
Expanding Roles of the E2F-RB-p53 Pathway in Tumor Suppression
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Classical Views of the RB-E2F-p53 Pathway
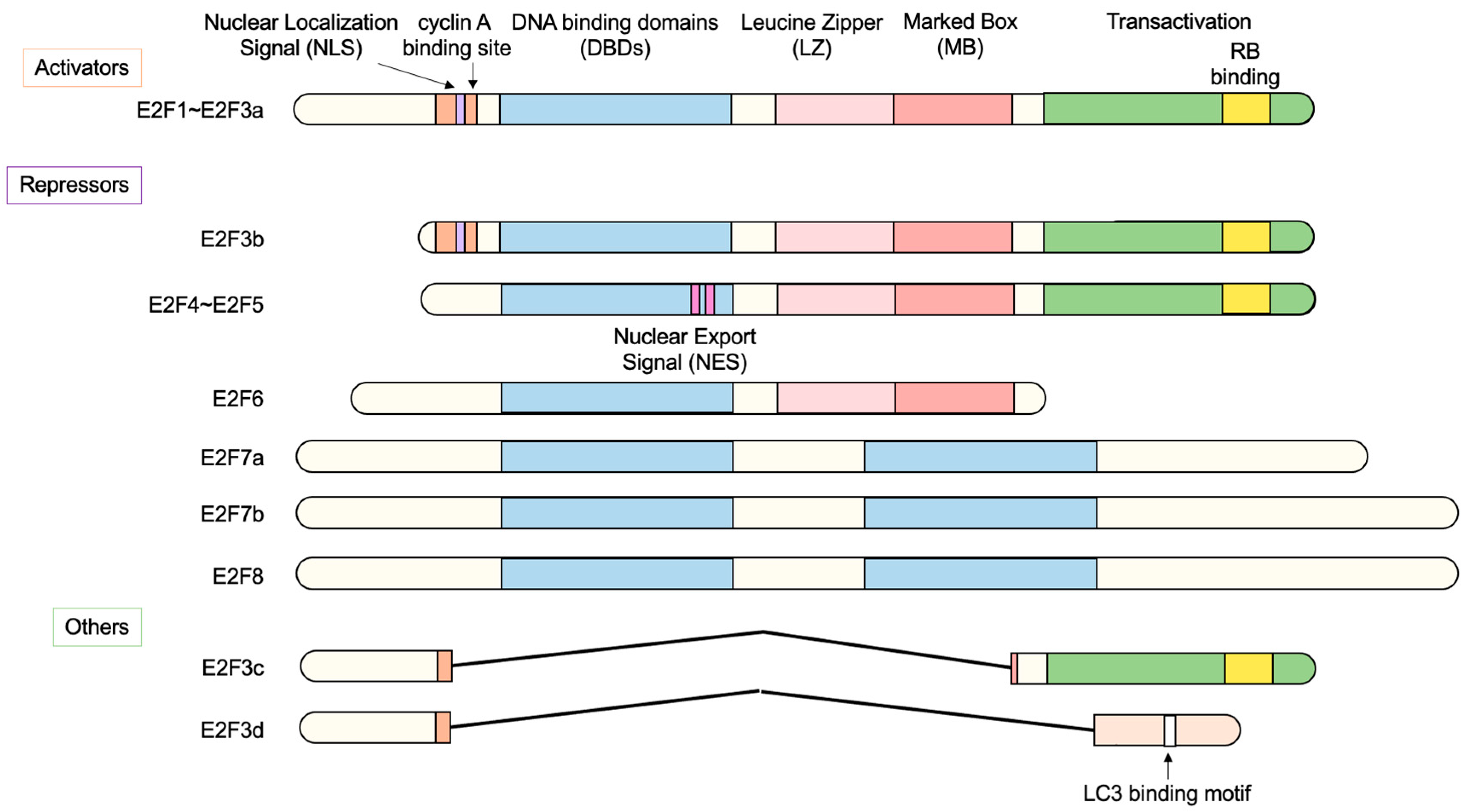
2.1. The E2F Family of Transcription Factors
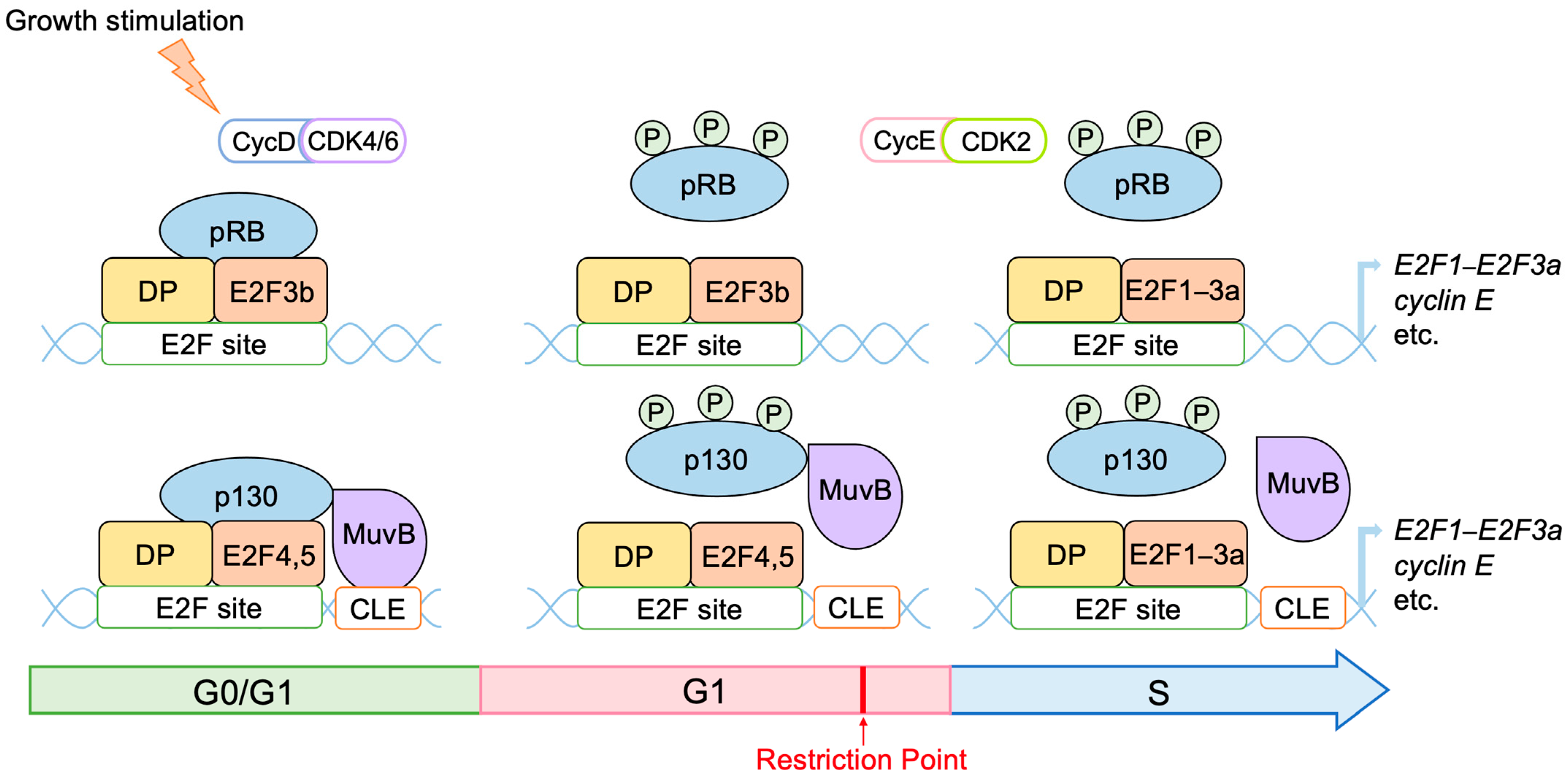
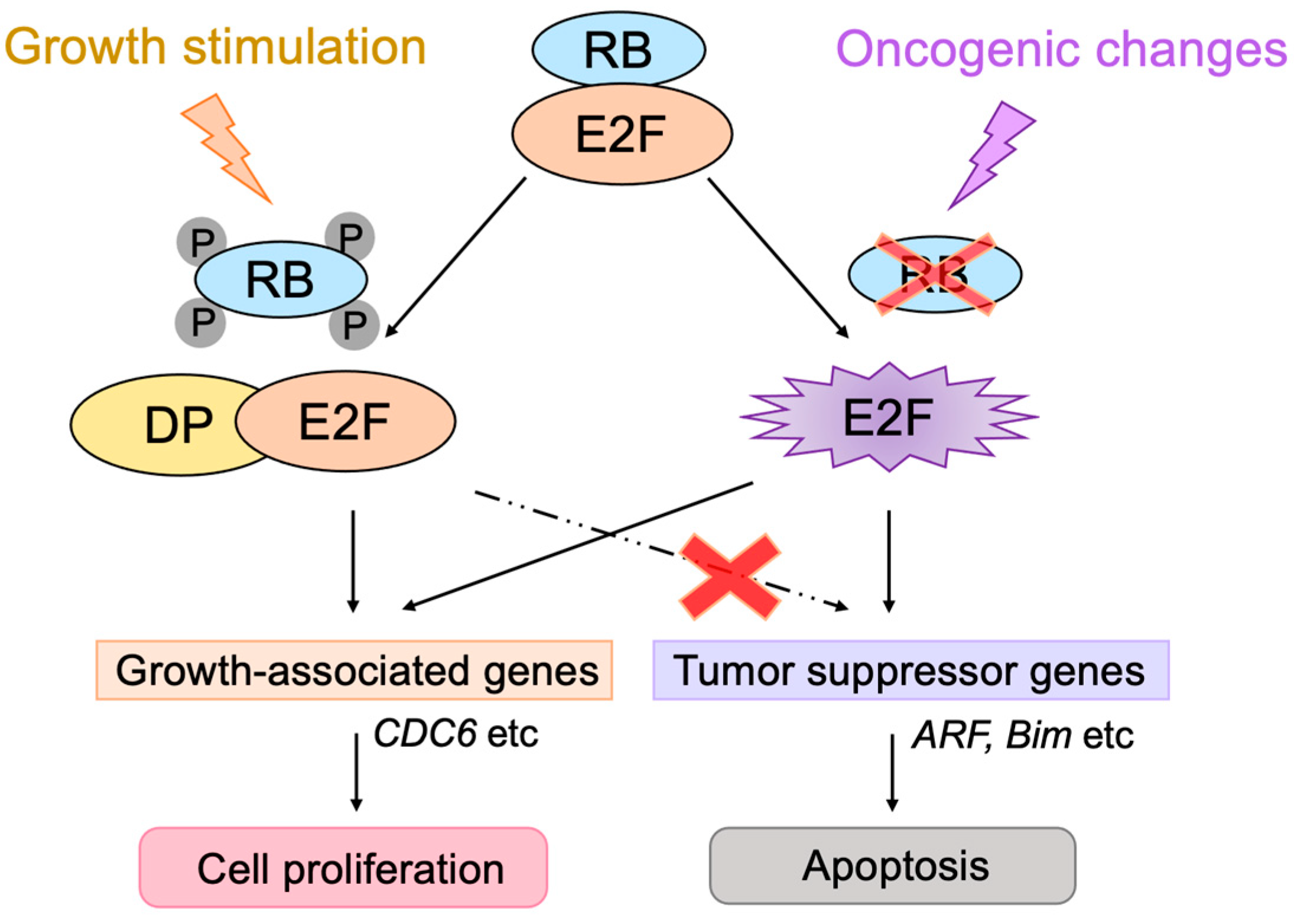
2.2. Roles of E2F in Cell Proliferation
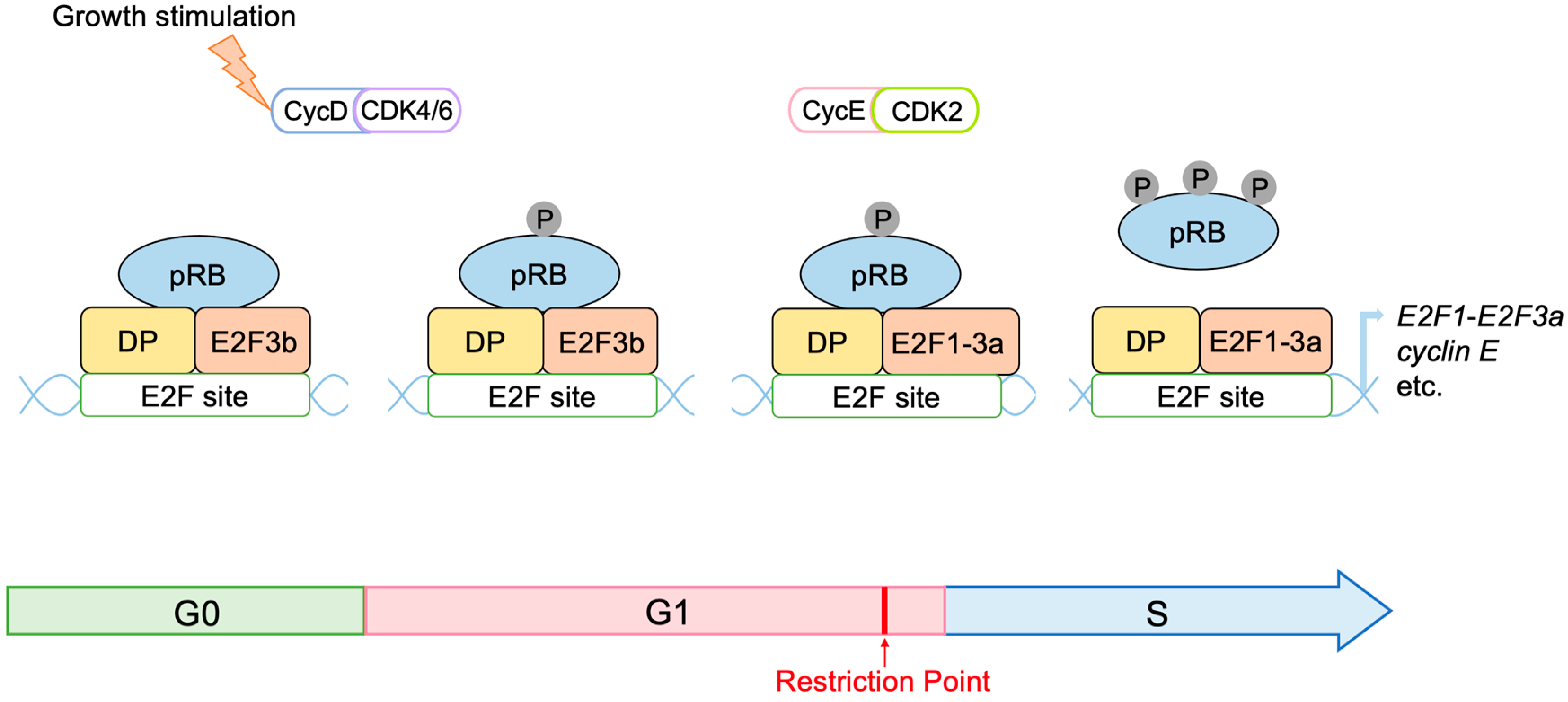
2.3. Regulation of E2F Activity by the RB Pathway

2.4. Defects in the RB Pathway in Cancer Cells
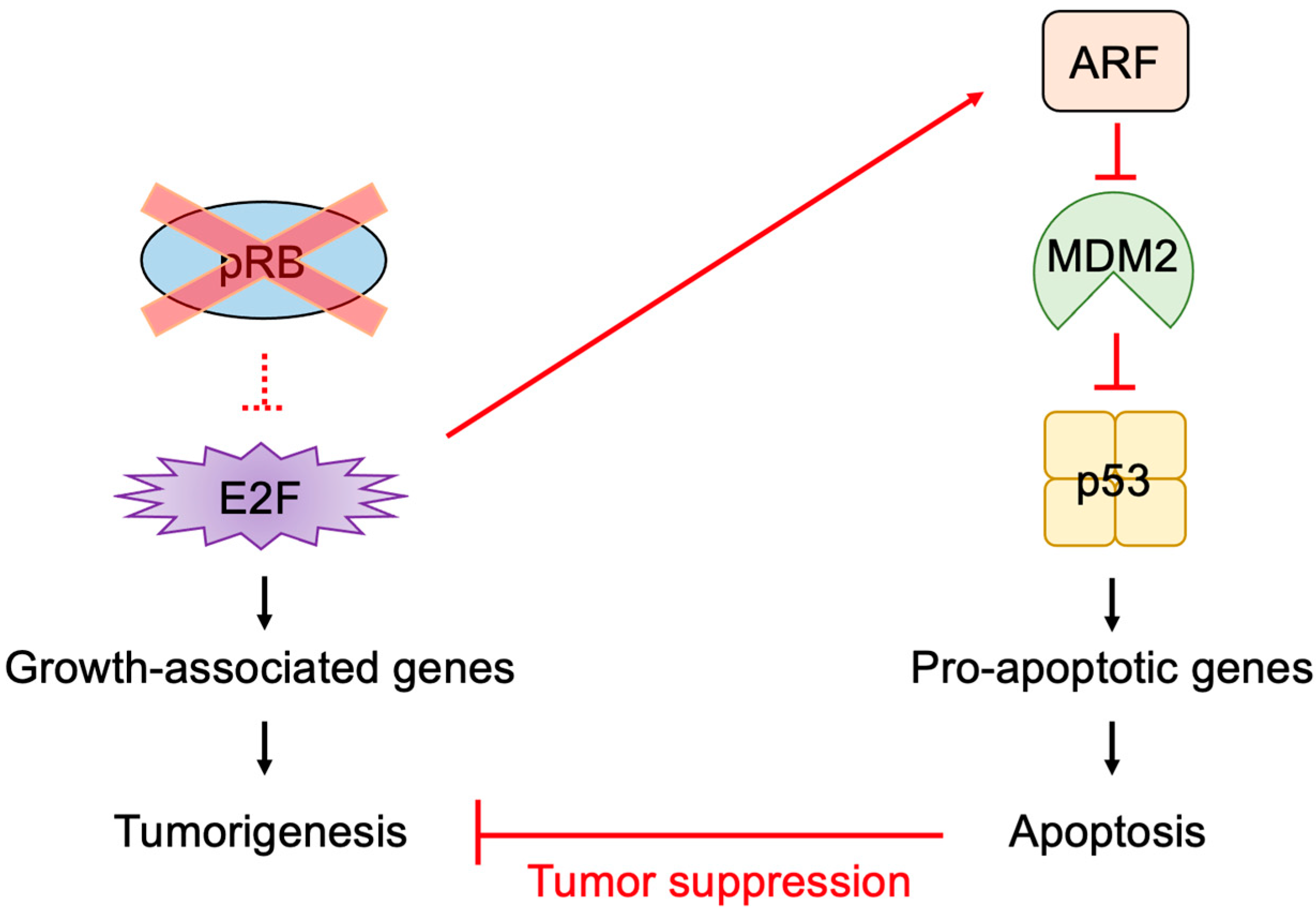
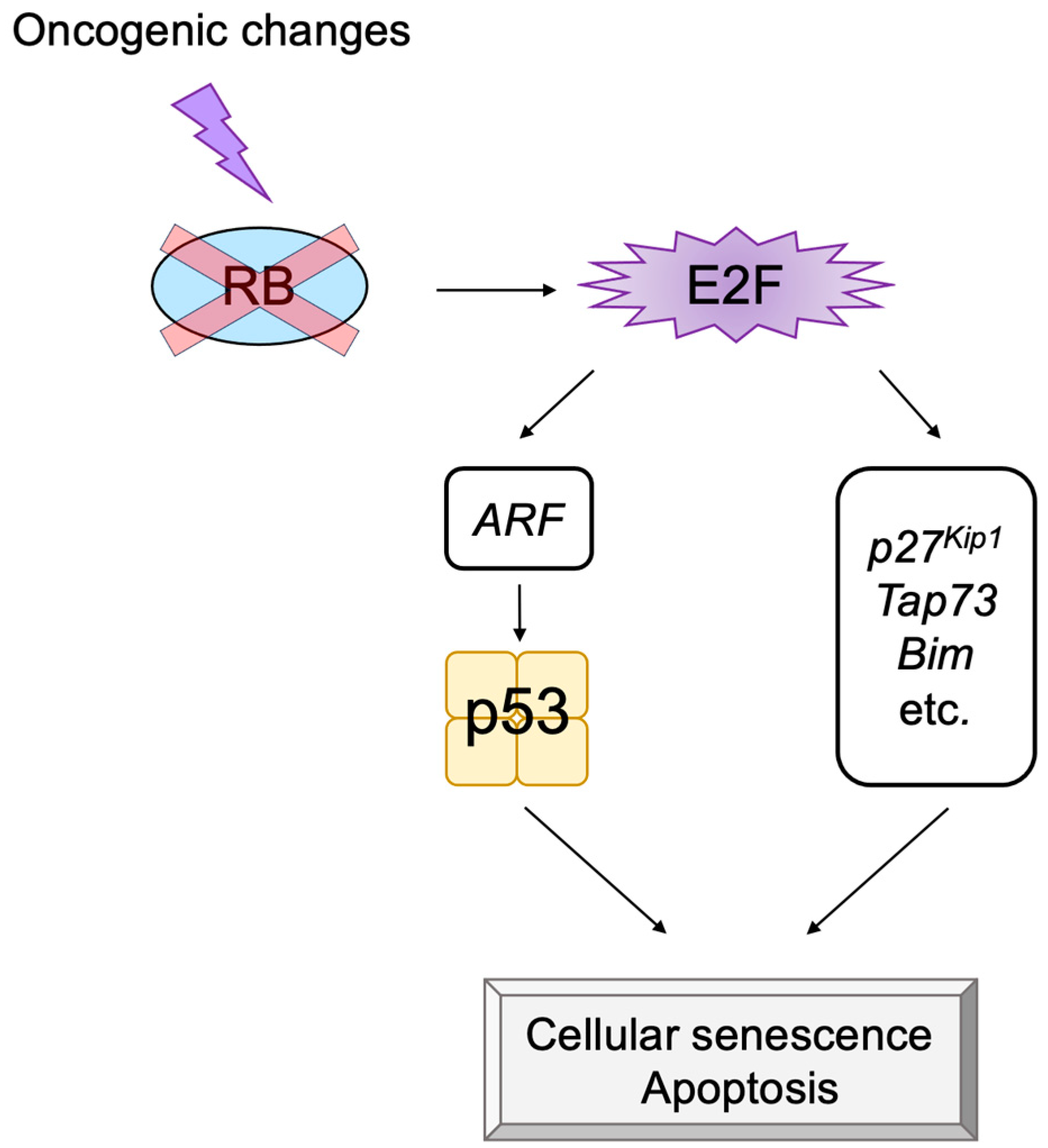
2.5. E2F Links the RB Pathway to the p53 Pathway
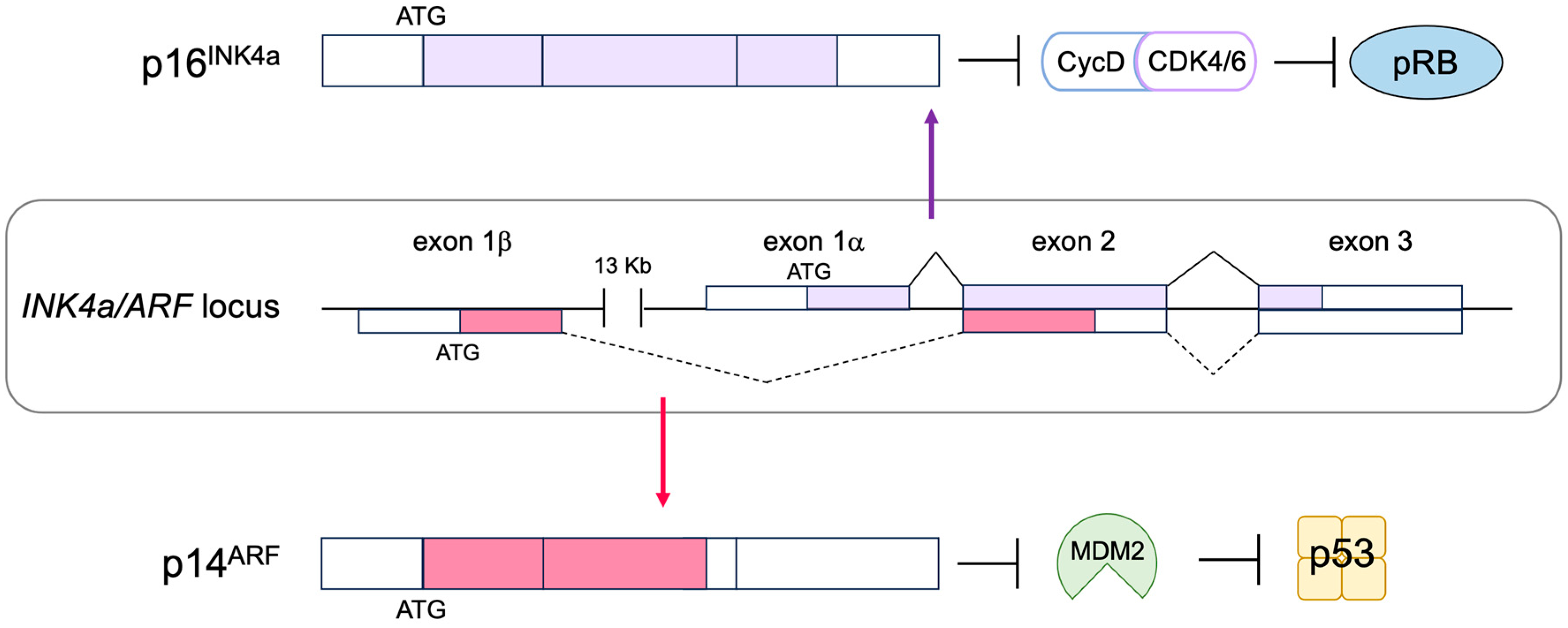
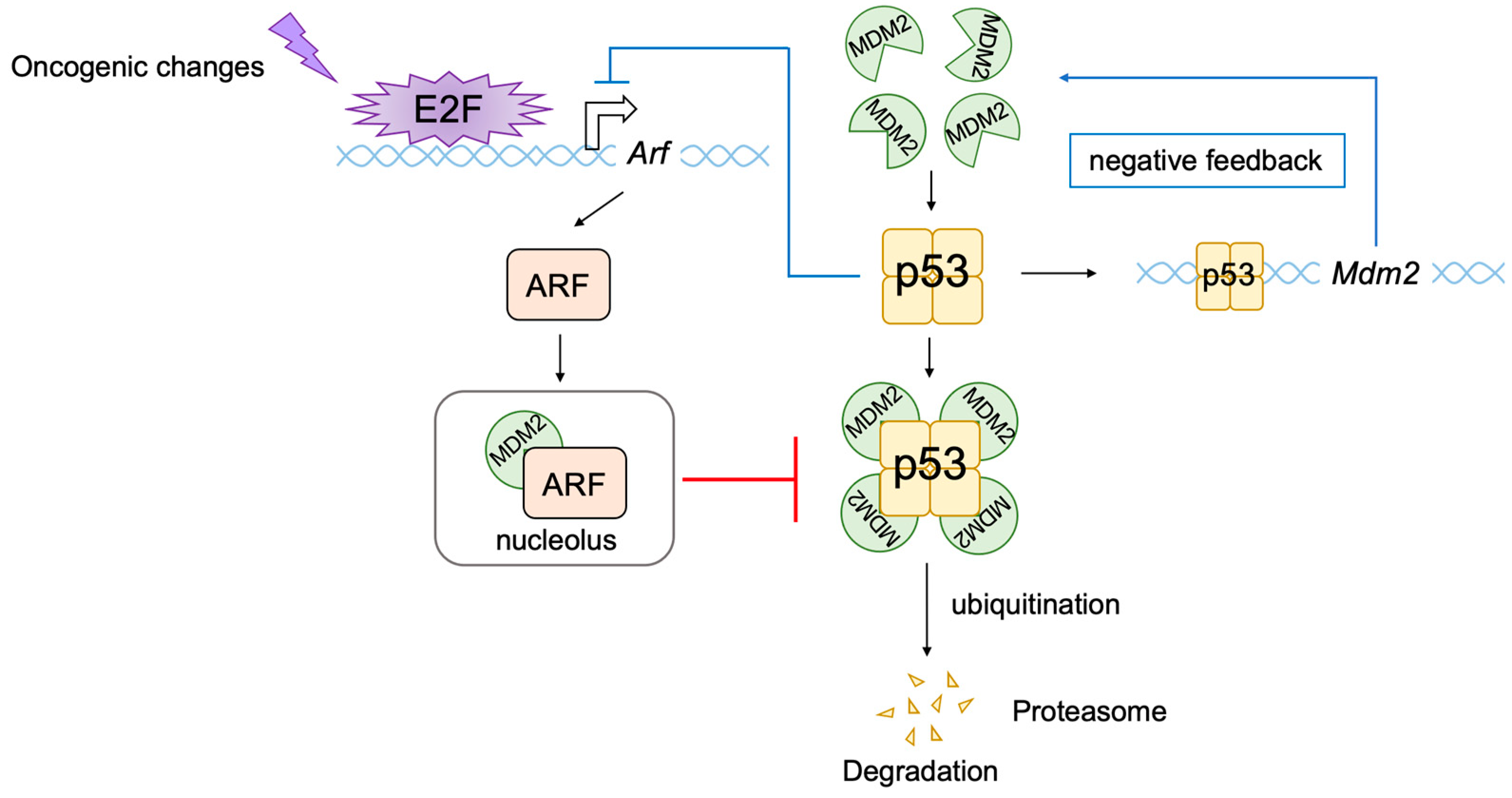
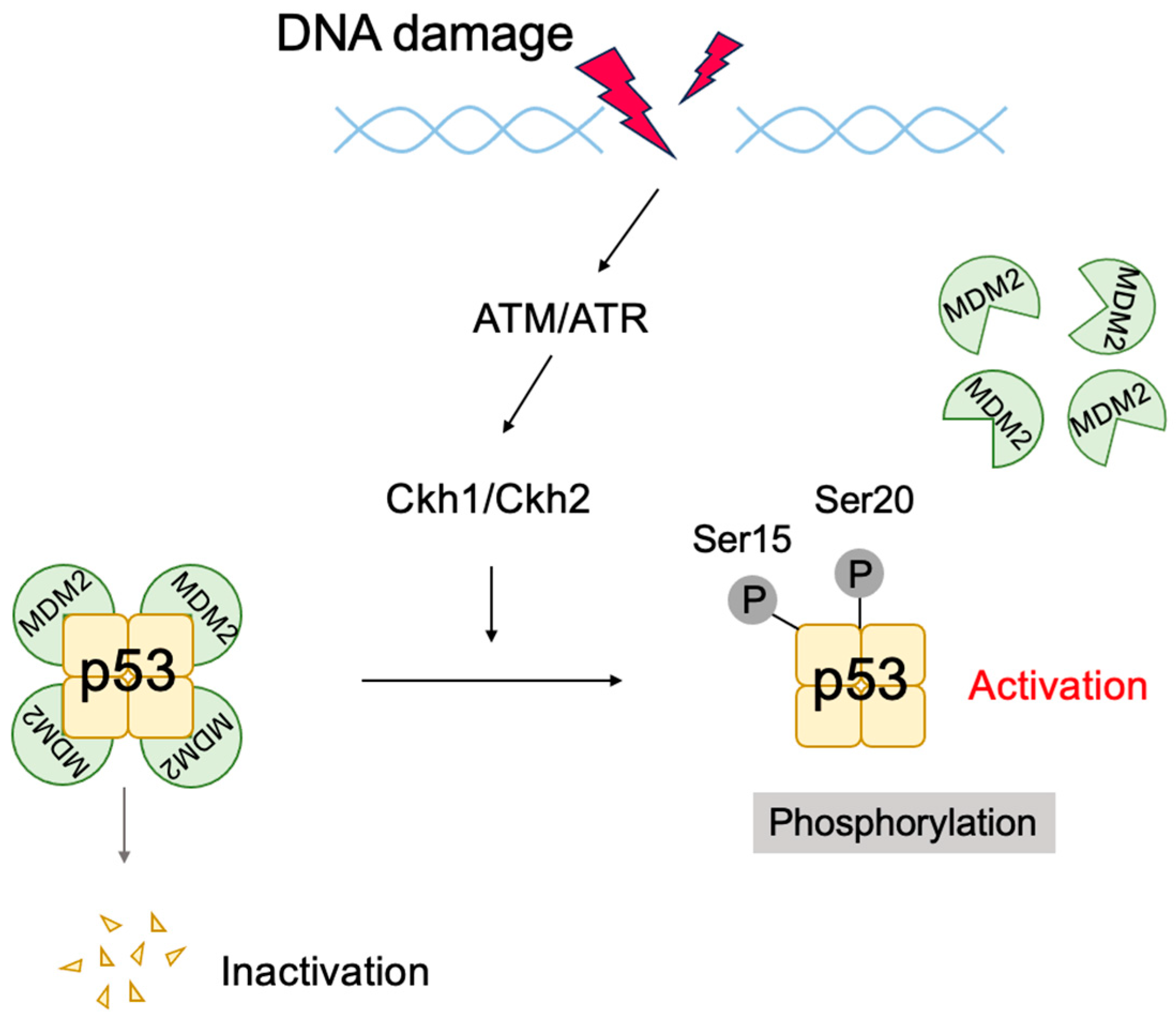
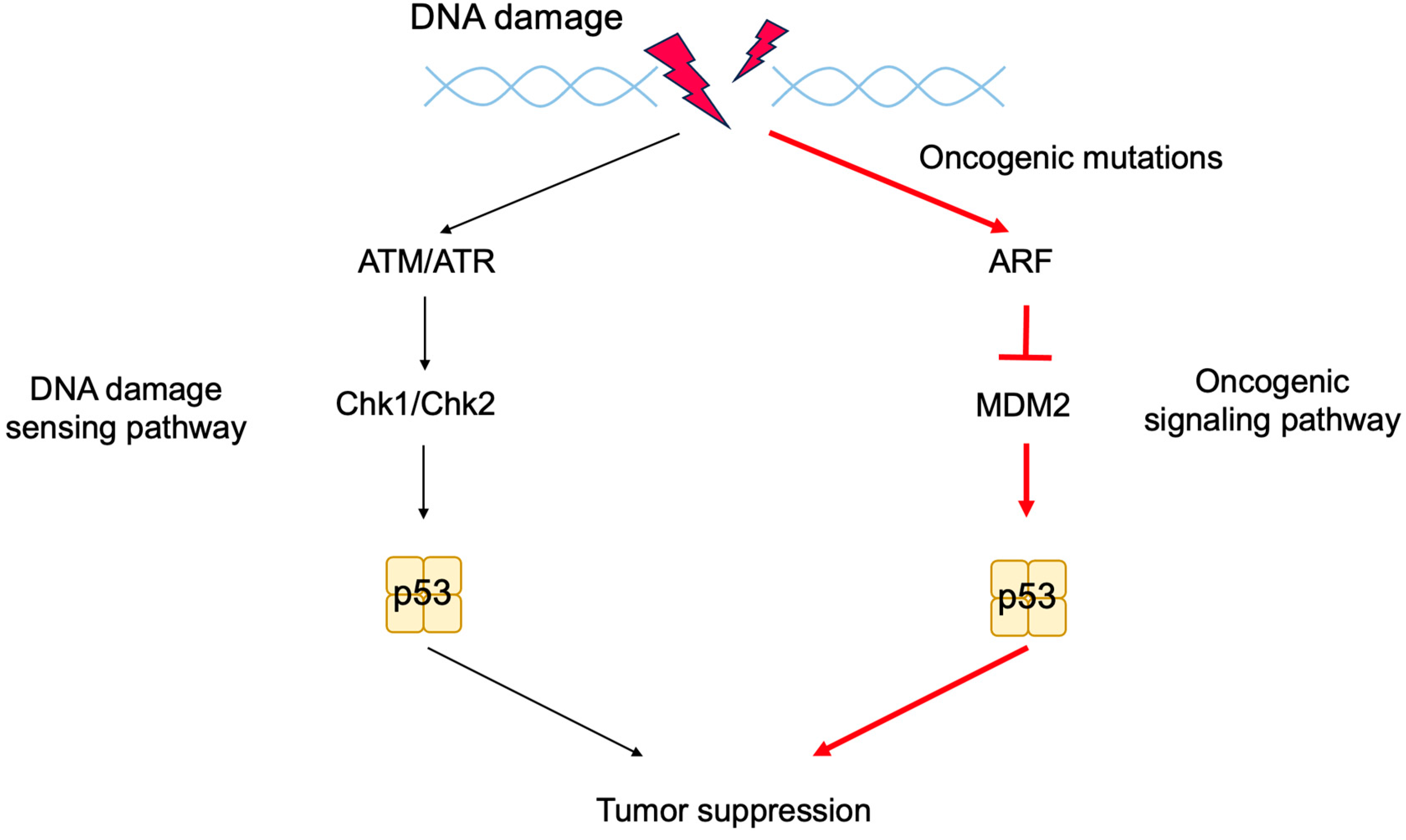
2.6. The p53 Pathway
3. Non-Classical Functions of Each Component of the RB-E2F-ARF-MDM2-p53 Pathway
3.1. Non-Classical Functions of RB
3.2. Non-Classical Functions of E2F
3.2.1. Unique Properties of E2F in Linking the RB Pathway to the p53 Pathway
3.2.2. E2F1 Targets Involved in p53-Independent Pathways for the Induction of Apoptosis
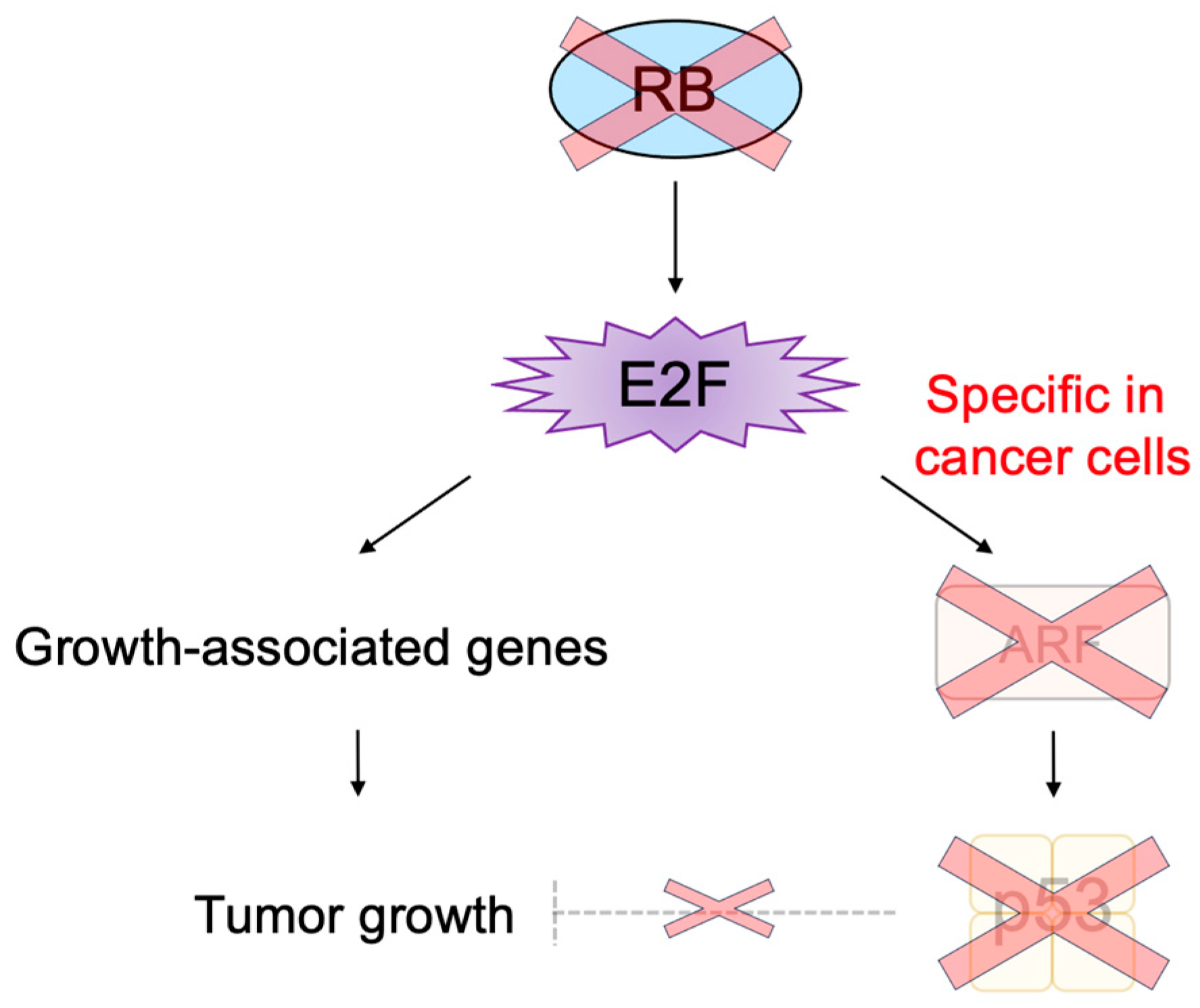
3.2.3. Cancer-Cell-Specific Deregulated E2F Activity as a Cancer-Cell-Specific Targeting Tool
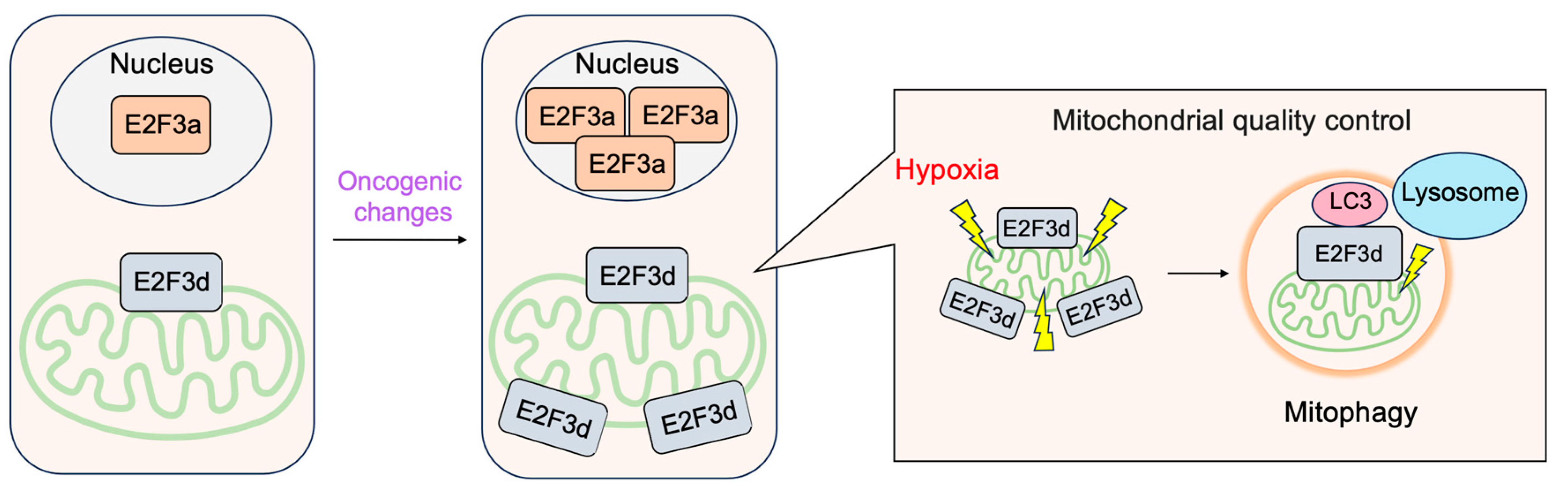
3.2.4. E2F3d, a Novel Member of the E2F3 Family, Mediates Hypoxia-Induced Mitophagy in Cancer Cells
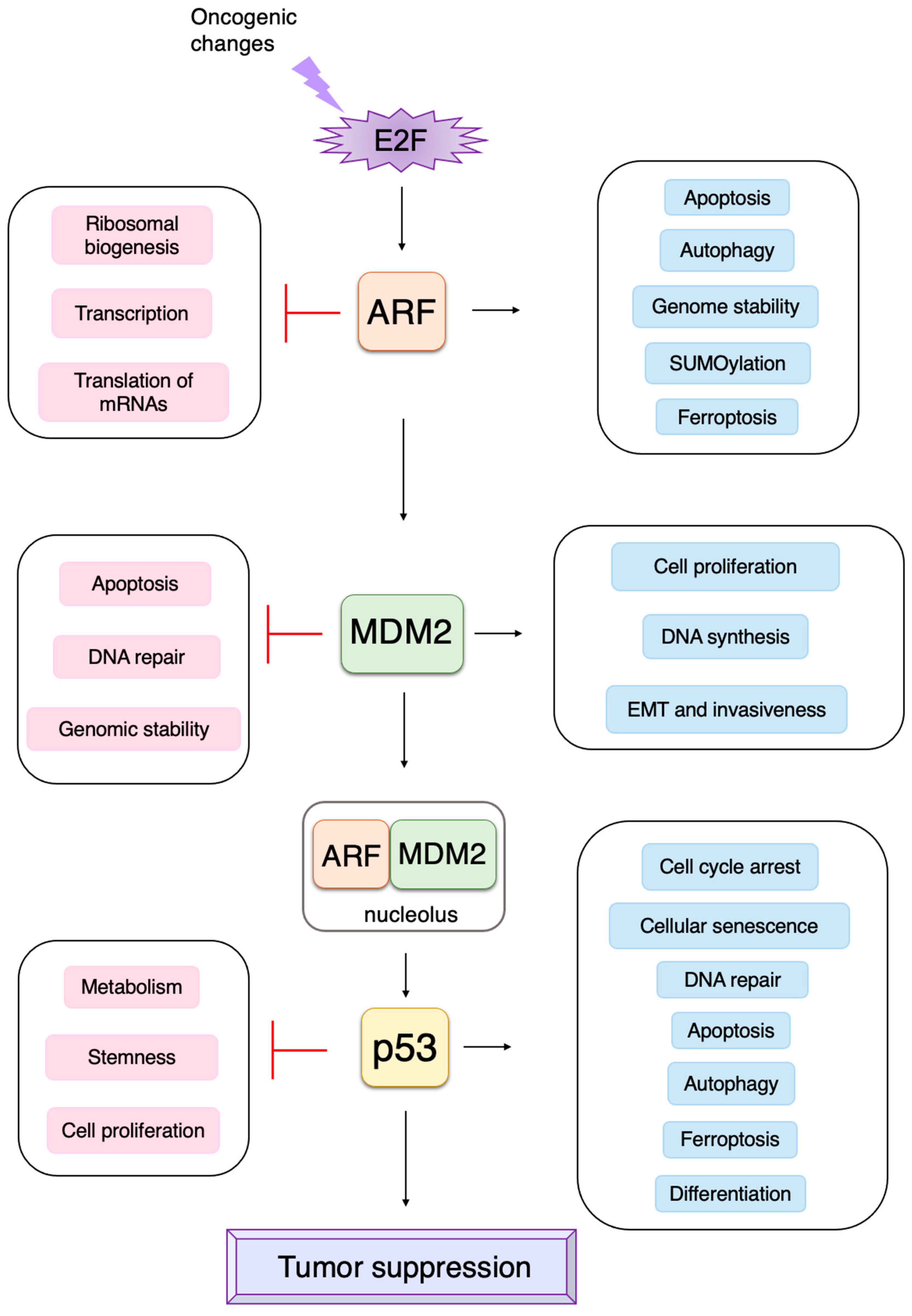
3.3. p53-Independent Functions of ARF in Tumor Suppression
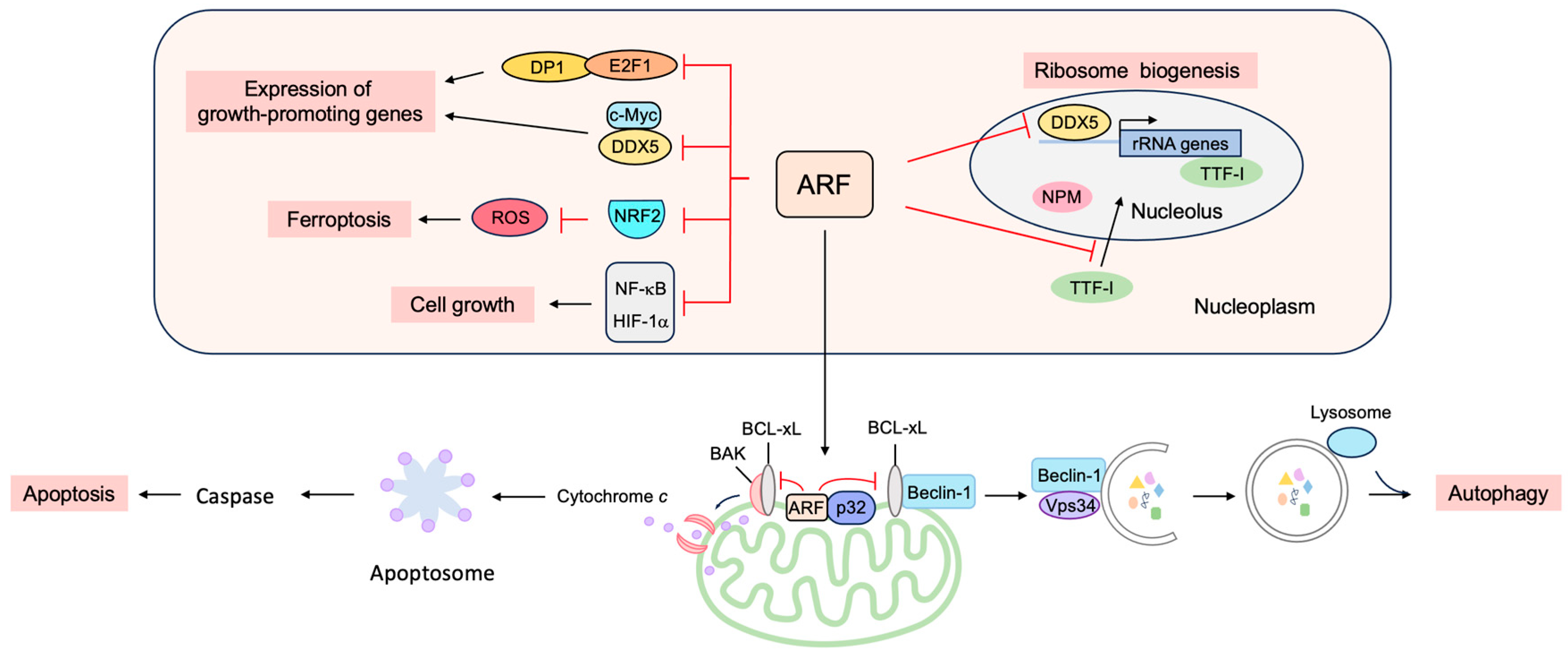
3.3.1. ARF Suppresses Ribosomal Biogenesis
3.3.2. ARF Suppresses the Expression of Growth-Promoting Genes
3.3.3. ARF Facilitates Apoptosis at the Mitochondria
3.3.4. ARF Facilitates Autophagy at the Mitochondria
3.3.5. ARF Contributes to Genome Stability
3.3.6. ARF Facilitates the SUMOylation of Interacting Proteins
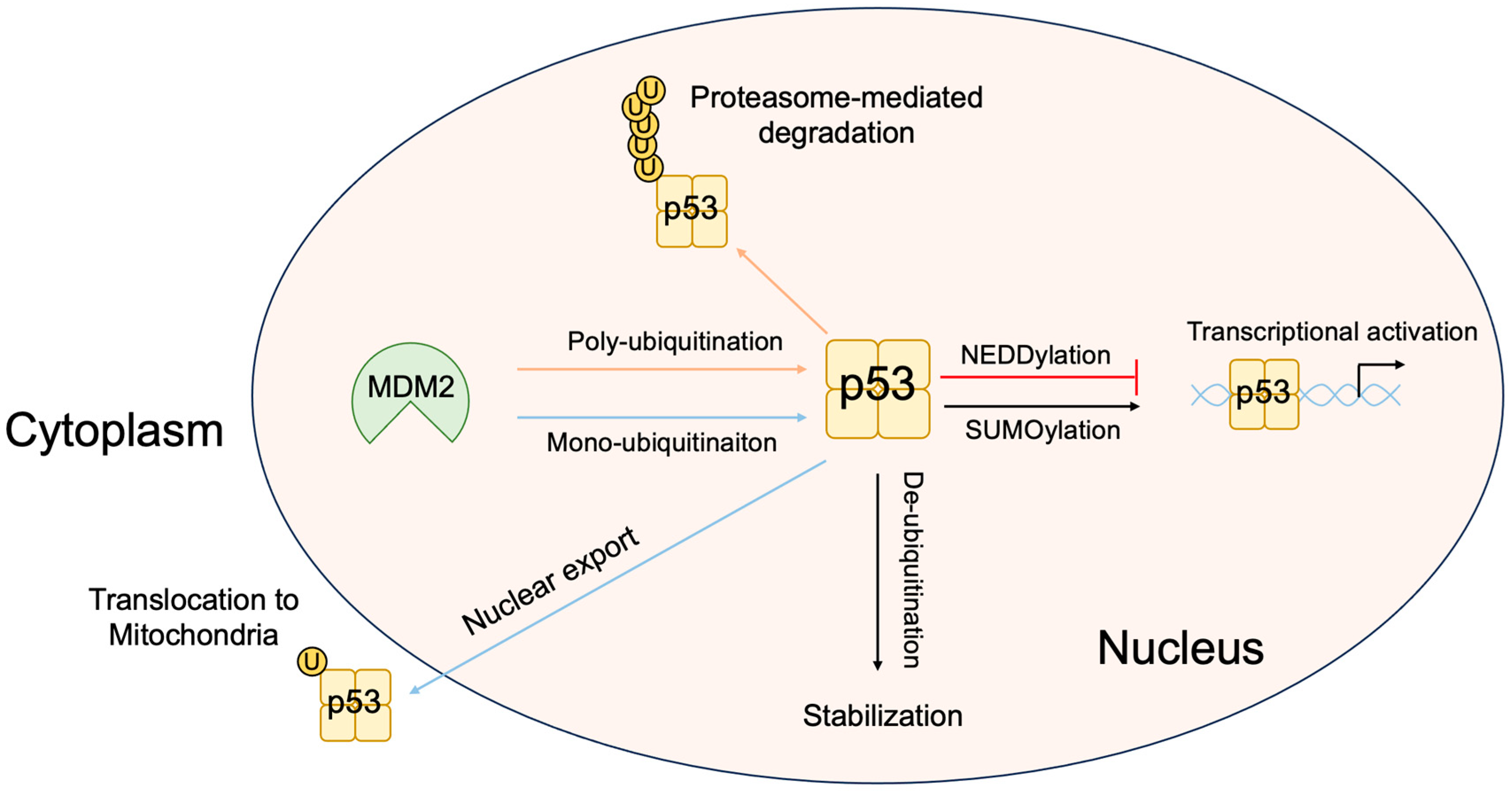
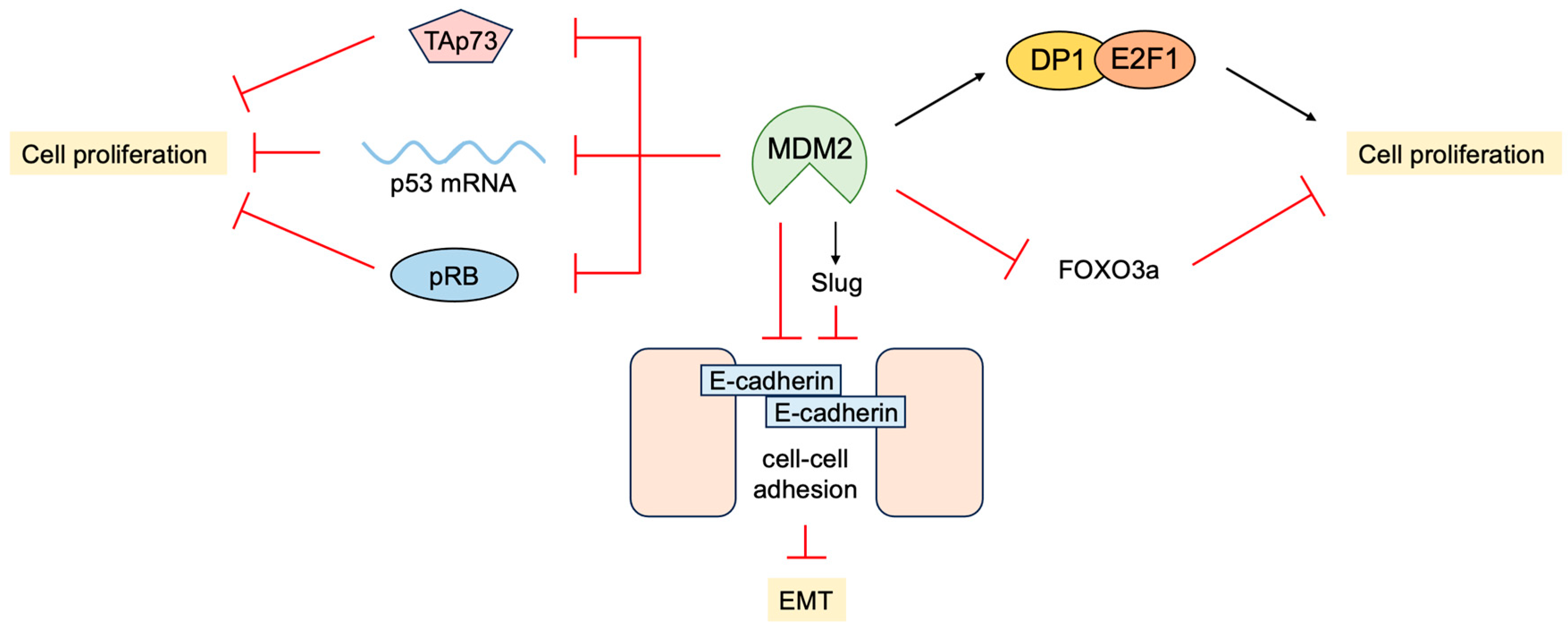
3.4. P53-Independent Functions of MDM2 in Tumor Promotion
3.5. Non-Classical Functions of p53
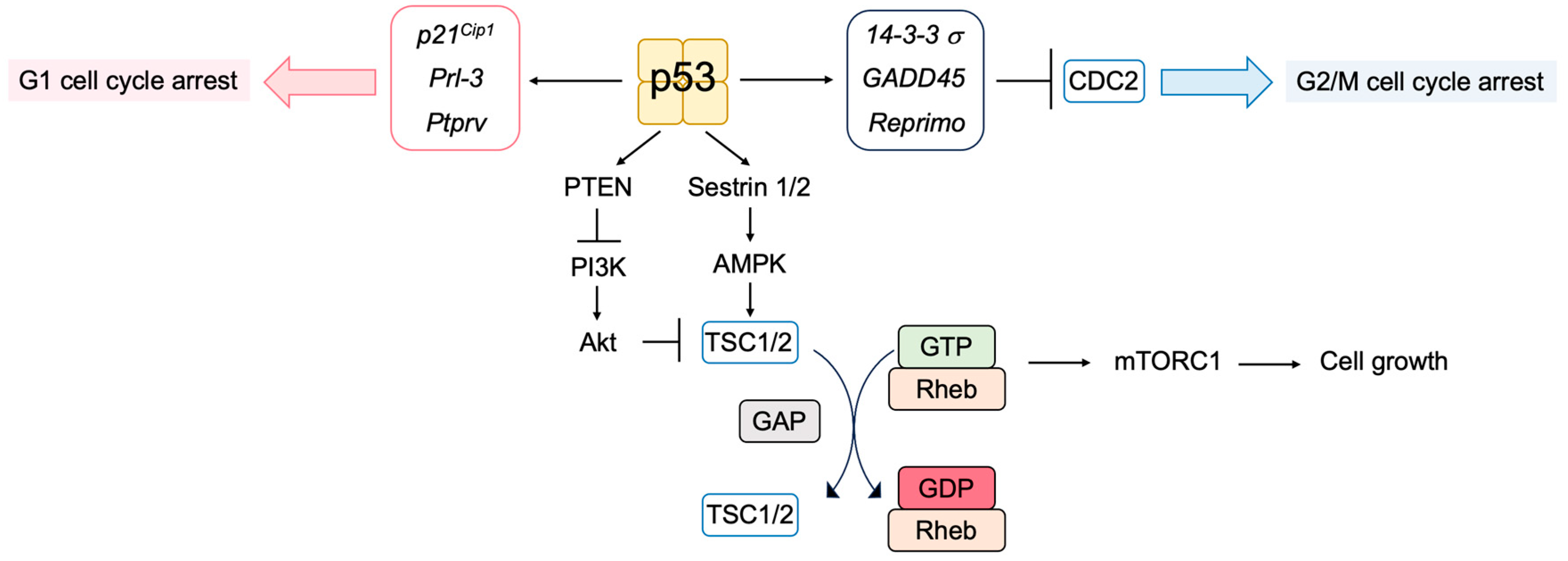
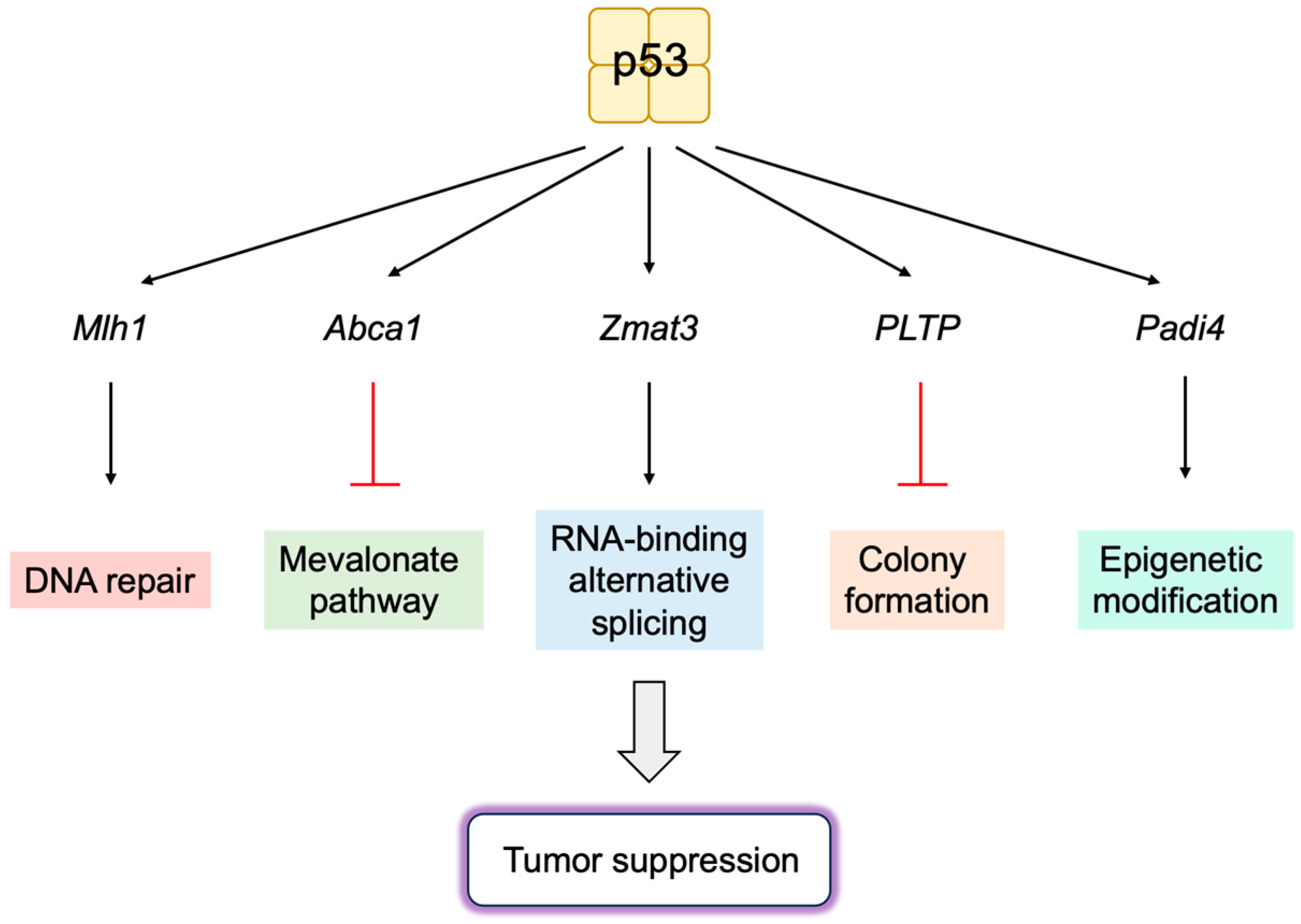
3.5.1. Novel p53 Targets Genes Important for Tumor Suppression

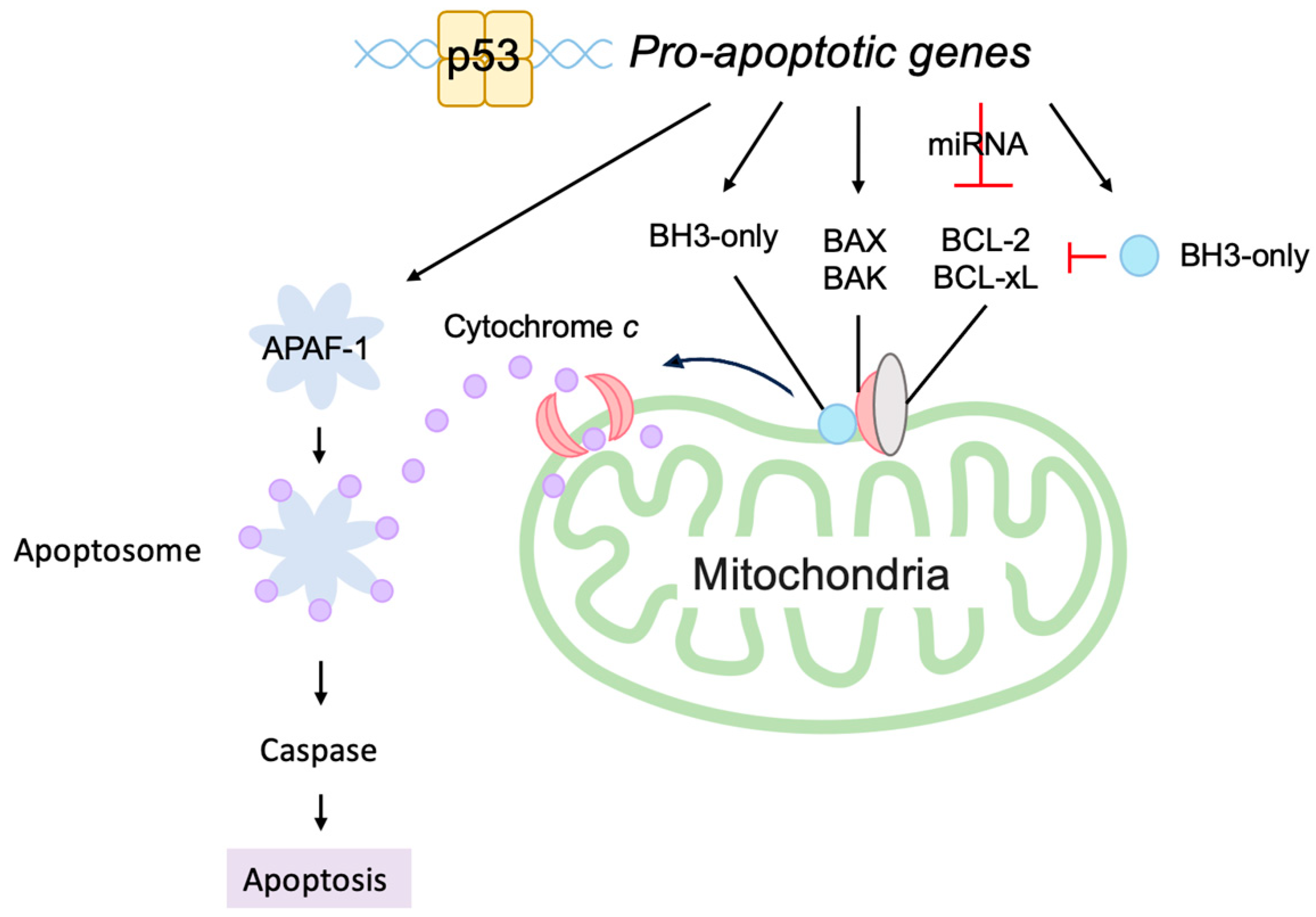
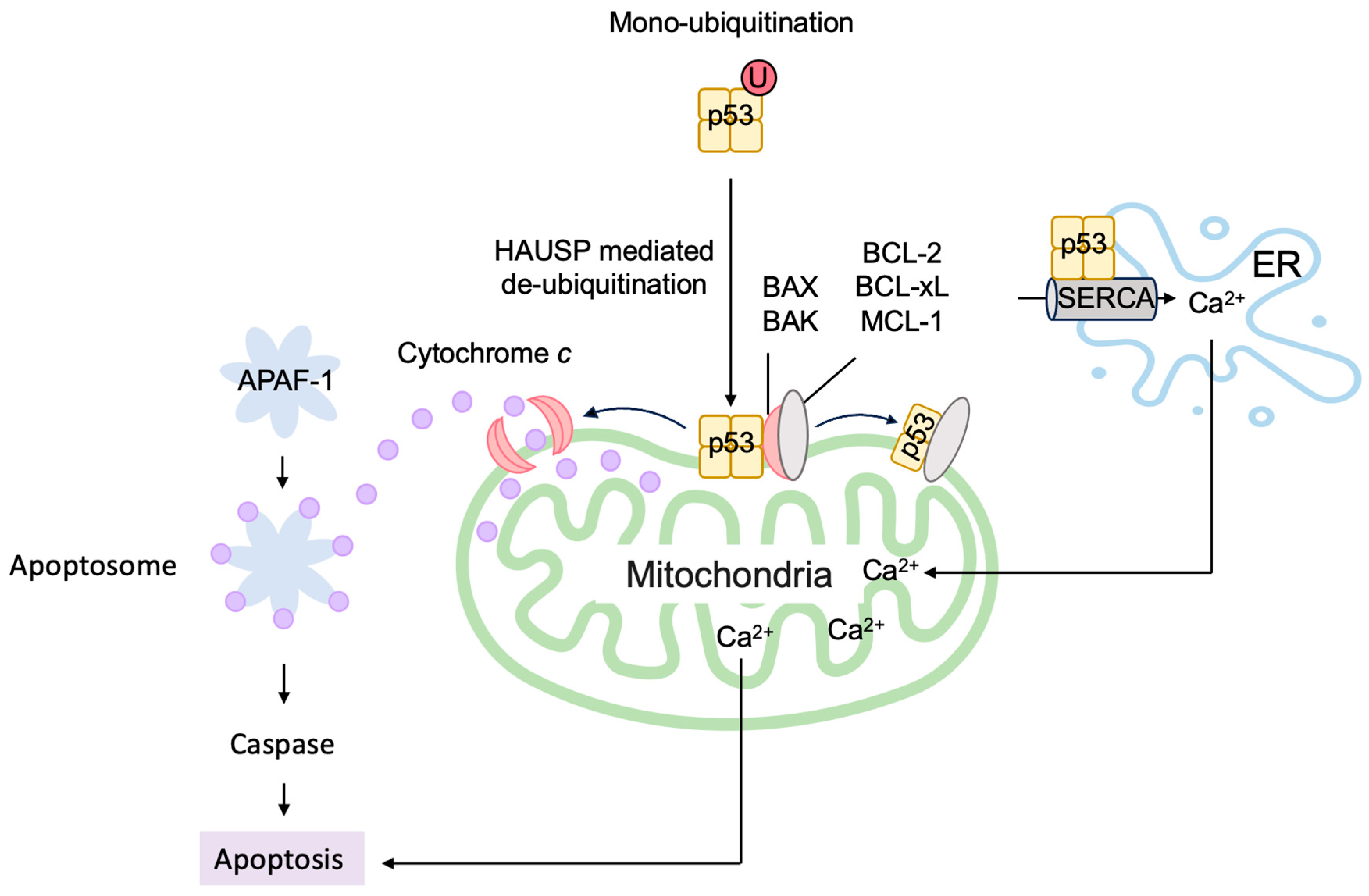
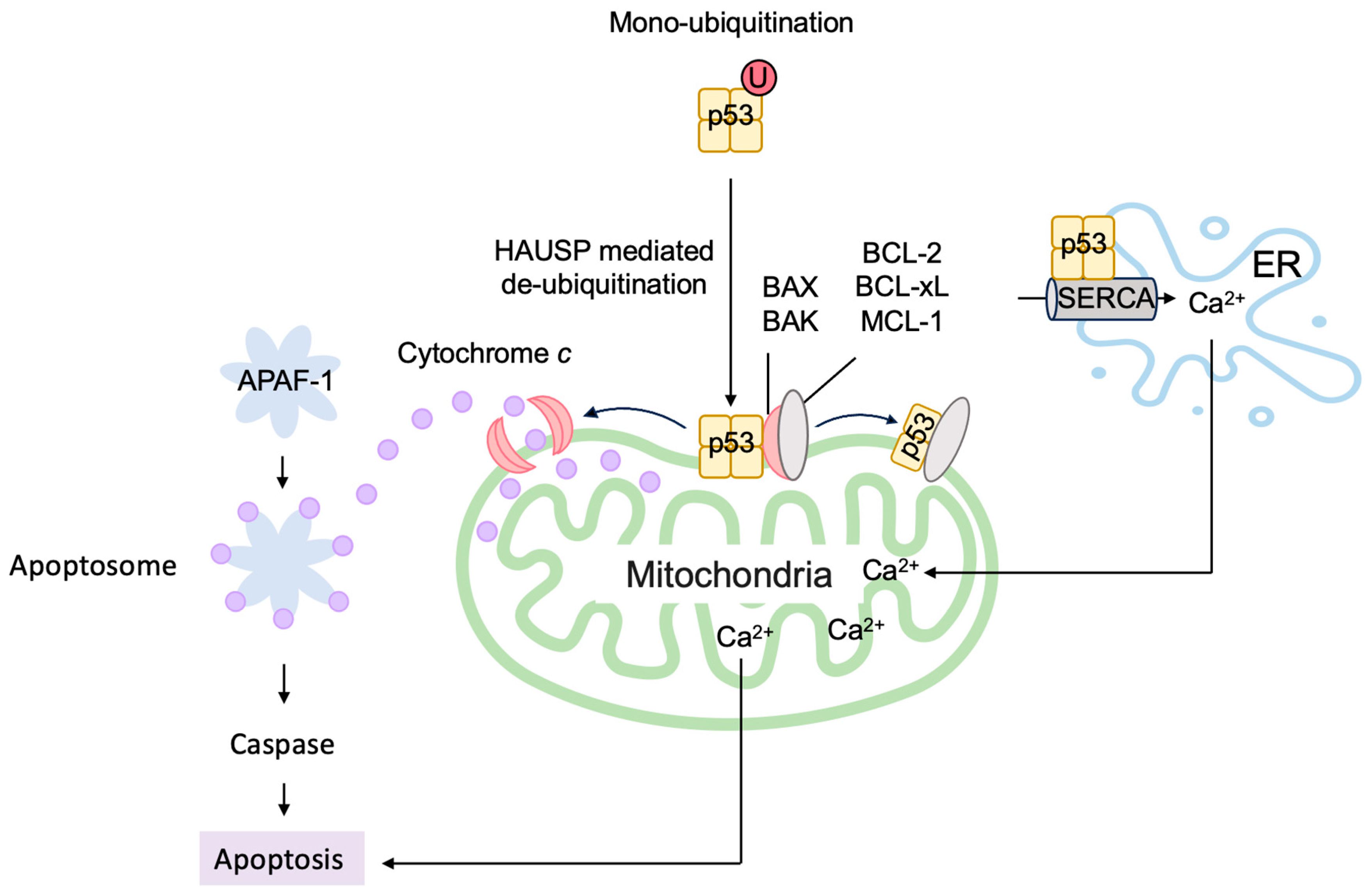
3.5.2. P53 Directly Induces Apoptosis at the Mitochondria
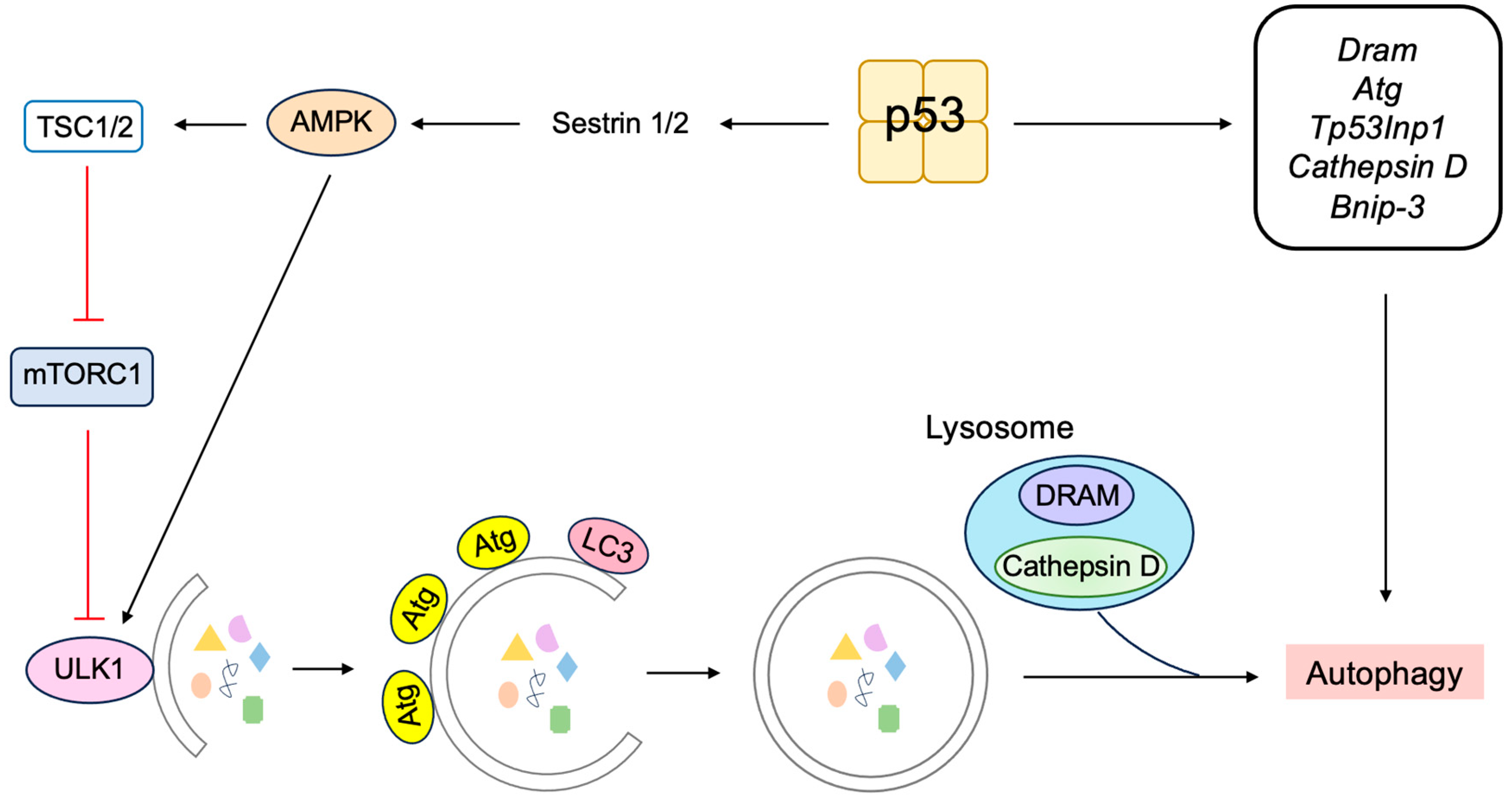
3.5.3. P53 Induces Autophagy to Suppress Tumorigenesis

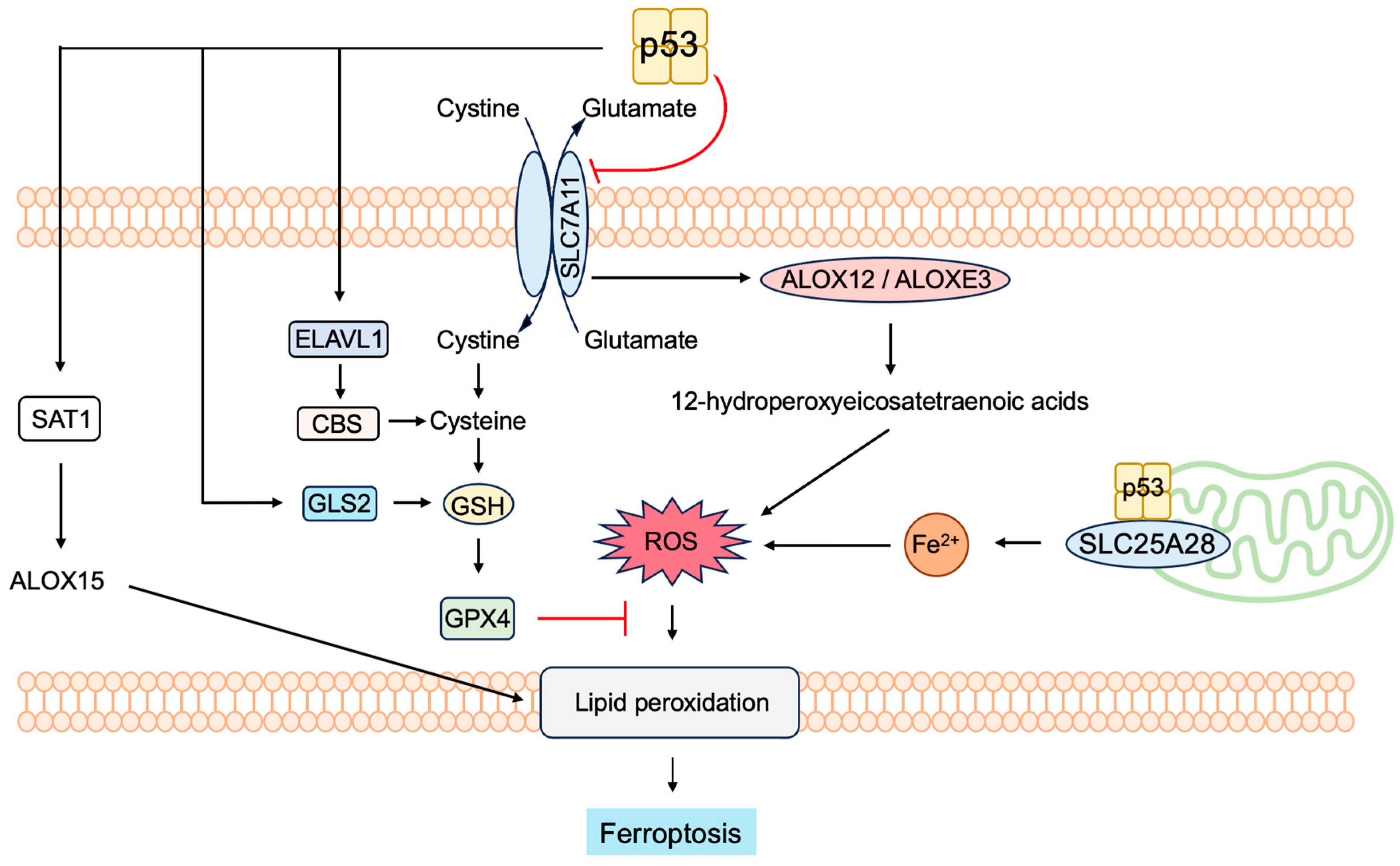
3.5.4. P53 Induces Ferroptosis to Suppress Tumorigenesis
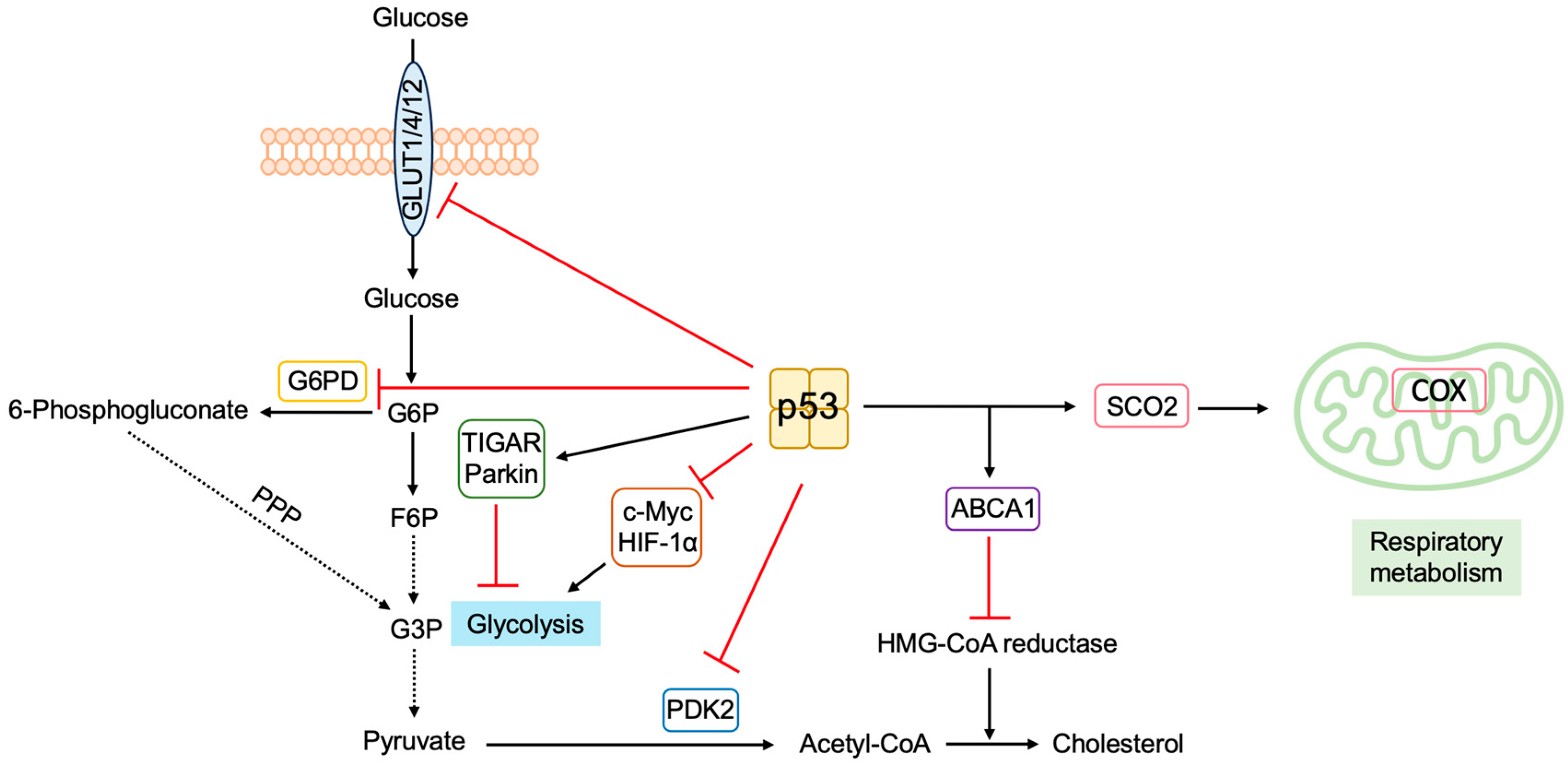
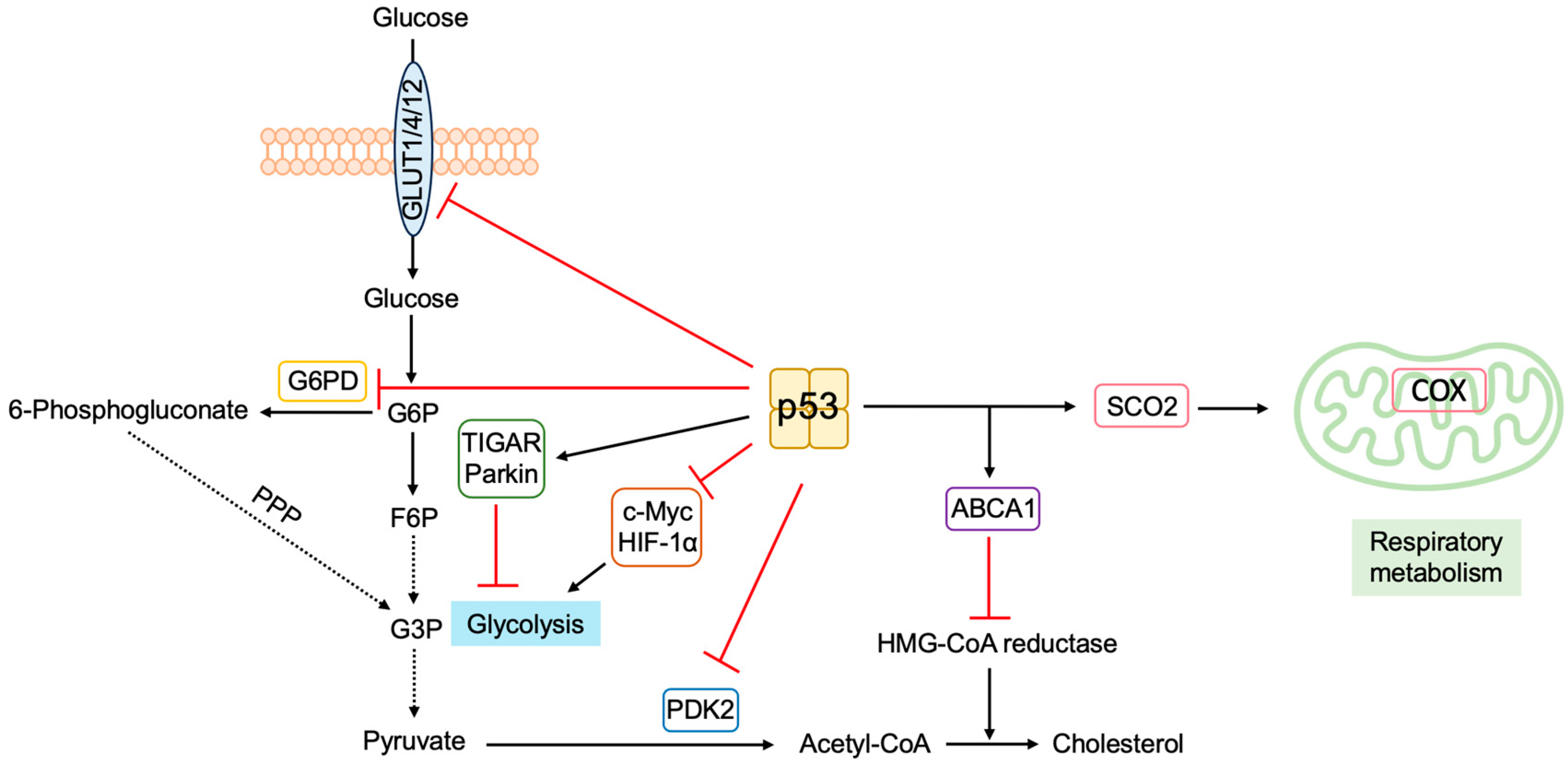
3.5.5. P53 Regulates Metabolism to Suppress Tumorigenesis
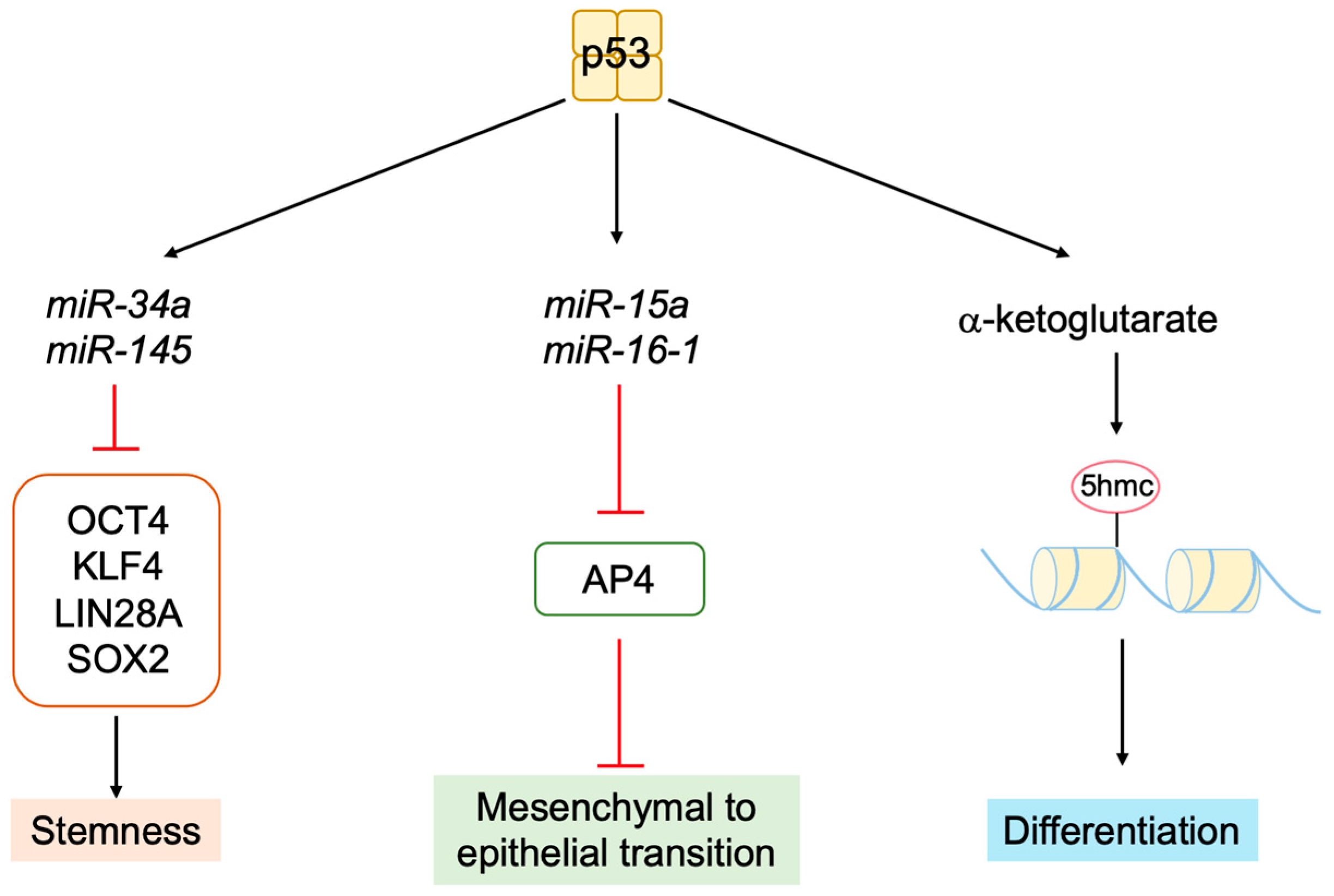
3.5.6. P53 Inhibits Stemness and Promotes Differentiation

4. Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef]
- Bates, S.; Phillips, A.C.; Clark, P.A.; Stott, F.; Peters, G.; Ludwig, R.L.; Vousden, K.H. p14ARF links the tumour suppressors RB and p53. Nature 1998, 395, 124–125. [Google Scholar] [CrossRef] [PubMed]
