
Fpr2−/− Mice Developed Exacerbated Alcohol-Associated Liver Disease
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Studies
2.2. Measurement of Plasma ALT Activity and Endotoxin Levels
2.3. Immunoassay of TNFα in Liver Tissue
2.4. Histopathological and Immunohistochemical Analysis of Liver Tissue
2.5. Liver Triglycerides Extraction and Measurement
2.6. RNA Isolation and Real-Time Quantitative Reverse Transcription PCR (RT-qPCR)
2.7. Flow Cytometry Analysis of Neutrophils
2.8. Measurement of Liver MPO Activity and H2O2 Levels
2.9. Flow Cytometry Analysis
2.10. BMDM and Neutrophil Co-Culture Experiment
2.11. Statistical Analysis
3. Results
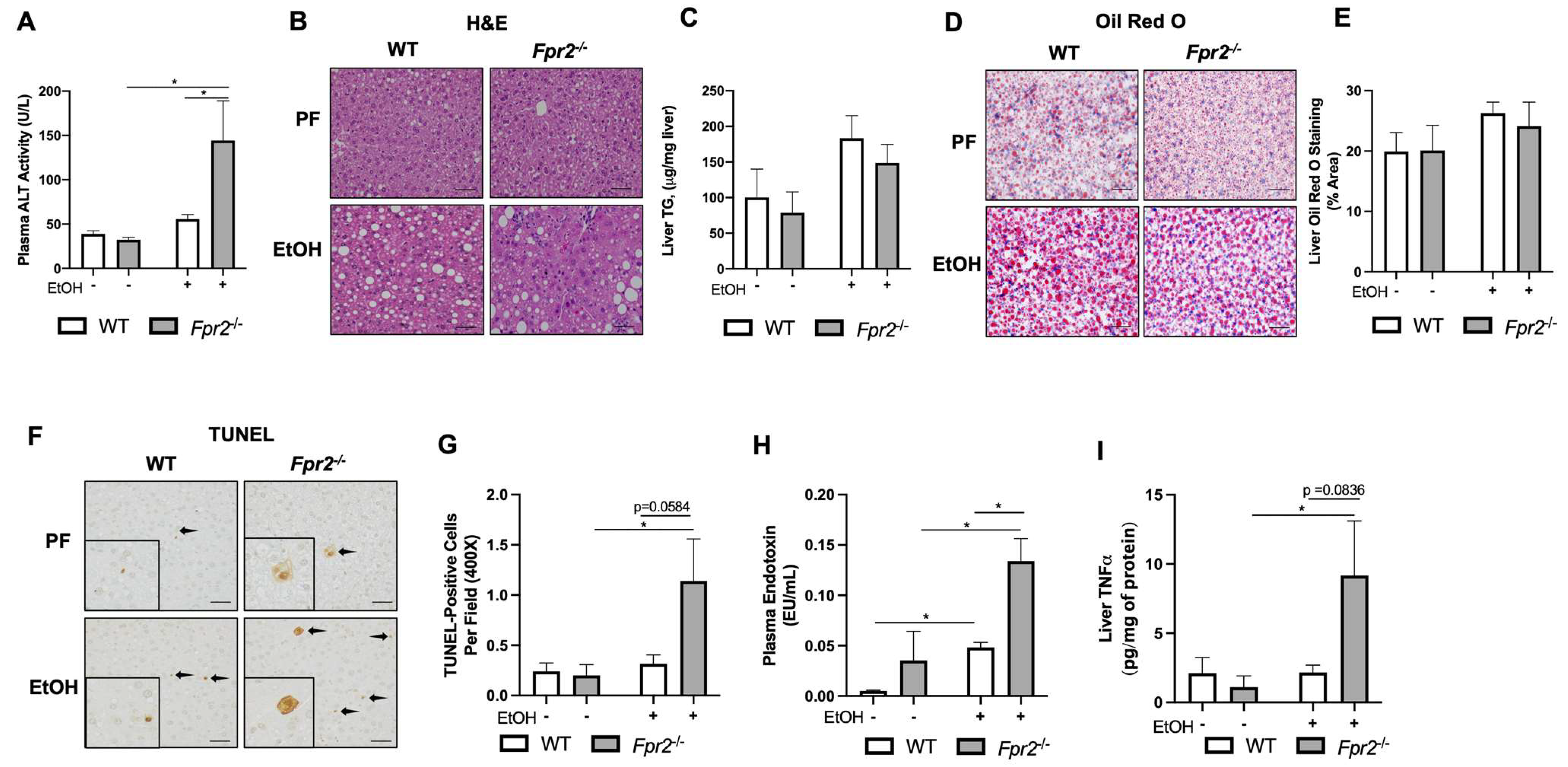
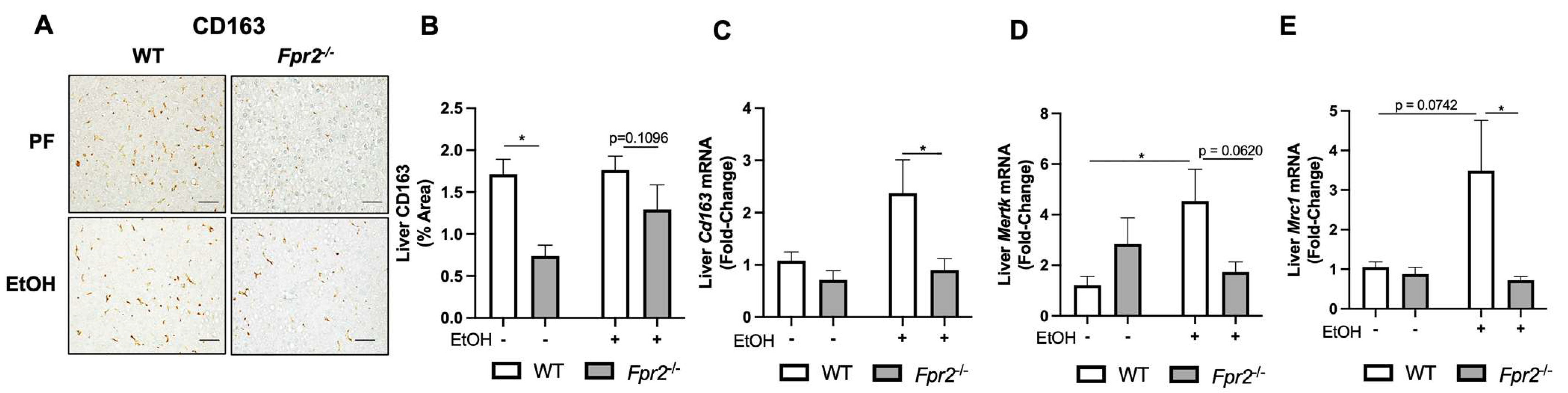
3.1. Fpr2−/− Mice Developed Exacerbated Acute-on-Chronic Alcohol-Induced Liver Injury and Inflammation
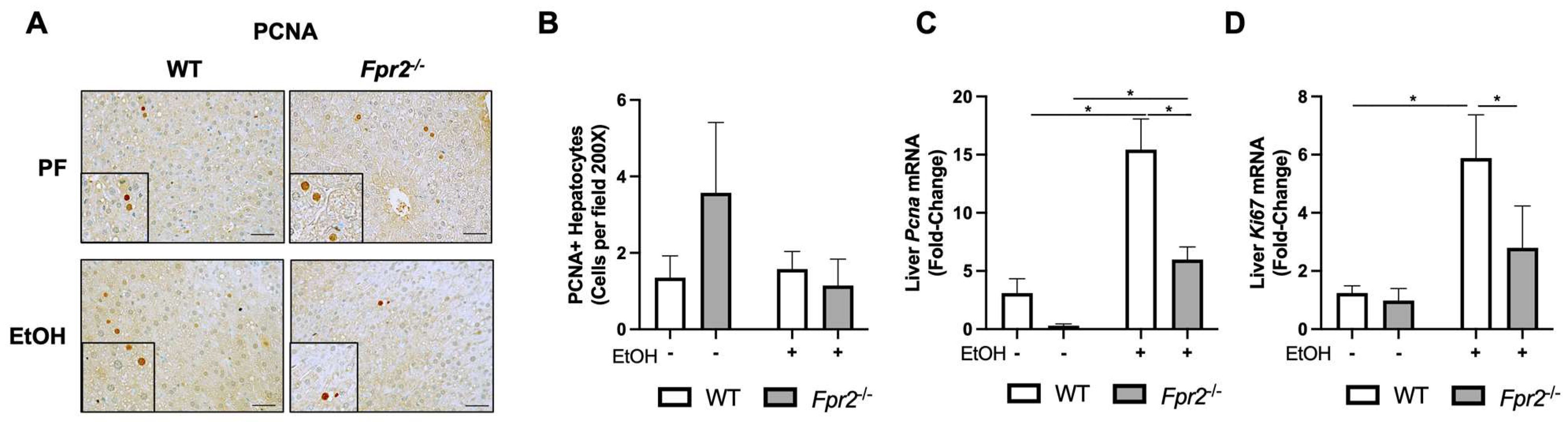
3.2. Effect of Fpr2 Genetic Ablation on Markers of Liver Regeneration in Response to EtOH
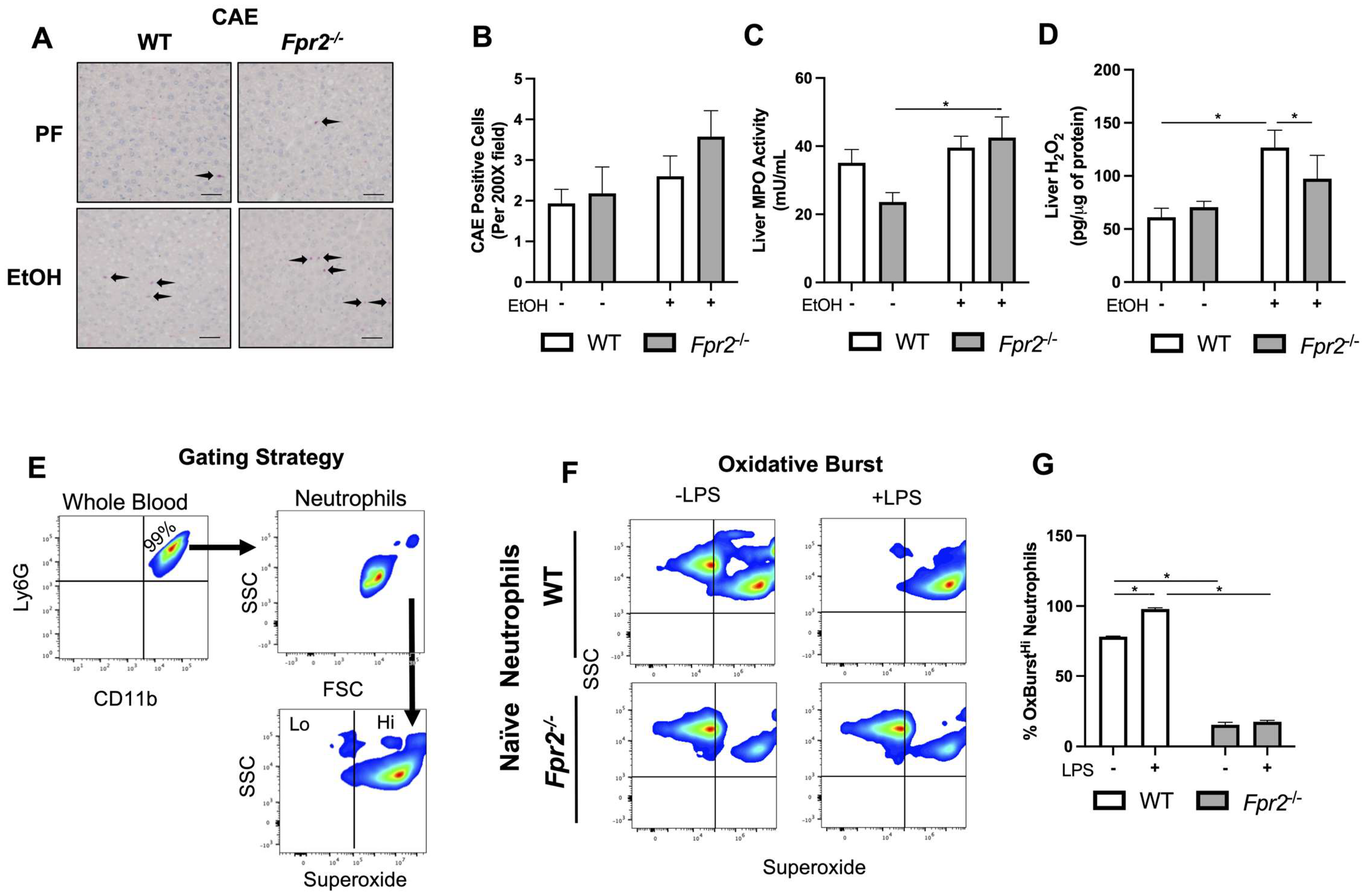
3.3. Liver Damage in EtOH-fed Fpr2−/− Mice was Associated with Alterations in Neutrophil Oxidative Burst
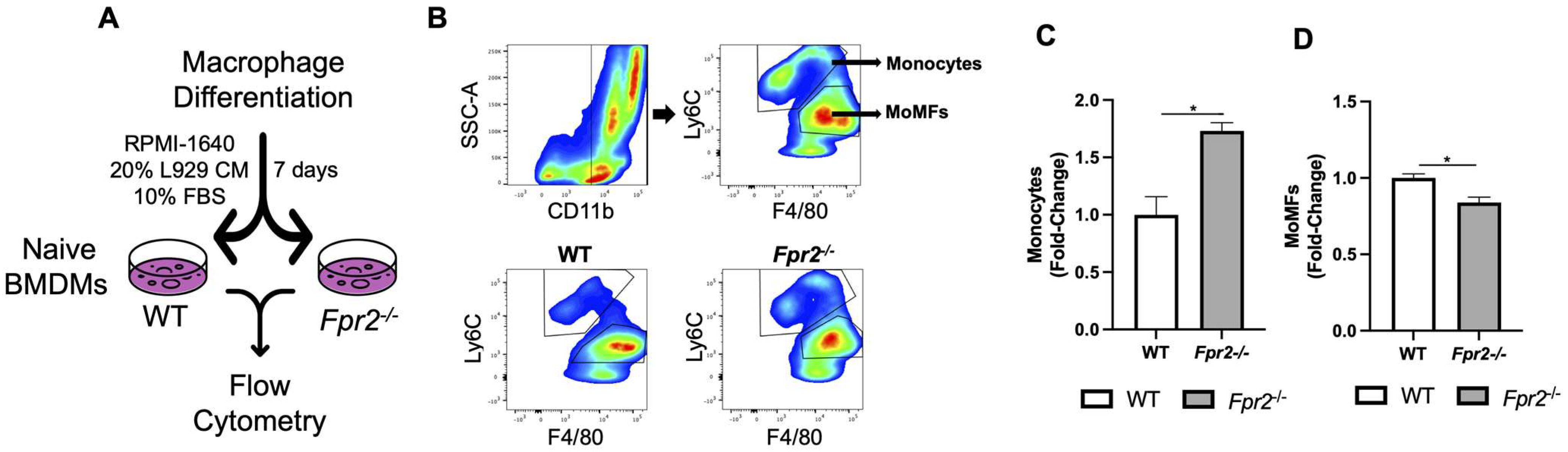
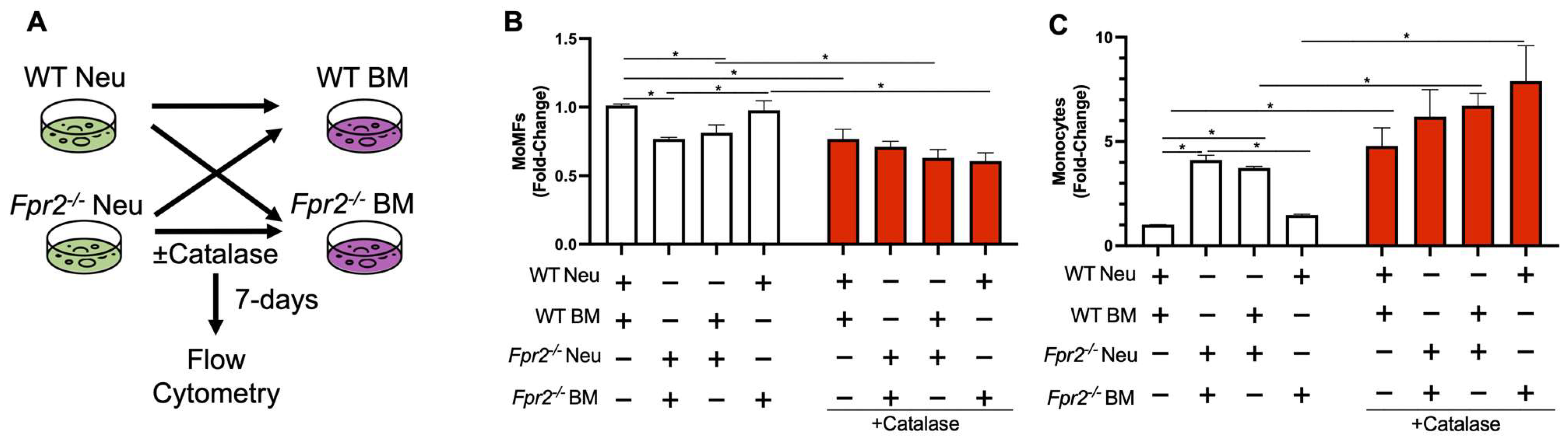
3.4. Fpr2−/− Bone Marrow Cells can Differentiate into MoMFs when Co-Cultured with WT Neutrophils
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Prim. 2018, 4, 16. [Google Scholar] [CrossRef]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Rao, R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 2009, 50, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.E. The Role of Innate Immunity in Alcoholic Liver Disease. Alcohol Res. 2015, 37, 237–250. [Google Scholar]
