
Immune Checkpoint Inhibitor Associated Myocarditis and Cardiomyopathy: A Translational Review

Abstract
Simple Summary
Abstract
1. Introduction
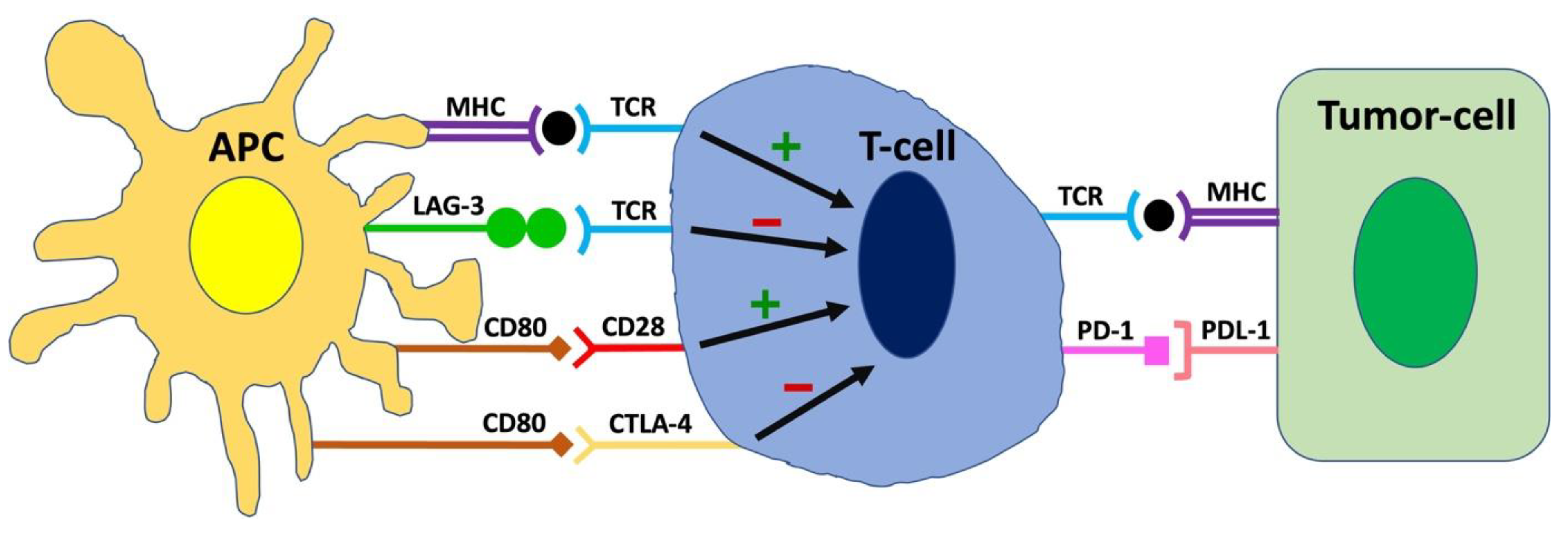
2. Molecular Mechanisms of Immune Checkpoints
3. Targeting Immune Checkpoints in Cancer Therapy
4. Immune-Related Adverse Events
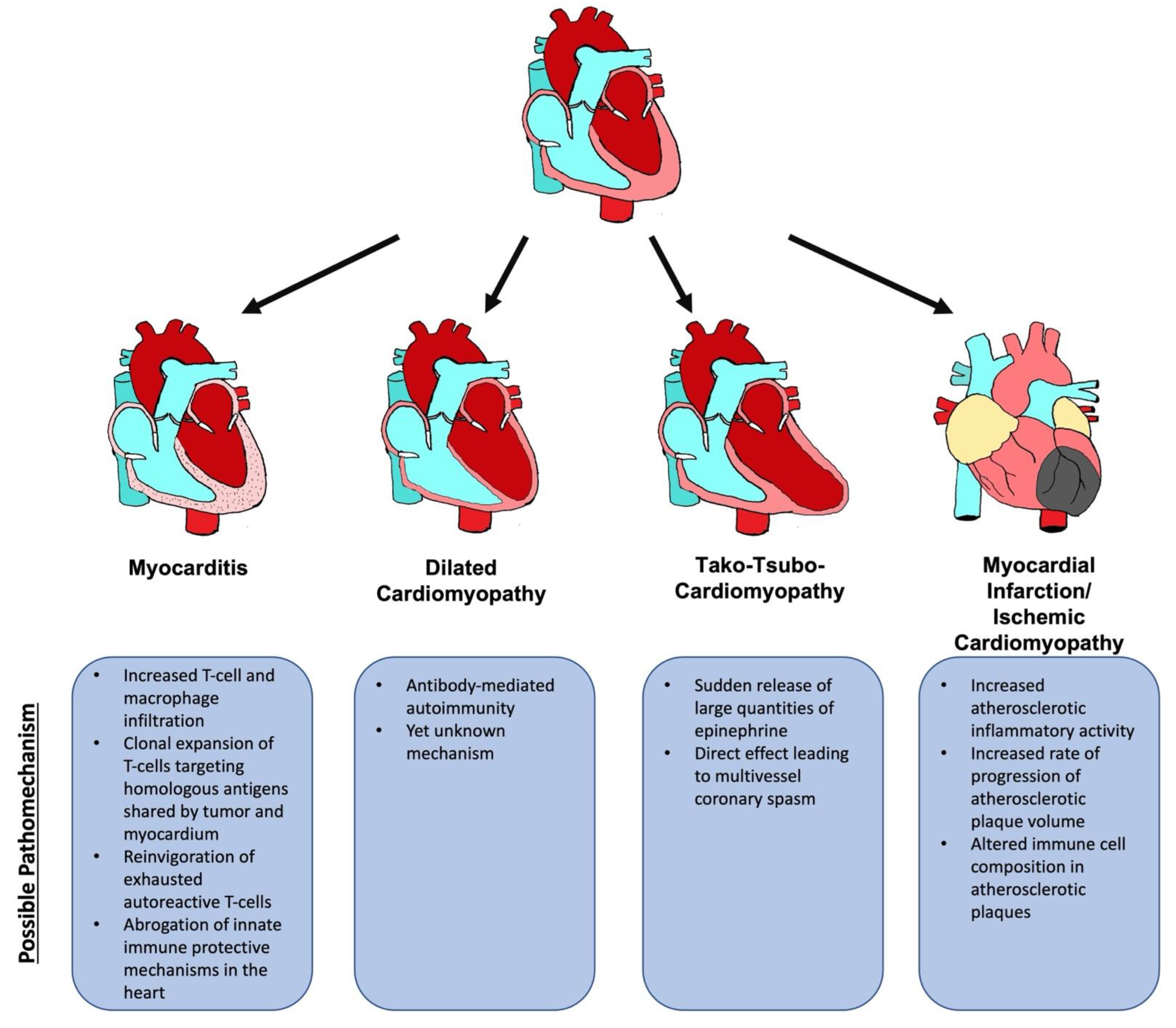
5. ICI-Associated Cardiotoxicity
5.1. ICI-Associated Myocarditis
5.1.1. Incidence and Risk Factors
5.1.2. Clinical Presentation
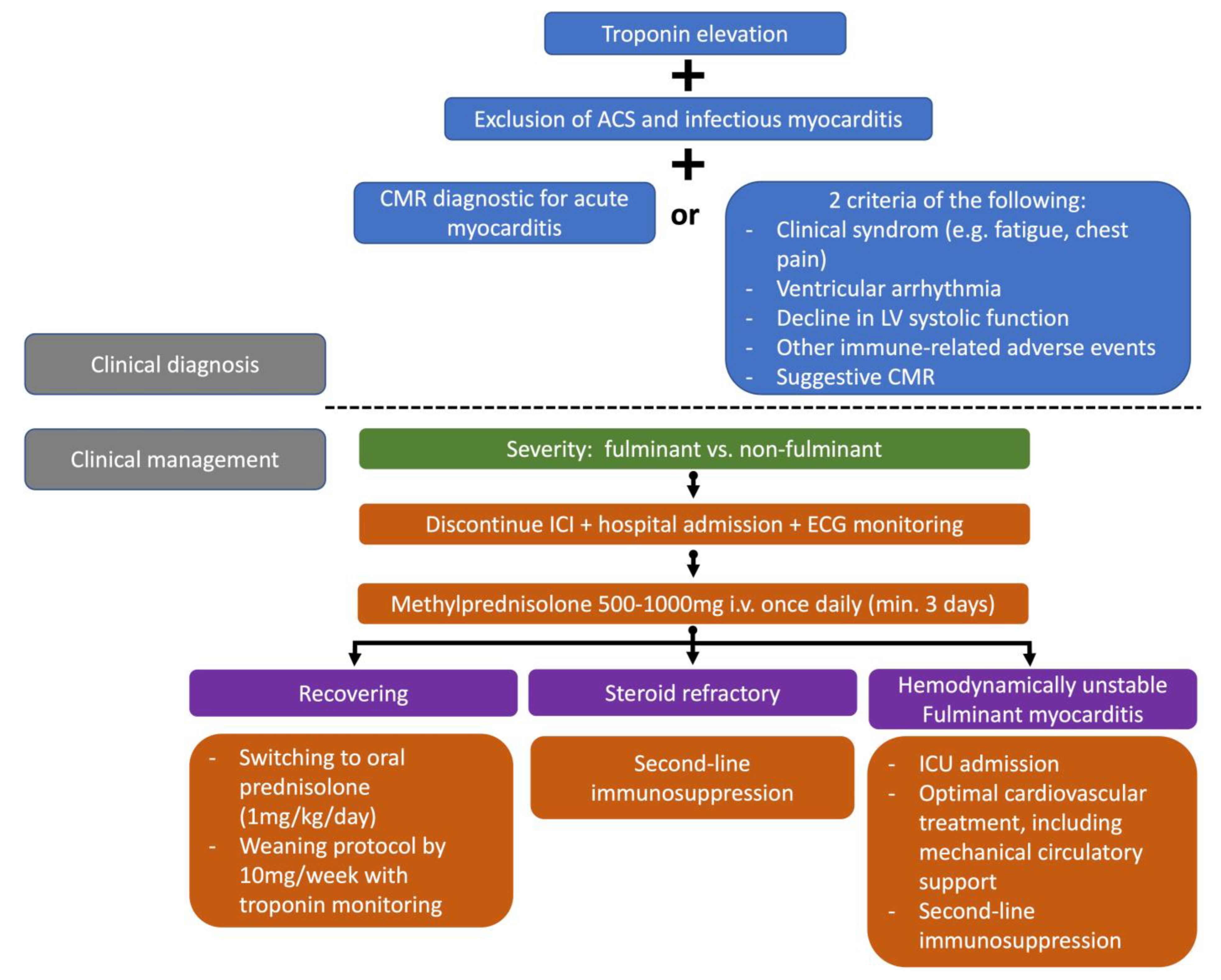
5.1.3. Diagnosis
5.1.4. Pathomechanism
5.1.5. Treatment
5.2. Non-Inflammatory Cardiomyopathy
5.2.1. Dilated Cardiomyopathy
5.2.2. Tako-Tsubo Cardiomyopathy
5.2.3. Ischemic Cardiomyopathy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beaver, J.A.; Pazdur, R. The Wild West of Checkpoint Inhibitor Development. N. Engl. J. Med. 2022, 386, 1297–1301. [Google Scholar] [CrossRef] [PubMed]
- Xin Yu, J.; Hubbard-Lucey, V.M.; Tang, J. Immuno-Oncology Drug Development Goes Global. Nat. Rev. Drug Discov. 2019, 18, 899–900. [Google Scholar] [CrossRef] [PubMed]
- Gilon, D.; Iakobishvili, Z.; Leibowitz, D. The Diagnosis and Management of Immune Checkpoint Inhibitor Cardiovascular Toxicity: Myocarditis and Beyond. Vaccines 2022, 10, 304. [Google Scholar] [CrossRef]
- Galluzzi, L.; Chan, T.A.; Kroemer, G.; Wolchok, J.D.; López-Soto, A. The Hallmarks of Successful Anticancer Immunotherapy. Sci. Transl. Med. 2018, 10, eaat7807. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-Intrinsic Resistance to Immune Checkpoint Blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Baxi, S.; Yang, A.; Gennarelli, R.L.; Khan, N.; Wang, Z.; Boyce, L.; Korenstein, D. Immune-Related Adverse Events for Anti-PD-1 and Anti-PD-L1 Drugs: Systematic Review and Meta-Analysis. BMJ 2018, 360, k793. [Google Scholar] [CrossRef] [PubMed]
- Shankar, B.; Zhang, J.; Naqash, A.R.; Forde, P.M.; Feliciano, J.L.; Marrone, K.A.; Ettinger, D.S.; Hann, C.L.; Brahmer, J.R.; Ricciuti, B.; et al. Multisystem Immune-Related Adverse Events Associated with Immune Checkpoint Inhibitors for Treatment of Non-Small Cell Lung Cancer. JAMA Oncol. 2020, 6, 1952–1956. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.R.; Florido, R.; Lipson, E.J.; Naidoo, J.; Ardehali, R.; Tocchetti, C.G.; Lyon, A.R.; Padera, R.F.; Johnson, D.B.; Moslehi, J. Cardiovascular Toxicities Associated with Immune Checkpoint Inhibitors. Cardiovasc. Res. 2019, 115, 854–868. [Google Scholar] [CrossRef]
- Salem, J.E.; Manouchehri, A.; Moey, M.; Lebrun-Vignes, B.; Bastarache, L.; Pariente, A.; Gobert, A.; Spano, J.P.; Balko, J.M.; Bonaca, M.P.; et al. Cardiovascular Toxicities Associated with Immune Checkpoint Inhibitors: An Observational, Retrospective, Pharmacovigilance Study. Lancet Oncol. 2018, 19, 1579–1589. [Google Scholar] [CrossRef]
- Drobni, Z.D.; Alvi, R.M.; Taron, J.; Zafar, A.; Murphy, S.P.; Rambarat, P.K.; Mosarla, R.C.; Lee, C.; Zlotoff, D.A.; Raghu, V.K.; et al. Association Between Immune Checkpoint Inhibitors With Cardiovascular Events and Atherosclerotic Plaque. Circulation 2020, 142, 2299–2311. [Google Scholar] [CrossRef]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+T Cells: Differentiation and Functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef]
- Berg, R.E.; Forman, J. The Role of CD8 T Cells in Innate Immunity and in Antigen Non-Specific Protection. Curr. Opin. Immunol. 2006, 18, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H. Mechanisms of Costimulation. Immunol. Rev. 2009, 229, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Harding, F.A.; McArthur, J.G.; Gross, J.A.; Raulet, D.H.; Allison, J.P. CD28-Mediated Signalling Co-Stimulates Murine T Cells and Prevents Induction of Anergy in T-Cell Clones. Nature 1992, 356, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Freeman, G.J. The B7-CD28 Superfamily. Nat. Rev. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Mir, M.A. Developing Costimulatory Molecules for Immunotherapy of Diseases. Dev. Costimulatory Mol. Immunother. Dis. 2015, 1–322. [Google Scholar] [CrossRef]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 Costimulation: From Mechanism to Therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef]