- Kovesdi, I.; Reichel, R.; Nevins, J.R. Identification of a cellular transcription factor involved in E1A trans-activation. Cell 1986, 45, 219–228. [Google Scholar] [CrossRef]
- Nevins, J.R.; Leone, G.; DeGregori, J.; Jakoi, L. Role of the Rb/E2F pathway in cell growth control. J. Cell. Physiol. 1997, 173, 233–236. [Google Scholar] [CrossRef]
- Fischer, M.; Schade, A.E.; Branigan, T.B.; Muller, G.A.; DeCaprio, J.A. Coordinating gene expression during the cell cycle. Trends Biochem. Sci. 2022, 47, 1009–1022. [Google Scholar] [CrossRef]
- Muller, H.; Bracken, A.P.; Vernell, R.; Moroni, M.C.; Christians, F.; Grassilli, E.; Prosperini, E.; Vigo, E.; Oliner, J.D.; Helin, K. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 2001, 15, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.C.; Vousden, K.H. E2F-1 induced apoptosis. Apoptosis 2001, 6, 173–182. [Google Scholar] [CrossRef]
- Polager, S.; Ginsberg, D. p53 and E2f: Partners in life and death. Nat. Rev. Cancer 2009, 9, 738–748. [Google Scholar] [CrossRef]
- Poppy Roworth, A.; Ghari, F.; La Thangue, N.B. To live or let die—Complexity within the E2F1 pathway. Mol. Cell. Oncol. 2015, 2, e970480. [Google Scholar] [CrossRef]
- Trimarchi, J.M.; Lees, J.A. Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 2002, 3, 11–20. [Google Scholar] [CrossRef]
- Iaquinta, P.J.; Lees, J.A. Life and death decisions by the E2F transcription factors. Curr. Opin. Cell Biol. 2007, 19, 649–657. [Google Scholar] [CrossRef]
- Chen, H.Z.; Tsai, S.Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef]
- Nicolay, B.N.; Dyson, N.J. The multiple connections between pRB and cell metabolism. Curr. Opin. Cell Biol. 2013, 25, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Schaal, C.; Pillai, S.; Chellappan, S.P. The Rb-E2F transcriptional regulatory pathway in tumor angiogenesis and metastasis. Adv. Cancer Res. 2014, 121, 147–182. [Google Scholar] [CrossRef] [PubMed]
- Benevolenskaya, E.V.; Frolov, M.V. Emerging links between E2F control and mitochondrial function. Cancer Res. 2015, 75, 619–623. [Google Scholar] [CrossRef]
- Kent, L.N.; Leone, G. The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 2019, 19, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Julian, L.M.; Blais, A. Transcriptional control of stem cell fate by E2Fs and pocket proteins. Front. Genet. 2015, 6, 161. [Google Scholar] [CrossRef]
- Jusino, S.; Saavedra, H.I. Role of E2Fs and mitotic regulators controlled by E2Fs in the epithelial to mesenchymal transition. Exp. Biol. Med. 2019, 244, 1419–1429. [Google Scholar] [CrossRef]
- Xie, D.; Pei, Q.; Li, J.; Wan, X.; Ye, T. Emerging Role of E2F Family in Cancer Stem Cells. Front. Oncol. 2021, 11, 723137. [Google Scholar] [CrossRef]
- Mandigo, A.C.; Yuan, W.; Xu, K.; Gallagher, P.; Pang, A.; Guan, Y.F.; Shafi, A.A.; Thangavel, C.; Sheehan, B.; Bogdan, D.; et al. RB/E2F1 as a Master Regulator of Cancer Cell Metabolism in Advanced Disease. Cancer Discov. 2021, 11, 2334–2353. [Google Scholar] [CrossRef]