- Wang, S.; Pacher, P.; De Lisle, R.C.; Huang, H.; Ding, W.X. A Mechanistic Review of Cell Death in Alcohol-Induced Liver Injury. Alcohol Clin. Exp. Res. 2016, 40, 1215–1223. [Google Scholar] [CrossRef]
- Wang, H.J.; Gao, B.; Zakhari, S.; Nagy, L.E. Inflammation in alcoholic liver disease. Annu. Rev. Nutr. 2012, 32, 343–368. [Google Scholar] [CrossRef]
- Lv, Y.; So, K.F.; Xiao, J. Liver regeneration and alcoholic liver disease. Ann. Transl. Med. 2020, 8, 567. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, N.; Ishac, E.J.; Gao, B. Liver regeneration is suppressed in alcoholic cirrhosis: Correlation with decreased STAT3 activation. Alcohol 2007, 41, 271–280. [Google Scholar] [CrossRef]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- Weiß, E.; Kretschmer, D. Formyl-Peptide Receptors in Infection, Inflammation, and Cancer. Trends Immunol. 2018, 39, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Hartt, J.K.; Barish, G.; Murphy, P.M.; Gao, J.L. N-formylpeptides induce two distinct concentration optima for mouse neutrophil chemotaxis by differential interaction with two N-formylpeptide receptor (FPR) subtypes. Molecular characterization of FPR2, a second mouse neutrophil FPR. J. Exp. Med. 1999, 190, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Cooray, S.N.; Gobbetti, T.; Montero-Melendez, T.; McArthur, S.; Thompson, D.; Clark, A.J.; Flower, R.J.; Perretti, M. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl. Acad. Sci. USA 2013, 110, 18232–18237. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.W.; Lee, S.M. Resolvin D1 protects the liver from ischemia/reperfusion injury by enhancing M2 macrophage polarization and efferocytosis. Biochim. Biophys. Acta 2016, 1861, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Oldekamp, S.; Pscheidl, S.; Kress, E.; Soehnlein, O.; Jansen, S.; Pufe, T.; Wang, J.M.; Tauber, S.C.; Brandenburg, L.O. Lack of formyl peptide receptor 1 and 2 leads to more severe inflammation and higher mortality in mice with of pneumococcal meningitis. Immunology 2014, 143, 447–461. [Google Scholar] [CrossRef]
- Lee, C.; Kim, J.; Han, J.; Oh, D.; Kim, M.; Jeong, H.; Kim, T.J.; Kim, S.W.; Kim, J.N.; Seo, Y.S.; et al. Formyl peptide receptor 2 determines sex-specific differences in the progression of nonalcoholic fatty liver disease and steatohepatitis. Nat. Commun. 2022, 13, 578. [Google Scholar] [CrossRef]
- Giebeler, A.; Streetz, K.L.; Soehnlein, O.; Neumann, U.; Wang, J.M.; Brandenburg, L.O. Deficiency of formyl peptide receptor 1 and 2 is associated with increased inflammation and enhanced liver injury after LPS-stimulation. PLoS ONE 2014, 9, e100522. [Google Scholar] [CrossRef] [PubMed]
- Gobbetti, T.; Coldewey, S.M.; Chen, J.; McArthur, S.; le Faouder, P.; Cenac, N.; Flower, R.J.; Thiemermann, C.; Perretti, M. Nonredundant protective properties of FPR2/ALX in polymicrobial murine sepsis. Proc. Natl. Acad. Sci. USA 2014, 111, 18685–18690. [Google Scholar] [CrossRef]
- Dufton, N.; Hannon, R.; Brancaleone, V.; Dalli, J.; Patel, H.B.; Gray, M.; D’Acquisto, F.; Buckingham, J.C.; Perretti, M.; Flower, R.J. Anti-inflammatory role of the murine formyl-peptide receptor 2: Ligand-specific effects on leukocyte responses and experimental inflammation. J. Immunol. 2010, 184, 2611–2619. [Google Scholar] [CrossRef]