- Linsley, P.S.; Clark, E.A.; Ledbetter, J.A. T-Cell Antigen CD28 Mediates Adhesion with B Cells by Interacting with Activation Antigen B7/BB-1. Proc. Natl. Acad. Sci. USA 1990, 87, 5031–5035. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 Have Opposing Effects on the Response of T Cells to Stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 Can Function as a Negative Regulator of T Cell Activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Collins, A.V.; Brodie, D.W.; Gilbert, R.J.C.; Iaboni, A.; Manso-Sancho, R.; Walse, B.; Stuart, D.I.; Van Der Merwe, P.A.; Davis, S.J. The Interaction Properties of Costimulatory Molecules Revisited. Immunity 2002, 17, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.J.; Sullivan, T.J.; Allison, J.P. CTLA-4 Overexpression Inhibits T Cell Responses through a CD28-B7-Dependent Mechanism. J. Immunol. 2006, 177, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Ronen, D.; Bsoul, A.; Lotem, M.; Abedat, S.; Yarkoni, M.; Amir, O.; Asleh, R. Exploring the Mechanisms Underlying the Cardiotoxic Effects of Immune Checkpoint Inhibitor Therapies. Vaccines 2022, 10, 540. [Google Scholar] [CrossRef]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 Control over Foxp3+ Regulatory T Cell Function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef]
- Rotte, A.; Jin, J.Y.; Lemaire, V. Mechanistic Overview of Immune Checkpoints to Support the Rational Design of Their Combinations in Cancer Immunotherapy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 71–83. [Google Scholar] [CrossRef]
- Eppihimer, M.J.; Gunn, J.; Freeman, G.J.; Greenfield, E.A.; Chernova, T.; Erickson, J.; Leonard, J.P. Expression and Regulation of the PD-L1 Immunoinhibitory Molecule on Microvascular Endothelial Cells. Microcirculation 2002, 9, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Tay, W.T.; Fang, Y.H.; Beh, S.T.; Liu, Y.W.; Hsu, L.W.; Yen, C.J.; Liu, P.Y. Programmed Cell Death-1: Programmed Cell Death-Ligand 1 Interaction Protects Human Cardiomyocytes Against T-Cell Mediated Inflammation and Apoptosis Response In Vitro. Int. J. Mol. Sci. 2020, 21, 2399. [Google Scholar] [CrossRef]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective Effects of PD-1 on Akt and Ras Pathways Regulate Molecular Components of the Cell Cycle and Inhibit T Cell Proliferation. Sci. Signal. 2012, 5, ra46. [Google Scholar] [CrossRef]
- Haxhinasto, S.; Mathis, D.; Benoist, C. The AKT-MTOR Axis Regulates de Novo Differentiation of CD4+Foxp3+ Cells. J. Exp. Med. 2008, 205, 565–574. [Google Scholar] [CrossRef]
- Carter, L.L.; Fouser, L.A.; Jussif, J.; Fitz, L.; Deng, B.; Wood, C.R.; Collins, M.; Honjo, T.; Freeman, G.J.; Carreno, B.M. PD-1:PD-L Inhibitory Pathway Affects Both CD4+ and CD8+ T Cells and Is Overcome by IL-2. Eur. J. Immunol. 2002, 32, 634–643. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The Diverse Functions of the PD1 Inhibitory Pathway. Nat. Rev. Immunol. 2017, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 Regulates the Development, Maintenance, and Function of Induced Regulatory T Cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef] [PubMed]
- Lenschow, D.J.; Walunas, T.L.; Bluestone, J.A. CD28/B7 System of T Cell Costimulation. Annu. Rev. Immunol. 1996, 14, 233–258. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.A.; Allison, J.P. Co-Stimulation in T Cell Responses. Curr. Opin. Immunol. 1997, 9, 396–404. [Google Scholar] [CrossRef]
- van der Heide, V.; Homann, D. CD28 Days Later: Resurrecting Costimulation for CD8(+) Memory T Cells. Eur. J. Immunol. 2016, 46, 1587–1591. [Google Scholar] [CrossRef]
- Ward-Kavanagh, L.K.; Lin, W.W.; Šedý, J.R.; Ware, C.F. The TNF Receptor Superfamily in Co-Stimulating and Co-Inhibitory Responses. Immunity 2016, 44, 1005–1019. [Google Scholar] [CrossRef]
- Watts, T.H. TNF/TNFR Family Members in Costimulation of T Cell Responses. Annu. Rev. Immunol. 2005, 23, 23–68. [Google Scholar] [CrossRef]