- Johnson, D.G.; Ohtani, K.; Nevins, J.R. Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev. 1994, 8, 1514–1525. [Google Scholar] [CrossRef]
- Sears, R.; Ohtani, K.; Nevins, J.R. Identification of positively and negatively acting elements regulating expression of the E2F2 gene in response to cell growth signals. Mol. Cell. Biol. 1997, 17, 5227–5235. [Google Scholar] [CrossRef]
- Adams, M.R.; Sears, R.; Nuckolls, F.; Leone, G.; Nevins, J.R. Complex transcriptional regulatory mechanisms control expression of the E2F3 locus. Mol. Cell. Biol. 2000, 20, 3633–3639. [Google Scholar] [CrossRef]
- Leone, G.; Nuckolls, F.; Ishida, S.; Adams, M.; Sears, R.; Jakoi, L.; Miron, A.; Nevins, J.R. Identification of a novel E2F3 product suggests a mechanism for determining specificity of repression by Rb proteins. Mol. Cell. Biol. 2000, 20, 3626–3632. [Google Scholar] [CrossRef]
- Ginsberg, D.; Vairo, G.; Chittenden, T.; Xiao, Z.X.; Xu, G.; Wydner, K.L.; DeCaprio, J.A.; Lawrence, J.B.; Livingston, D.M. E2F-4, a new member of the E2F transcription factor family, interacts with p107. Genes Dev. 1994, 8, 2665–2679. [Google Scholar] [CrossRef]
- Hijmans, E.M.; Voorhoeve, P.M.; Beijersbergen, R.L.; van’t Veer, L.J.; Bernards, R. E2F-5, a new E2F family member that interacts with p130 in vivo. Mol. Cell. Biol. 1995, 15, 3082–3089. [Google Scholar] [CrossRef]
- Sardet, C.; Vidal, M.; Cobrinik, D.; Geng, Y.; Onufryk, C.; Chen, A.; Weinberg, R.A. E2F-4 and E2F-5, two members of the E2F family, are expressed in the early phases of the cell cycle. Proc. Natl. Acad. Sci. USA 1995, 92, 2403–2407. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, J.M.; Fairchild, B.; Verona, R.; Moberg, K.; Andon, N.; Lees, J.A. E2F-6, a member of the E2F family that can behave as a transcriptional repressor. Proc. Natl. Acad. Sci. USA 1998, 95, 2850–2855. [Google Scholar] [CrossRef]
- de Bruin, A.; Maiti, B.; Jakoi, L.; Timmers, C.; Buerki, R.; Leone, G. Identification and characterization of E2F7, a novel mammalian E2F family member capable of blocking cellular proliferation. J. Biol. Chem. 2003, 278, 42041–42049. [Google Scholar] [CrossRef]
- Christensen, J.; Cloos, P.; Toftegaard, U.; Klinkenberg, D.; Bracken, A.P.; Trinh, E.; Heeran, M.; Di Stefano, L.; Helin, K. Characterization of E2F8, a novel E2F-like cell-cycle regulated repressor of E2F-activated transcription. Nucleic Acids Res. 2005, 33, 5458–5470. [Google Scholar] [CrossRef]
- Lammens, T.; Li, J.; Leone, G.; De Veylder, L. Atypical E2Fs: New players in the E2F transcription factor family. Trends Cell Biol. 2009, 19, 111–118. [Google Scholar] [CrossRef]
- Magae, J.; Wu, C.L.; Illenye, S.; Harlow, E.; Heintz, N.H. Nuclear localization of DP and E2F transcription factors by heterodimeric partners and retinoblastoma protein family members. J. Cell Sci. 1996, 109 Pt 7, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.; Moroni, M.C.; Vigo, E.; Petersen, B.O.; Bartek, J.; Helin, K. Induction of S-phase entry by E2F transcription factors depends on their nuclear localization. Mol. Cell. Biol. 1997, 17, 5508–5520. [Google Scholar] [CrossRef] [PubMed]