- Warner, D.R.; Warner, J.B.; Hardesty, J.E.; Song, Y.L.; Chen, C.Y.; Chen, Z.; Kang, J.X.; McClain, C.J.; Kirpich, I.A. Beneficial effects of an endogenous enrichment in n3-PUFAs on Wnt signaling are associated with attenuation of alcohol-mediated liver disease in mice. FASEB J. 2021, 35, e21377. [Google Scholar] [CrossRef]
- Kirpich, I.A.; Gobejishvili, L.N.; Bon Homme, M.; Waigel, S.; Cave, M.; Arteel, G.; Barve, S.S.; McClain, C.J.; Deaciuc, I.V. Integrated hepatic transcriptome and proteome analysis of mice with high-fat diet-induced nonalcoholic fatty liver disease. J. Nutr. Biochem. 2011, 22, 38–45. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Warner, J.; Hardesty, J.; Song, Y.; Sun, R.; Deng, Z.; Xu, R.; Yin, X.; Zhang, X.; McClain, C.; Warner, D.; et al. Fat-1 Transgenic Mice with Augmented n3-Polyunsaturated Fatty Acids Are Protected from Liver Injury Caused by Acute-On-Chronic Ethanol Administration. Front. Pharmacol. 2021, 12, 711590. [Google Scholar] [CrossRef]
- Bailey, J.D.; Shaw, A.; McNeill, E.; Nicol, T.; Diotallevi, M.; Chuaiphichai, S.; Patel, J.; Hale, A.; Channon, K.M.; Crabtree, M.J. Isolation and culture of murine bone marrow-derived macrophages for nitric oxide and redox biology. Nitric Oxide 2020, 100–101, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Allenbach, C.; Zufferey, C.; Perez, C.; Launois, P.; Mueller, C.; Tacchini-Cottier, F. Macrophages induce neutrophil apoptosis through membrane TNF, a process amplified by Leishmania major. J. Immunol. 2006, 176, 6656–6664. [Google Scholar] [CrossRef] [PubMed]