- Hehlgans, T.; Pfeffer, K. The Intriguing Biology of the Tumour Necrosis Factor/Tumour Necrosis Factor Receptor Superfamily: Players, Rules and the Games. Immunology 2005, 115, 1–20. [Google Scholar] [CrossRef]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The Surface Protein TIGIT Suppresses T Cell Activation by Promoting the Generation of Mature Immunoregulatory Dendritic Cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a Novel Lymphocyte Activation Gene Closely Related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-Inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Khong, H.T.; Restifo, N.P. Natural Selection of Tumor Variants in the Generation of “Tumor Escape” Phenotypes. Nat. Immunol. 2002, 3, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massagué, J. TGF-Beta Directly Targets Cytotoxic T Cell Functions during Tumor Evasion of Immune Surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.; Gajewski, T.F.; Mackensen, A. Interaction of PD-L1 on Tumor Cells with PD-1 on Tumor-Specific T Cells as a Mechanism of Immune Evasion: Implications for Tumor Immunotherapy. Cancer Immunol. Immunother. 2005, 54, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Drake, C.G.; Jaffee, E.; Pardoll, D.M. Mechanisms of Immune Evasion by Tumors. Adv. Immunol. 2006, 90, 51–81. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive Strategies That Are Mediated by Tumor Cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on Tumor Cells in the Escape from Host Immune System and Tumor Immunotherapy by PD-L1 Blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Cameron, F.; Whiteside, G.; Perry, C. Ipilimumab: First Global Approval. Drugs 2011, 71, 1093–1104. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Galluzzi, L.; Humeau, J.; Buqué, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with Chemotherapy in the Era of Immune Checkpoint Inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Chargari, C.; Galluzzi, L.; Kroemer, G. Optimising Efficacy and Reducing Toxicity of Anticancer Radioimmunotherapy. Lancet Oncol. 2019, 20, e452–e463. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Ulas, E.B.; Dickhoff, C.; Schneiders, F.L.; Senan, S.; Bahce, I. Neoadjuvant Immune Checkpoint Inhibitors in Resectable Non-Small-Cell Lung Cancer: A Systematic Review. ESMO Open 2021, 6, 100244. [Google Scholar] [CrossRef]
- Petroni, G.; Buqué, A.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Immunomodulation by Targeted Anticancer Agents. Cancer Cell 2021, 39, 310–345. [Google Scholar] [CrossRef] [PubMed]
- Ghisoni, E.; Wicky, A.; Bouchaab, H.; Imbimbo, M.; Delyon, J.; Gautron Moura, B.; Gérard, C.L.; Latifyan, S.; Özdemir, B.C.; Caikovski, M.; et al. Late-Onset and Long-Lasting Immune-Related Adverse Events from Immune Checkpoint-Inhibitors: An Overlooked Aspect in Immunotherapy. Eur. J. Cancer 2021, 149, 153–164. [Google Scholar] [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Khoja, L.; Day, D.; Wei-Wu Chen, T.; Siu, L.L.; Hansen, A.R. Tumour- and Class-Specific Patterns of Immune-Related Adverse Events of Immune Checkpoint Inhibitors: A Systematic Review. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 2377–2385. [Google Scholar] [CrossRef]
- Darnell, E.P.; Mooradian, M.J.; Baruch, E.N.; Yilmaz, M.; Reynolds, K.L. Immune-Related Adverse Events (IrAEs): Diagnosis, Management, and Clinical Pearls. Curr. Oncol. Rep. 2020, 22, 1–11. [Google Scholar] [CrossRef]
- Okiyama, N.; Tanaka, R. Immune-Related Adverse Events in Various Organs Caused by Immune Checkpoint Inhibitors. Allergol. Int. 2022, 71, 169–178. [Google Scholar] [CrossRef]
- Johnson, D.B.; Reynolds, K.L.; Sullivan, R.J.; Balko, J.M.; Patrinely, J.R.; Cappelli, L.C.; Naidoo, J.; Moslehi, J.J. Immune Checkpoint Inhibitor Toxicities: Systems-Based Approaches to Improve Patient Care and Research. Lancet Oncol. 2020, 21, e398–e404. [Google Scholar] [CrossRef]
- Wang, D.Y.; Salem, J.E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-Analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Michel, L.; Totzeck, M.; Lehmann, L.; Finke, D. Emerging Role of Immune Checkpoint Inhibitors and Their Relevance for the Cardiovascular System. Herz 2020, 45, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Balko, J.M.; Compton, M.L.; Chalkias, S.; Gorham, J.; Xu, Y.; Hicks, M.; Puzanov, I.; Alexander, M.R.; Bloomer, T.L.; et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N. Engl. J. Med. 2016, 375, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, M.; Nielsen, D.; Svane, I.M.; Iversen, K.; Rasmussen, P.V.; Madelaire, C.; Fosbøl, E.; Køber, L.; Gustafsson, F.; Andersson, C.; et al. The Risk of Cardiac Events in Patients Receiving Immune Checkpoint Inhibitors: A Nationwide Danish Study. Eur. Heart J. 2021, 42, 1621–1631. [Google Scholar] [CrossRef]
- Waheed, N.; Fradley, M.G.; DeRemer, D.L.; Mahmoud, A.; Shah, C.P.; Langaee, T.Y.; Lipori, G.P.; March, K.; Pepine, C.J.; Cooper-DeHoff, R.M.; et al. Newly Diagnosed Cardiovascular Disease in Patients Treated with Immune Checkpoint Inhibitors: A Retrospective Analysis of Patients at an Academic Tertiary Care Center. Cardio-Oncol. 2021, 7, 1–7. [Google Scholar] [CrossRef]