- Krek, W.; Ewen, M.E.; Shirodkar, S.; Arany, Z.; Kaelin, W.G., Jr.; Livingston, D.M. Negative regulation of the growth-promoting transcription factor E2F-1 by a stably bound cyclin A-dependent protein kinase. Cell 1994, 78, 161–172. [Google Scholar] [CrossRef]
- Krek, W.; Xu, G.; Livingston, D.M. Cyclin A-kinase regulation of E2F-1 DNA binding function underlies suppression of an S phase checkpoint. Cell 1995, 83, 1149–1158. [Google Scholar] [CrossRef]
- Verona, R.; Moberg, K.; Estes, S.; Starz, M.; Vernon, J.P.; Lees, J.A. E2F activity is regulated by cell cycle-dependent changes in subcellular localization. Mol. Cell. Biol. 1997, 17, 7268–7282. [Google Scholar] [CrossRef] [PubMed]
- Gaubatz, S.; Lees, J.A.; Lindeman, G.J.; Livingston, D.M. E2F4 is exported from the nucleus in a CRM1-dependent manner. Mol. Cell. Biol. 2001, 21, 1384–1392. [Google Scholar] [CrossRef]
- Helin, K.; Wu, C.L.; Fattaey, A.R.; Lees, J.A.; Dynlacht, B.D.; Ngwu, C.; Harlow, E. Heterodimerization of the transcription factors E2F-1 and DP-1 leads to cooperative trans-activation. Genes Dev. 1993, 7, 1850–1861. [Google Scholar] [CrossRef]
- Di Stefano, L.; Jensen, M.R.; Helin, K. E2F7, a novel E2F featuring DP-independent repression of a subset of E2F-regulated genes. EMBO J. 2003, 22, 6289–6298. [Google Scholar] [CrossRef]
- Maiti, B.; Li, J.; de Bruin, A.; Gordon, F.; Timmers, C.; Opavsky, R.; Patil, K.; Tuttle, J.; Cleghorn, W.; Leone, G. Cloning and characterization of mouse E2F8, a novel mammalian E2F family member capable of blocking cellular proliferation. J. Biol. Chem. 2005, 280, 18211–18220. [Google Scholar] [CrossRef]
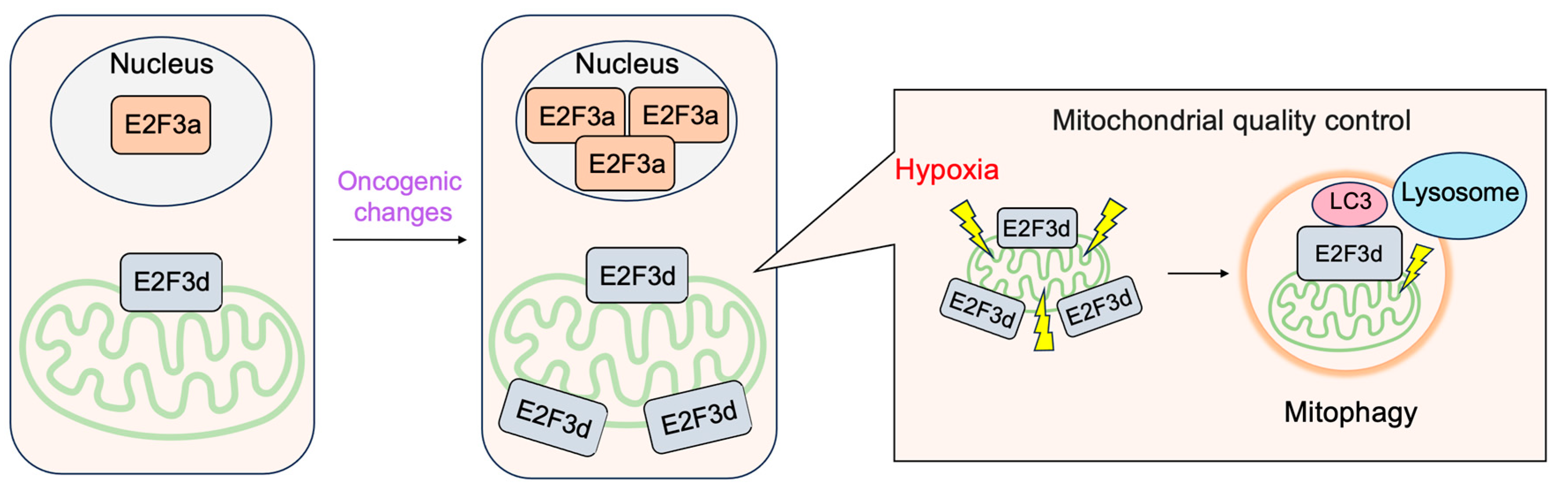
- Araki, K.; Kawauchi, K.; Sugimoto, W.; Tsuda, D.; Oda, H.; Yoshida, R.; Ohtani, K. Mitochondrial protein E2F3d, a distinctive E2F3 product, mediates hypoxia-induced mitophagy in cancer cells. Commun. Biol. 2019, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Dryja, T.P. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986, 323, 643–646. [Google Scholar] [CrossRef]
- Chellappan, S.P.; Hiebert, S.; Mudryj, M.; Horowitz, J.M.; Nevins, J.R. The E2F transcription factor is a cellular target for the RB protein. Cell 1991, 65, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Sadasivam, S.; DeCaprio, J.A. The DREAM complex: Master coordinator of cell cycle-dependent gene expression. Nat. Rev. Cancer 2013, 13, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Muller, G.A. Cell cycle transcription control: DREAM/MuvB and RB-E2F complexes. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 638–662. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Schade, A.E.; Oser, M.G.; Nicholson, H.E.; DeCaprio, J.A. Cyclin D-CDK4 relieves cooperative repression of proliferation and cell cycle gene expression by DREAM and RB. Oncogene 2019, 38, 4962–4976. [Google Scholar] [CrossRef]
- Kato, J.; Matsushime, H.; Hiebert, S.W.; Ewen, M.E.; Sherr, C.J. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993, 7, 331–342. [Google Scholar] [CrossRef]