- Triantafyllou, E.; Woollard, K.J.; McPhail, M.J.W.; Antoniades, C.G.; Possamai, L.A. The Role of Monocytes and Macrophages in Acute and Acute-on-Chronic Liver Failure. Front. Immunol. 2018, 9, 2948. [Google Scholar] [CrossRef]
- Calvente, C.J.; Tameda, M.; Johnson, C.D.; Del Pilar, H.; Lin, Y.C.; Adronikou, N.; De Mollerat Du Jeu, X.; Llorente, C.; Boyer, J.; Feldstein, A.E. Neutrophils contribute to spontaneous resolution of liver inflammation and fibrosis via microRNA-223. J. Clin. Investig. 2019, 129, 4091–4109. [Google Scholar] [CrossRef]
- Triantafyllou, E.; Pop, O.T.; Possamai, L.A.; Wilhelm, A.; Liaskou, E.; Singanayagam, A.; Bernsmeier, C.; Khamri, W.; Petts, G.; Dargue, R.; et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut 2018, 67, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Marim, F.M.; Silveira, T.N.; Lima, D.S., Jr.; Zamboni, D.S. A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PLoS ONE 2010, 5, e15263. [Google Scholar] [CrossRef]
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun. 2019, 10, 1076. [Google Scholar] [CrossRef]
- Mookerjee, R.P.; Stadlbauer, V.; Lidder, S.; Wright, G.A.; Hodges, S.J.; Davies, N.A.; Jalan, R. Neutrophil dysfunction in alcoholic hepatitis superimposed on cirrhosis is reversible and predicts the outcome. Hepatology 2007, 46, 831–840. [Google Scholar] [CrossRef]
- Choi, H.K.; Kim, T.H.; Jhon, G.J.; Lee, S.Y. Reactive oxygen species regulate M-CSF-induced monocyte/macrophage proliferation through SHP1 oxidation. Cell. Signal. 2011, 23, 1633–1639. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef]