- Rubio-Infante, N.; Ramírez-Flores, Y.A.; Castillo, E.C.; Lozano, O.; García-Rivas, G.; Torre-Amione, G. Cardiotoxicity Associated with Immune Checkpoint Inhibitor Therapy: A Meta-Analysis. Eur. J. Heart Fail. 2021, 23, 1739–1747. [Google Scholar] [CrossRef]
- Dolladille, C.; Akroun, J.; Morice, P.M.; Dompmartin, A.; Ezine, E.; Sassier, M.; Da-Silva, A.; Plane, A.F.; Legallois, D.; L’Orphelin, J.M.; et al. Cardiovascular Immunotoxicities Associated with Immune Checkpoint Inhibitors: A Safety Meta-Analysis. Eur. Heart J. 2021, 42, 4964–4977. [Google Scholar] [CrossRef]
- Lyon, A.R.; López-Fernández, T.; Couch, L.S.; Asteggiano, R.; Aznar, M.C.; Bergler-Klein, J.; Boriani, G.; Cardinale, D.; Cordoba, R.; Cosyns, B.; et al. 2022 ESC Guidelines on Cardio-Oncology Developed in Collaboration with the European Hematology Association (EHA), the European Society for Therapeutic Radiology and Oncology (ESTRO) and the International Cardio-Oncology Society (IC-OS). Eur. Heart J. 2022, 43, 4229–4361. [Google Scholar] [CrossRef]
- Cooper, L.T. Myocarditis. N. Engl. J. Med. 2009, 360, 1526–1538. [Google Scholar] [CrossRef]
- Mahmaljy, H.; Yelamanchili, V.S.; Singhal, M. Dilated Cardiomyopathy; StatPearls Publishing: Orlando, FL, USA, 2022. [Google Scholar]
- Groarke, J.D.; Cheng, S.; Moslehi, J. Cancer-Drug Discovery and Cardiovascular Surveillance. N. Engl. J. Med. 2013, 369, 1779–1781. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, S.S.; Fradley, M.G.; Cohen, J.V.; Nohria, A.; Reynolds, K.L.; Heinzerling, L.M.; Sullivan, R.J.; Damrongwatanasuk, R.; Chen, C.L.; Gupta, D.; et al. Myocarditis in Patients Treated With Immune Checkpoint Inhibitors. J. Am. Coll. Cardiol. 2018, 71, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Moslehi, J.J.; Salem, J.E.; Sosman, J.A.; Lebrun-Vignes, B.; Johnson, D.B. Increased Reporting of Fatal Immune Checkpoint Inhibitor-Associated Myocarditis. Lancet 2018, 391, 933. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Galdos, F.X.; Lee, D.; Waliany, S.; Huang, Y.V.; Ryan, J.; Dang, K.; Neal, J.W.; Wakelee, H.A.; Reddy, S.A.; et al. Identification of Pathogenic Immune Cell Subsets Associated With Checkpoint Inhibitor-Induced Myocarditis. Circulation 2022, 146, 316–335. [Google Scholar] [CrossRef] [PubMed]
- Reinke, S.; Geissler, S.; Taylor, W.R.; Schmidt-Bleek, K.; Juelke, K.; Schwachmeyer, V.; Dahne, M.; Hartwig, T.; Akyüz, L.; Meisel, C.; et al. Terminally Differentiated CD8+ T Cells Negatively Affect Bone Regeneration in Humans. Sci. Transl. Med. 2013, 5, 177ra36. [Google Scholar] [CrossRef] [PubMed]
- Awadalla, M.; Golden, D.L.A.; Mahmood, S.S.; Alvi, R.M.; Mercaldo, N.D.; Hassan, M.Z.O.; Banerji, D.; Rokicki, A.; Mulligan, C.; Murphy, S.P.T.; et al. Influenza Vaccination and Myocarditis among Patients Receiving Immune Checkpoint Inhibitors. J. Immunother. Cancer 2019, 7, 1–10. [Google Scholar] [CrossRef]
- Dal’bo, N.; Patel, R.; Parikh, R.; Shah, S.P.; Guha, A.; Dani, S.S.; Ganatra, S. Cardiotoxicity of Contemporary Anticancer Immunotherapy. Curr. Treat. Options Cardiovasc. Med. 2020, 22, 1–5. [Google Scholar] [CrossRef]
- Escudier, M.; Cautela, J.; Malissen, N.; Ancedy, Y.; Orabona, M.; Pinto, J.; Monestier, S.; Grob, J.J.; Scemama, U.; Jacquier, A.; et al. Clinical Features, Management, and Outcomes of Immune Checkpoint Inhibitor-Related Cardiotoxicity. Circulation 2017, 136, 2085–2087. [Google Scholar] [CrossRef]
- Norwood, T.G.; Westbrook, B.C.; Johnson, D.B.; Litovsky, S.H.; Terry, N.L.; McKee, S.B.; Gertler, A.S.; Moslehi, J.J.; Conry, R.M. Smoldering Myocarditis Following Immune Checkpoint Blockade. J. Immunother. Cancer 2017, 5, 1–6. [Google Scholar] [CrossRef]
- Kindermann, I.; Barth, C.; Mahfoud, F.; Ukena, C.; Lenski, M.; Yilmaz, A.; Klingel, K.; Kandolf, R.; Sechtem, U.; Cooper, L.T.; et al. Update on Myocarditis. J. Am. Coll. Cardiol. 2012, 59, 779–792. [Google Scholar] [CrossRef]