- Ewen, M.E.; Sluss, H.K.; Sherr, C.J.; Matsushime, H.; Kato, J.; Livingston, D.M. Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell 1993, 73, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, K.; DeGregori, J.; Nevins, J.R. Regulation of the cyclin E gene by transcription factor E2F1. Proc. Natl. Acad. Sci. USA 1995, 92, 12146–12150. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Eaton, E.N.; Picon, M.; Roberts, J.M.; Lundberg, A.S.; Gifford, A.; Sardet, C.; Weinberg, R.A. Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene 1996, 12, 1173–1180. [Google Scholar] [PubMed]
- Ohtani, K.; Tsujimoto, A.; Ikeda, M.; Nakamura, M. Regulation of cell growth-dependent expression of mammalian CDC6 gene by the cell cycle transcription factor E2F. Oncogene 1998, 17, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; DeGregori, J.; Shohet, R.; Leone, G.; Stillman, B.; Nevins, J.R.; Williams, R.S. Cdc6 is regulated by E2F and is essential for DNA replication in mammalian cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3603–3608. [Google Scholar] [CrossRef] [PubMed]
- Rubin, S.M.; Sage, J.; Skotheim, J.M. Integrating Old and New Paradigms of G1/S Control. Mol. Cell 2020, 80, 183–192. [Google Scholar] [CrossRef]
- Yao, G.; Lee, T.J.; Mori, S.; Nevins, J.R.; You, L. A bistable Rb-E2F switch underlies the restriction point. Nat. Cell Biol. 2008, 10, 476–482. [Google Scholar] [CrossRef]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell. Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef]
- Stallaert, W.; Kedziora, K.M.; Chao, H.X.; Purvis, J.E. Bistable switches as integrators and actuators during cell cycle progression. FEBS Lett. 2019, 593, 2805–2816. [Google Scholar] [CrossRef]
- Gill, R.M.; Hamel, P.A.; Zhe, J.; Zacksenhaus, E.; Gallie, B.L.; Phillips, R.A. Characterization of the human RB1 promoter and of elements involved in transcriptional regulation. Cell Growth Differ. 1994, 5, 467–474. [Google Scholar]
- Shan, B.; Chang, C.Y.; Jones, D.; Lee, W.H. The transcription factor E2F-1 mediates the autoregulation of RB gene expression. Mol. Cell. Biol. 1994, 14, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, D.L.; Ngai, L.K.; Roake, C.M.; Viatour, P.; Thangavel, C.; Ho, V.M.; Knudsen, E.S.; Sage, J. Regulation of RB transcription in vivo by RB family members. Mol. Cell. Biol. 2010, 30, 1729–1745. [Google Scholar] [CrossRef] [PubMed]
- Henley, S.A.; Dick, F.A. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.A.; Jakoi, L.; Nevins, J.R. A unique role for the Rb protein in controlling E2F accumulation during cell growth and differentiation. Proc. Natl. Acad. Sci. USA 1996, 93, 3215–3220. [Google Scholar] [CrossRef] [PubMed]