- Guilliams, M.; Bonnardel, J.; Haest, B.; Vanderborght, B.; Wagner, C.; Remmerie, A.; Bujko, A.; Martens, L.; Thoné, T.; Browaeys, R.; et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 2022, 185, 379–396.e38. [Google Scholar] [CrossRef]
- He, H.Q.; Ye, R.D. The Formyl Peptide Receptors: Diversity of Ligands and Mechanism for Recognition. Molecules 2017, 22, 455. [Google Scholar] [CrossRef] [PubMed]
- Dubuquoy, L.; Louvet, A.; Lassailly, G.; Truant, S.; Boleslawski, E.; Artru, F.; Maggiotto, F.; Gantier, E.; Buob, D.; Leteurtre, E.; et al. Progenitor cell expansion and impaired hepatocyte regeneration in explanted livers from alcoholic hepatitis. Gut 2015, 64, 1949–1960. [Google Scholar] [CrossRef]
- Owumi, S.E.; Corthals, S.M.; Uwaifo, A.O.; Kamendulis, L.M.; Klaunig, J.E. Depletion of Kupffer cells modulates ethanol-induced hepatocyte DNA synthesis in C57Bl/6 mice. Environ. Toxicol. 2014, 29, 867–875. [Google Scholar] [CrossRef] [PubMed]
- García, R.A.; Lupisella, J.A.; Ito, B.R.; Hsu, M.Y.; Fernando, G.; Carson, N.L.; Allocco, J.J.; Ryan, C.S.; Zhang, R.; Wang, Z.; et al. Selective FPR2 Agonism Promotes a Proresolution Macrophage Phenotype and Improves Cardiac Structure-Function Post Myocardial Infarction. JACC Basic Transl. Sci. 2021, 6, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Dal-Secco, D.; Wang, J.; Zeng, Z.; Kolaczkowska, E.; Wong, C.H.; Petri, B.; Ransohoff, R.M.; Charo, I.F.; Jenne, C.N.; Kubes, P. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J. Exp. Med. 2015, 212, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Caso, V.M.; Manzo, V.; Pecchillo Cimmino, T.; Conti, V.; Caso, P.; Esposito, G.; Russo, V.; Filippelli, A.; Ammendola, R.; Cattaneo, F. Regulation of Inflammation and Oxidative Stress by Formyl Peptide Receptors in Cardiovascular Disease Progression. Life 2021, 11, 243. [Google Scholar] [CrossRef] [PubMed]