- Hauck, A.J.; Kearney, D.L.; Edwards, W.D. Evaluation of Postmortem Endomyocardial Biopsy Specimens from 38 Patients with Lymphocytic Myocarditis: Implications for Role of Sampling Error. Mayo Clin. Proc. 1989, 64, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Bonaca, M.P.; Olenchock, B.A.; Salem, J.E.; Wiviott, S.D.; Ederhy, S.; Cohen, A.; Stewart, G.C.; Choueiri, T.K.; Di Carli, M.; Allenbach, Y.; et al. Myocarditis in the Setting of Cancer Therapeutics: Proposed Case Definitions for Emerging Clinical Syndromes in Cardio-Oncology. Circulation 2019, 140, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Berliner, D.; Beutel, G.; Bauersachs, J. Echocardiography and Biomarkers for the Diagnosis of Cardiotoxicity. Herz 2020, 45, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Rassaf, T.; Totzeck, M.; Backs, J.; Bokemeyer, C.; Hallek, M.; Hilfiker-Kleiner, D.; Hochhaus, A.; Lüftner, D.; Müller, O.J.; Neudorf, U.; et al. Onco-Cardiology: Consensus Paper of the German Cardiac Society, the German Society for Pediatric Cardiology and Congenital Heart Defects and the German Society for Hematology and Medical Oncology. Clin. Res. Cardiol. 2020, 109, 1197–1222. [Google Scholar] [CrossRef] [PubMed]
- Grabie, N.; Lichtman, A.H.; Padera, R. T Cell Checkpoint Regulators in the Heart. Cardiovasc. Res. 2019, 115, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The Immune System in Cardiac Homeostasis and Disease. Nat. Rev. Immunol. 2018, 18, 733–744. [Google Scholar] [CrossRef]
- Grabie, N.; Gotsman, I.; DaCosta, R.; Pang, H.; Stavrakis, G.; Butte, M.J.; Keir, M.E.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Endothelial Programmed Death-1 Ligand 1 (PD-L1) Regulates CD8+ T-Cell Mediated Injury in the Heart. Circulation 2007, 116, 2062–2071. [Google Scholar] [CrossRef]
- Kania, G.; Siegert, S.; Behnke, S.; Prados-Rosales, R.; Casadevall, A.; Lüscher, T.F.; Luther, S.A.; Kopf, M.; Eriksson, U.; Blyszczuk, P. Innate Signaling Promotes Formation of Regulatory Nitric Oxide-Producing Dendritic Cells Limiting T-Cell Expansion in Experimental Autoimmune Myocarditis. Circulation 2013, 127, 2285–2294. [Google Scholar] [CrossRef]
- Caforio, A.L.P.; Grazzini, M.; Mann, J.M.; Keeling, P.J.; Bottazzo, G.F.; McKenna, W.J.; Schiaffino, S. Identification of Alpha- and Beta-Cardiac Myosin Heavy Chain Isoforms as Major Autoantigens in Dilated Cardiomyopathy. Circulation 1992, 85, 1734–1742. [Google Scholar] [CrossRef]
- Lauer, B.; Schannwell, M.; Kühl, U.; Strauer, B.E.; Schultheiss, H.P. Antimyosin Autoantibodies Are Associated with Deterioration of Systolic and Diastolic Left Ventricular Function in Patients with Chronic Myocarditis. J. Am. Coll. Cardiol. 2000, 35, 11–18. [Google Scholar] [CrossRef]
- Lv, H.J.; Havari, E.; Pinto, S.; Gottumukkala, R.V.; Cornivelli, L.; Raddassi, K.; Matsui, T.; Rosenzweig, A.; Bronson, R.T.; Smith, R.; et al. Impaired Thymic Tolerance to α-Myosin Directs Autoimmunity to the Heart in Mice and Humans. J. Clin. Investig. 2011, 121, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Ganatra, S.; Neilan, T.G. Immune Checkpoint Inhibitor-Associated Myocarditis. Oncologist 2018, 23, 879–886. [Google Scholar] [CrossRef]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 Leads to Massive Lymphoproliferation and Fatal Multiorgan Tissue Destruction, Revealing a Critical Negative Regulatory Role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative Disorders with Early Lethality in Mice Deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Yang, L.; Qiao, G.; Li, Z.; Zhang, L.; Yin, F.; Xie, D.; Zhang, J. Cutting Edge: CTLA-4--B7 Interaction Suppresses Th17 Cell Differentiation. J. Immunol. 2010, 185, 1375–1378. [Google Scholar] [CrossRef] [PubMed]
- Love, V.A.; Grabie, N.; Duramad, P.; Stavrakis, G.; Sharpe, A.; Lichtman, A. CTLA-4 Ablation and Interleukin-12 Driven Differentiation Synergistically Augment Cardiac Pathogenicity of Cytotoxic T Lymphocytes. Circ. Res. 2007, 101, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune Dilated Cardiomyopathy in PD-1 Receptor-Deficient Mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Tanaka, Y.; Nishio, R.; Mitsuiye, T.; Mizoguchi, A.; Wang, J.; Ishida, M.; Hiai, H.; Matsumori, A.; Minato, N.; et al. Autoantibodies against Cardiac Troponin I Are Responsible for Dilated Cardiomyopathy in PD-1-Deficient Mice. Nat. Med. 2003, 9, 1477–1483. [Google Scholar] [CrossRef]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of Lupus-like Autoimmune Diseases by Disruption of the PD-1 Gene Encoding an ITIM Motif-Carrying Immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Keir, M.E.; Liang, S.C.; Guleria, I.; Latchman, Y.E.; Qipo, A.; Albacker, L.A.; Koulmanda, M.; Freeman, G.J.; Sayegh, M.H.; Sharpe, A.H. Tissue Expression of PD-L1 Mediates Peripheral T Cell Tolerance. J. Exp. Med. 2006, 203, 883–895. [Google Scholar] [CrossRef]