- Clijsters, L.; Hoencamp, C.; Calis, J.J.A.; Marzio, A.; Handgraaf, S.M.; Cuitino, M.C.; Rosenberg, B.R.; Leone, G.; Pagano, M. Cyclin F Controls Cell-Cycle Transcriptional Outputs by Directing the Degradation of the Three Activator E2Fs. Mol. Cell 2019, 74, 1264–1277.e1267. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, M.J.; Enrico, T.P.; Mouery, R.D.; Wasserman, D.; Nachum, S.; Tzur, A. Complex Cartography: Regulation of E2F Transcription Factors by Cyclin F and Ubiquitin. Trends Cell Biol. 2020, 30, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Burdova, K.; Yang, H.; Faedda, R.; Hume, S.; Chauhan, J.; Ebner, D.; Kessler, B.M.; Vendrell, I.; Drewry, D.H.; Wells, C.I.; et al. E2F1 proteolysis via SCF-cyclin F underlies synthetic lethality between cyclin F loss and Chk1 inhibition. EMBO J. 2019, 38, e101443. [Google Scholar] [CrossRef] [PubMed]
- Grana, X.; Reddy, E.P. Cell cycle control in mammalian cells: Role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene 1995, 11, 211–219. [Google Scholar]
- Boekhout, M.; Yuan, R.; Wondergem, A.P.; Segeren, H.A.; van Liere, E.A.; Awol, N.; Jansen, I.; Wolthuis, R.M.; de Bruin, A.; Westendorp, B. Feedback regulation between atypical E2Fs and APC/CCdh1 coordinates cell cycle progression. EMBO Rep. 2016, 17, 414–427. [Google Scholar] [CrossRef]
- Nevins, J.R. The Rb/E2F pathway and cancer. Hum. Mol. Genet. 2001, 10, 699–703. [Google Scholar] [CrossRef]
- Lee, W.H.; Shew, J.Y.; Hong, F.D.; Sery, T.W.; Donoso, L.A.; Young, L.J.; Bookstein, R.; Lee, E.Y. The retinoblastoma susceptibility gene encodes a nuclear phosphoprotein associated with DNA binding activity. Nature 1987, 329, 642–645. [Google Scholar] [CrossRef]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.; Peters, G. Cyclin D1 as a cellular proto-oncogene. Semin. Cancer Biol. 1995, 6, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of Oncogenes and Tumor-suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020, 40, 6009–6015. [Google Scholar] [CrossRef]
- Punekar, S.R.; Velcheti, V.; Neel, B.G.; Wong, K.K. The current state of the art and future trends in RAS-targeted cancer therapies. Nat. Rev. Clin. Oncol. 2022, 19, 637–655. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene—The grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.A.; Ryan, K.M. Life and death decisions by E2F-1. Cell Death Differ. 2004, 11, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Brauer, C.; Brauer, P.M.; Chen, Y.J.; Pimkina, J.; Raychaudhuri, P. Tumor suppression by ARF: Gatekeeper and caretaker. Cell Cycle 2010, 9, 86–89. [Google Scholar] [CrossRef]
- Ruas, M.; Peters, G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim. Biophys. Acta 1998, 1378, F115–F177. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Tumor surveillance via the ARF-p53 pathway. Genes Dev. 1998, 12, 2984–2991. [Google Scholar] [CrossRef]
- Sharpless, N.E.; DePinho, R.A. The INK4A/ARF locus and its two gene products. Curr. Opin. Genet Dev. 1999, 9, 22–30. [Google Scholar] [CrossRef]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gyuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 1993, 362, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Midgley, C.A.; Lane, D.P. p53 protein stability in tumour cells is not determined by mutation but is dependent on Mdm2 binding. Oncogene 1997, 15, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.; Dobbelstein, M.; Freedman, D.A.; Shenk, T.; Levine, A.J. Nucleo-cytoplasmic shuttling of the hdm2 oncoprotein regulates the levels of the p53 protein via a pathway used by the human immunodeficiency virus rev protein. EMBO J. 1998, 17, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Kulikov, R.; Letienne, J.; Kaur, M.; Grossman, S.R.; Arts, J.; Blattner, C. Mdm2 facilitates the association of p53 with the proteasome. Proc. Natl. Acad. Sci. USA 2010, 107, 10038–10043. [Google Scholar] [CrossRef]
- Tao, W.; Levine, A.J. P19(ARF) stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc. Natl. Acad. Sci. USA 1999, 96, 6937–6941. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiong, Y. Mutations in human ARF exon 2 disrupt its nucleolar localization and impair its ability to block nuclear export of MDM2 and p53. Mol. Cell 1999, 3, 579–591. [Google Scholar] [CrossRef]
- Rizos, H.; Darmanian, A.P.; Mann, G.J.; Kefford, R.F. Two arginine rich domains in the p14ARF tumour suppressor mediate nucleolar localization. Oncogene 2000, 19, 2978–2985. [Google Scholar] [CrossRef]
- Pomerantz, J.; Schreiber-Agus, N.; Liegeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.W.; et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef]
- Kamijo, T.; Weber, J.D.; Zambetti, G.; Zindy, F.; Roussel, M.F.; Sherr, C.J. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc. Natl. Acad. Sci. USA 1998, 95, 8292–8297. [Google Scholar] [CrossRef]
- Lohrum, M.A.; Ashcroft, M.; Kubbutat, M.H.; Vousden, K.H. Contribution of two independent MDM2-binding domains in p14(ARF) to p53 stabilization. Curr. Biol. 2000, 10, 539–542. [Google Scholar] [CrossRef]
- Weber, J.D.; Kuo, M.L.; Bothner, B.; DiGiammarino, E.L.; Kriwacki, R.W.; Roussel, M.F.; Sherr, C.J. Cooperative signals governing ARF-mdm2 interaction and nucleolar localization of the complex. Mol. Cell. Biol. 2000, 20, 2517–2528. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.D.; Taylor, L.J.; Roussel, M.F.; Sherr, C.J.; Bar-Sagi, D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat. Cell Biol. 1999, 1, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Llanos, S.; Clark, P.A.; Rowe, J.; Peters, G. Stabilization of p53 by p14ARF without relocation of MDM2 to the nucleolus. Nat. Cell Biol. 2001, 3, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Yasuda, H. Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 1999, 18, 22–27. [Google Scholar] [CrossRef]
- Midgley, C.A.; Desterro, J.M.; Saville, M.K.; Howard, S.; Sparks, A.; Hay, R.T.; Lane, D.P. An N-terminal p14ARF peptide blocks Mdm2-dependent ubiquitination in vitro and can activate p53 in vivo. Oncogene 2000, 19, 2312–2323. [Google Scholar] [CrossRef]
- Barak, Y.; Juven, T.; Haffner, R.; Oren, M. mdm2 expression is induced by wild type p53 activity. EMBO J. 1993, 12, 461–468. [Google Scholar] [CrossRef]
- Zeng, Y.; Kotake, Y.; Pei, X.H.; Smith, M.D.; Xiong, Y. p53 binds to and is required for the repression of Arf tumor suppressor by HDAC and polycomb. Cancer Res. 2011, 71, 2781–2792. [Google Scholar] [CrossRef]
