk, S. Engineered Cytokine Signaling to Improve CAR T Cell Effector Function. Front. Immunol. 2021, 12, 684642. [Google Scholar] [CrossRef] [PubMed]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 Secreting Tumor-Targeted Chimeric Antigen Receptor T Cells Eradicate Ovarian Tumors in Vivo. Oncoimmunology 2015, 4, e994446. [Google Scholar] [CrossRef]
- Hombach, A.; Barden, M.; Hannappel, L.; Chmielewski, M.; Rappl, G.; Sachinidis, A.; Abken, H. IL12 Integrated into the CAR Exodomain Converts CD8+ T Cells to Poly-Functional NK-like Cells with Superior Killing of Antigen-Loss Tumors. Mol. Ther. 2022, 30, 593–605. [Google Scholar] [CrossRef]
- Li, L.; Li, Q.; Yan, Z.-X.; Sheng, L.-S.; Fu, D.; Xu, P.; Wang, L.; Zhao, W.-L. Transgenic Expression of IL-7 Regulates CAR-T Cell Metabolism and Enhances in Vivo Persistence against Tumor Cells. Sci. Rep. 2022, 12, 12506. [Google Scholar] [CrossRef] [PubMed]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef]
- Sockolosky, J.T.; Trotta, E.; Parisi, G.; Picton, L.; Su, L.L.; Le, A.C.; Chhabra, A.; Silveria, S.L.; George, B.M.; King, I.C.; et al. Selective Targeting of Engineered T Cells Using Orthogonal IL-2 Cytokine-Receptor Complexes. Science 2018, 359, 1037–1042. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhuang, Q.; Wang, F.; Zhang, C.; Xu, C.; Gu, A.; Zhong, W.H.; Hu, Y.; Zhong, X. Co-Expression IL-15 Receptor Alpha with IL-15 Reduces Toxicity via Limiting IL-15 Systemic Exposure during CAR-T Immunotherapy. J. Transl. Med. 2022, 20, 432. [Google Scholar] [CrossRef]
- Rowley, J.; Monie, A.; Hung, C.F.; Wu, T.C. Expression of IL-15RA or an IL-15/IL-15RA Fusion on CD8+ T Cells Modifies Adoptively Transferred T-Cell Function in Cis. Eur. J. Immunol. 2009, 39, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, Q.; Han, Z.; Zhu, Y.; Shen, H.; Liu, Z.; Zhou, Z.; Ding, W.; Han, S.; He, J.; et al. IL-7 and CCR2b Co-Expression-Mediated Enhanced CAR-T Survival and Infiltration in Solid Tumors. Front. Oncol. 2021, 11, 734593. [Google Scholar] [CrossRef]
- Goto, S.; Sakoda, Y.; Adachi, K.; Sekido, Y.; Yano, S.; Eto, M.; Tamada, K. Enhanced Anti-Tumor Efficacy of IL-7/CCL19-Producing Human CAR-T Cells in Orthotopic and Patient-Derived Xenograft Tumor Models. Cancer Immunol. Immunother. 2021, 70, 2503–2515. [Google Scholar] [CrossRef]
- Tan, A.H.J.; Vinanica, N.; Campana, D. Chimeric Antigen Receptor-T Cells with Cytokine Neutralizing Capacity. Blood Adv. 2020, 4, 1419–1431. [Google Scholar] [CrossRef]
- Qiu, Y.; Su, M.; Liu, L.; Tang, Y.; Pan, Y.; Sun, J. Clinical Application of Cytokines in Cancer Immunotherapy. Drug Des. Devel. Ther. 2021, 15, 2269–2287. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in Cancer Immunotherapy. Oncoimmunology 2016, 5, e1163462. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).