- Tarrio, M.L.; Grabie, N.; Bu, D.; Sharpe, A.H.; Lichtman, A.H. PD-1 Protects against Inflammation and Myocyte Damage in T Cell-Mediated Myocarditis. J. Immunol. 2012, 188, 4876–4884. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.A.; Menke, J.; Rabacal, W.A.; Schoen, F.J.; Sharpe, A.H.; Kelley, V.R. Programmed Death Ligand 1 Regulates a Critical Checkpoint for Autoimmune Myocarditis and Pneumonitis in MRL Mice. J. Immunol. 2008, 181, 2513–2521. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Okazaki, I.M.; Yoshida, T.; Chikuma, S.; Kato, Y.; Nakaki, F.; Hiai, H.; Honjo, T.; Okazaki, T. PD-1 Deficiency Results in the Development of Fatal Myocarditis in MRL Mice. Int. Immunol. 2010, 22, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Okazaki, I.M.; Wang, J.; Sugiura, D.; Nakaki, F.; Yoshida, T.; Kato, Y.; Fagarasan, S.; Muramatsu, M.; Eto, T.; et al. PD-1 and LAG-3 Inhibitory Co-Receptors Act Synergistically to Prevent Autoimmunity in Mice. J. Exp. Med. 2011, 208, 395–407. [Google Scholar] [CrossRef]
- Wei, S.C.; Meijers, W.C.; Axelrod, M.L.; Anang, N.-A.A.S.; Screever, E.M.; Wescott, E.C.; Johnson, D.B.; Whitley, E.; Lehmann, L.; Courand, P.-Y.; et al. A Genetic Mouse Model Recapitulates Immune Checkpoint Inhibitor-Associated Myocarditis and Supports a Mechanism-Based Therapeutic Intervention. Cancer Discov. 2021, 11, 614–625. [Google Scholar] [CrossRef]
- Hardy, T.; Yin, M.; Chavez, J.A.; Ivanov, I.; Chen, W.; Nadasdy, T.; Brodsky, S.V. Acute Fatal Myocarditis after a Single Dose of Anti-PD-1 Immunotherapy, Autopsy Findings: A Case Report. Cardiovasc. Pathol. 2020, 46, 107202. [Google Scholar] [CrossRef]
- Baban, B.; Liu, J.Y.; Qin, X.; Weintraub, N.L.; Mozaffari, M.S. Upregulation of Programmed Death-1 and Its Ligand in Cardiac Injury Models: Interaction with GADD153. PLoS ONE 2015, 10, e0124059. [Google Scholar] [CrossRef]
- Kushnareva, E.; Kushnarev, V.; Artemyeva, A.; Mitrofanova, L.; Moiseeva, O. Myocardial PD-L1 Expression in Patients With Ischemic and Non-Ischemic Heart Failure. Front. Cardiovasc. Med. 2021, 8, 759972. [Google Scholar] [CrossRef]
- Tian, Y.; Babor, M.; Lane, J.; Schulten, V.; Patil, V.S.; Seumois, G.; Rosales, S.L.; Fu, Z.; Picarda, G.; Burel, J.; et al. Unique Phenotypes and Clonal Expansions of Human CD4 Effector Memory T Cells Re-Expressing CD45RA. Nat. Commun. 2017, 8, 1473. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.J.; Naidoo, J.; Santomasso, B.D.; Lacchetti, C.; Adkins, S.; Anadkat, M.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; et al. Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: ASCO Guideline Update. J. Clin. Oncol. 2021, 39, 4073–4126. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Frigerio, M.; Adler, E.D.; Basso, C.; Birnie, D.H.; Brambatti, M.; Friedrich, M.G.; Klingel, K.; Lehtonen, J.; Moslehi, J.J.; et al. Management of Acute Myocarditis and Chronic Inflammatory Cardiomyopathy: An Expert Consensus Document. Circ. Heart Fail. 2020, 13, E007405. [Google Scholar] [CrossRef] [PubMed]
- Thuny, F.; Alexandre, J.; Salem, J.E.; Mirabel, M.; Dolladille, C.; Cohen-Solal, A.; Cohen, A.; Ederhy, S.; Cautela, J. Management of Immune Checkpoint Inhibitor-Induced Myocarditis: The French Working Group’s Plea for a Pragmatic Approach. JACC CardioOncol. 2021, 3, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Lenihan, D.; Fradley, M.; Ganatra, S.; Barac, A.; Blaes, A.; Herrmann, J.; Porter, C.; Lyon, A.R.; Lancellotti, P.; et al. Management of Cardiac Disease in Cancer Patients throughout Oncological Treatment: ESMO Consensus Recommendations. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 171–190. [Google Scholar] [CrossRef]
- Safi, M.; Ahmed, H.; Al-Azab, M.; Xia, Y.; Shan, X.; Al-radhi, M.; Al-danakh, A.; Shopit, A.; Liu, J. PD-1/PDL-1 Inhibitors and Cardiotoxicity; Molecular, Etiological and Management Outlines. J. Adv. Res. 2020, 29, 45–54. [Google Scholar] [CrossRef]
- Roth, M.E.; Muluneh, B.; Jensen, B.C.; Madamanchi, C.; Lee, C.B. Left Ventricular Dysfunction After Treatment With Ipilimumab for Metastatic Melanoma. Am. J. Ther. 2016, 23, e1925–e1928. [Google Scholar] [CrossRef]
- Michel, L.; Korste, S.; Spomer, A.; Hendgen-Cotta, U.B.; Rassaf, T.; Totzeck, M. PD1 Deficiency Modifies Cardiac Immunity during Baseline Conditions and in Reperfused Acute Myocardial Infarction. Int. J. Mol. Sci. 2022, 23, 7533. [Google Scholar] [CrossRef]