- Iimuro, Y.; Frankenberg, M.V.; Arteel, G.E.; Bradford, B.U.; Wall, C.A.; Thurman, R.G. Female rats exhibit greater susceptibility to early alcohol-induced liver injury than males. Am. J. Physiol. 1997, 272 Pt 1, G1186–G1194. [Google Scholar] [CrossRef]
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol Res. 2017, 38, 147–161. [Google Scholar] [PubMed]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hardesty, J.E.; Warner, J.B.; Song, Y.L.; Floyd, A.; McClain, C.J.; Warner, D.R.; Kirpich, I.A. Fpr2−/− Mice Developed Exacerbated Alcohol-Associated Liver Disease. Biology 2023, 12, 639. https://doi.org/10.3390/biology12050639
Hardesty JE, Warner JB, Song YL, Floyd A, McClain CJ, Warner DR, Kirpich IA. Fpr2−/− Mice Developed Exacerbated Alcohol-Associated Liver Disease. Biology. 2023; 12(5):639. https://doi.org/10.3390/biology12050639
Chicago/Turabian StyleHardesty, Josiah E., Jeffrey B. Warner, Ying L. Song, Alison Floyd, Craig J. McClain, Dennis R. Warner, and Irina A. Kirpich. 2023. "Fpr2−/− Mice Developed Exacerbated Alcohol-Associated Liver Disease" Biology 12, no. 5: 639. https://doi.org/10.3390/biology12050639
APA StyleHardesty, J. E., Warner, J. B., Song, Y. L., Floyd, A., McClain, C. J., Warner, D. R., & Kirpich, I. A. (2023). Fpr2−/− Mice Developed Exacerbated Alcohol-Associated Liver Disease. Biology, 12(5), 639. https://doi.org/10.3390/biology12050639