- Templin, C.; Ghadri, J.R.; Diekmann, J.; Napp, L.C.; Bataiosu, D.R.; Jaguszewski, M.; Cammann, V.L.; Sarcon, A.; Geyer, V.; Neumann, C.A.; et al. Clinical Features and Outcomes of Takotsubo (Stress) Cardiomyopathy. N. Engl. J. Med. 2015, 373, 929–938. [Google Scholar] [CrossRef]
- Napp, L.C.; Bauersachs, J. Takotsubo Syndrome: Between Evidence, Myths, and Misunderstandings. Herz 2020, 45, 252–266. [Google Scholar] [CrossRef]
- Geisler, B.P.; Raad, R.A.; Esaian, D.; Sharon, E.; Schwartz, D.R. Apical Ballooning and Cardiomyopathy in a Melanoma Patient Treated with Ipilimumab: A Case of Takotsubo-like Syndrome. J. Immunother. Cancer 2015, 3, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Serzan, M.; Rapisuwon, S.; Krishnan, J.; Chang, I.C.; Barac, A. Takotsubo Cardiomyopathy Associated With Checkpoint Inhibitor Therapy: Endomyocardial Biopsy Provides Pathological Insights to Dual Diseases. JACC CardioOncol. 2021, 3, 330–334. [Google Scholar] [CrossRef]
- Lyon, A.R.; Yousaf, N.; Battisti, N.M.L.; Moslehi, J.; Larkin, J. Immune Checkpoint Inhibitors and Cardiovascular Toxicity. Lancet Oncol. 2018, 19, e447–e458. [Google Scholar] [CrossRef] [PubMed]
- Rivard, A.; Andrés, V. Vascular Smooth Muscle Cell Proliferation in the Pathogenesis of Atherosclerotic Cardiovascular Diseases. Histol. Histopathol. 2000, 15, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Deuse, T.; Stubbendorff, M.; Chernogubova, E.; Erben, R.G.; Eken, S.M.; Jin, H.; Li, Y.; Busch, A.; Heeger, C.H.; et al. Local MicroRNA Modulation Using a Novel Anti-MIR-21-Eluting Stent Effectively Prevents Experimental in-Stent Restenosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1945–1963. [Google Scholar] [CrossRef] [PubMed]
- Bar, J.; Markel, G.; Gottfried, T.; Percik, R.; Leibowitz-Amit, R.; Berger, R.; Golan, T.; Daher, S.; Taliansky, A.; Dudnik, E.; et al. Acute Vascular Events as a Possibly Related Adverse Event of Immunotherapy: A Single-Institute Retrospective Study. Eur. J. Cancer 2019, 120, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in Atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Fernandez, D.M.; Rahman, A.H.; Fernandez, N.F.; Chudnovskiy, A.; Amir, E.A.; Amadori, L.; Khan, N.S.; Wong, C.K.; Shamailova, R.; Hill, C.A.; et al. Single-Cell Immune Landscape of Human Atherosclerotic Plaques. Nat. Med. 2019, 25, 1576–1588. [Google Scholar] [CrossRef]
- Gotsman, I.; Grabie, N.; Dacosta, R.; Sukhova, G.; Sharpe, A.; Lichtman, A.H. Proatherogenic Immune Responses Are Regulated by the PD-1/PD-L Pathway in Mice. J. Clin. Investig. 2007, 117, 2974–2982. [Google Scholar] [CrossRef]
- Bu, D.X.; Tarrio, M.; Maganto-Garcia, E.; Stavrakis, G.; Tajima, G.; Lederer, J.; Jarolim, P.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Impairment of the Programmed Cell Death-1 Pathway Increases Atherosclerotic Lesion Development and Inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1100–1107. [Google Scholar] [CrossRef]
- Cochain, C.; Chaudhari, S.M.; Koch, M.; Wiendl, H.; Eckstein, H.H.; Zernecke, A. Programmed Cell Death-1 Deficiency Exacerbates T Cell Activation and Atherogenesis despite Expansion of Regulatory T Cells in Atherosclerosis-Prone Mice. PLoS ONE 2014, 9, e93280. [Google Scholar] [CrossRef] [PubMed]
- Poels, K.; van Leent, M.M.T.; Boutros, C.; Tissot, H.; Roy, S.; Meerwaldt, A.E.; Toner, Y.C.A.; Reiche, M.E.; Kusters, P.J.H.; Malinova, T.; et al. Immune Checkpoint Inhibitor Therapy Aggravates T Cell–Driven Plaque Inflammation in Atherosclerosis. JACC CardioOncol. 2020, 2, 599–610. [Google Scholar] [CrossRef]
- Poels, K.; van Leent, M.M.T.; Reiche, M.E.; Kusters, P.J.H.; Huveneers, S.; de Winther, M.P.J.; Mulder, W.J.M.; Lutgens, E.; Seijkens, T.T.P. Antibody-Mediated Inhibition of CTLA4 Aggravates Atherosclerotic Plaque Inflammation and Progression in Hyperlipidemic Mice. Cells 2020, 9, 1987. [Google Scholar] [CrossRef] [PubMed]
- Calabretta, R.; Hoeller, C.; Pichler, V.; Mitterhauser, M.; Karanikas, G.; Haug, A.; Li, X.; Hacker, M. Immune Checkpoint Inhibitor Therapy Induces Inflammatory Activity in Large Arteries. Circulation 2020, 142, 2396–2398. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.L.; Stone, J.R. Immune Checkpoint Inhibition Alters the Inflammatory Cell Composition of Human Coronary Artery Atherosclerosis. Cardiovasc. Pathol. 2019, 43, 107148. [Google Scholar] [CrossRef]
- de Wall, C.; Bauersachs, J.; Berliner, D. Cardiooncology—Dealing with Modern Drug Treatment, Long-Term Complications, and Cancer Survivorship. Clin. Exp. Metastasis 2021, 38, 361–371. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug | Target | Approval | Indications |
|---|---|---|---|
| Ipilimumab (Yervoy®) | CTLA-4 | 2011 | Melanoma, renal cell carcinoma, colorectal cancer, hepatocellular carcinoma, non-small cell lung cancer, malignant pleural mesothelioma, esophageal cancer |
| Nivolumab (Opdivo®) | PD-1 | 2014 | Melanoma, non-small cell lung cancer, malignant pleural mesothelioma, renal cell carcinoma, classical Hodgkin lymphoma, squamous cell carcinoma of the head and neck, urothelial carcinoma, colorectal cancer, hepatocellular carcinoma, esophageal cancer, gastric cancer, gastroesophageal junction cancer, esophageal adenocarcinoma |
| Pembrolizumab (Keytruda®) | PD-1 | 2014 | Melanoma, non-small cell lung cancer, head and neck squamous cell carcinoma, classical Hodgkin lymphoma, primary mediastinal large B-cell lymphoma, urothelial carcinoma, non-muscle invasive bladder cancer, colorectal cancer, gastric cancer, esophageal cancer, cervical cancer, hepatocellular carcinoma, Merkel cell carcinoma, renal cell carcinoma, endometrial carcinoma, cutaneous squamous cell carcinoma, triple-negative breast cancer |
| Atezolizumab (Tecentriq®) | PD-L1 | 2016 | Non-small cell lung cancer, small cell lung cancer, hepatocellular carcinoma, melanoma, alveolar soft part sarcoma |
| Durvalumab (Imfinzi®) | PD-L1 | 2017 | Non-small cell lung cancer, small cell lung cancer, biliary tract cancer, hepatocellular carcinoma |
| Avelumab (Bavencio®) | PD-L1 | 2017 | Merkel cell carcinoma, urothelial carcinoma, renal cell carcinoma |
| Cemiplimab (Libtayo®) | PD-1 | 2019 | Cutaneous squamous cell carcinoma, basal cell carcinoma, non-small cell lung cancer |
| Dostarlimab (Jemperli®) | PD-1 | 2021 | Endometrial cancer |
| Relatlimab (Opdualag®, combination with Nivolumab) | LAG-3 | 2022 | Melanoma |
| Symptoms | Diagnosis | Treatment | |
|---|---|---|---|
| Myocarditis | Dyspnea Chest pain Fatigue Palpitations Decreased exercise tolerance Syncope | Troponin Echocardiography ECG Cardio-MRI PET (if CMR is not available or contraindicated) EMB (if suspected, but not confirmed by non-invasive diagnostic) | High dose methylprednisolone If steroid refractory, switch to second-line immunosuppression Cessation of ICI ECG monitoring Complications should be treated as per specific ESC Guidelines |
| Dilated cardiomyopathy | Dyspnea/ orthopnea Edema Fatigue | Echocardiography ECG Exclusion of myocarditis, TTC, ACS | No indication for immunosuppression Treatment according to ESC guidelines for heart failure Interruption of ICI depending on multidisciplinary decision |
| Tako-Tsubo cardiomyopathy | Chest pain Shortness of breath Palpitations Syncope | Echocardiography Troponin, CK, CK-MB ECG Coronary angiography (exclusion of ACS) | No indication for immunosuppression Treatment according to ESC guidelines for heart failure Interruption of ICI depending on multidisciplinary decision |
| Myocardial infarction | Chest pain Shortness of breath Palpitations Syncope | Troponin, CK, CK-MB ECG Echocardiography Coronary angiography | No indication for immunosuppression Treatment according to ESC guidelines for acute coronary syndrome Interruption of ICI depending on multidisciplinary decision |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Bauersachs, J.; Berliner, D. Immune Checkpoint Inhibitor Associated Myocarditis and Cardiomyopathy: A Translational Review. Biology 2023, 12, 472. https://doi.org/10.3390/biology12030472
Wang D, Bauersachs J, Berliner D. Immune Checkpoint Inhibitor Associated Myocarditis and Cardiomyopathy: A Translational Review. Biology. 2023; 12(3):472. https://doi.org/10.3390/biology12030472
Chicago/Turabian StyleWang, Dong, Johann Bauersachs, and Dominik Berliner. 2023. "Immune Checkpoint Inhibitor Associated Myocarditis and Cardiomyopathy: A Translational Review" Biology 12, no. 3: 472. https://doi.org/10.3390/biology12030472
APA StyleWang, D., Bauersachs, J., & Berliner, D. (2023). Immune Checkpoint Inhibitor Associated Myocarditis and Cardiomyopathy: A Translational Review. Biology, 12(3), 472. https://doi.org/10.3390/biology12030472