- Zacchi, P.; Gostissa, M.; Uchida, T.; Salvagno, C.; Avolio, F.; Volinia, S.; Ronai, Z.; Blandino, G.; Schneider, C.; Del Sal, G. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature 2002, 419, 853–857. [Google Scholar] [CrossRef]
- Zheng, H.; You, H.; Zhou, X.Z.; Murray, S.A.; Uchida, T.; Wulf, G.; Gu, L.; Tang, X.; Lu, K.P.; Xiao, Z.X. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 2002, 419, 849–853. [Google Scholar] [CrossRef]
- el-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef]
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Parant, J.; Chavez-Reyes, A.; Little, N.A.; Yan, W.; Reinke, V.; Jochemsen, A.G.; Lozano, G. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat. Genet. 2001, 29, 92–95. [Google Scholar] [CrossRef]
- Kawai, H.; Lopez-Pajares, V.; Kim, M.M.; Wiederschain, D.; Yuan, Z.M. RING domain-mediated interaction is a requirement for MDM2’s E3 ligase activity. Cancer Res. 2007, 67, 6026–6030. [Google Scholar] [CrossRef]
- Yang, J.; Jin, A.; Han, J.; Chen, X.; Zheng, J.; Zhang, Y. MDMX Recruits UbcH5c to Facilitate MDM2 E3 Ligase Activity and Subsequent p53 Degradation In Vivo. Cancer Res. 2021, 81, 898–909. [Google Scholar] [CrossRef]
- Huang, L.; Yan, Z.; Liao, X.; Li, Y.; Yang, J.; Wang, Z.G.; Zuo, Y.; Kawai, H.; Shadfan, M.; Ganapathy, S.; et al. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 12001–12006. [Google Scholar] [CrossRef] [PubMed]
- Szwarc, M.M.; Guarnieri, A.L.; Joshi, M.; Duc, H.N.; Laird, M.C.; Pandey, A.; Khanal, S.; Dohm, E.; Bui, A.K.; Sullivan, K.D.; et al. FAM193A is a positive regulator of p53 activity. Cell Rep. 2023, 42, 112230. [Google Scholar] [CrossRef]
- Li, M.; Chen, D.; Shiloh, A.; Luo, J.; Nikolaev, A.Y.; Qin, J.; Gu, W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 2002, 416, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Brooks, C.L.; Kon, N.; Gu, W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell 2004, 13, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Cummins, J.M.; Rago, C.; Kohli, M.; Kinzler, K.W.; Lengauer, C.; Vogelstein, B. Tumour suppression: Disruption of HAUSP gene stabilizes p53. Nature 2004, 428, 1–2. [Google Scholar] [CrossRef]
- Brooks, C.L.; Li, M.; Hu, M.; Shi, Y.; Gu, W. The p53--Mdm2--HAUSP complex is involved in p53 stabilization by HAUSP. Oncogene 2007, 26, 7262–7266. [Google Scholar] [CrossRef] [PubMed]
- Xirodimas, D.P.; Chisholm, J.; Desterro, J.M.; Lane, D.P.; Hay, R.T. P14ARF promotes accumulation of SUMO-1 conjugated (H)Mdm2. FEBS Lett. 2002, 528, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, J. MDM2-ARF complex regulates p53 sumoylation. Oncogene 2003, 22, 5348–5357. [Google Scholar] [CrossRef]
- Stindt, M.H.; Carter, S.; Vigneron, A.M.; Ryan, K.M.; Vousden, K.H. MDM2 promotes SUMO-2/3 modification of p53 to modulate transcriptional activity. Cell Cycle 2011, 10, 3176–3188. [Google Scholar] [CrossRef]
- Ivanschitz, L.; Takahashi, Y.; Jollivet, F.; Ayrault, O.; Le Bras, M.; de The, H. PML IV/ARF interaction enhances p53 SUMO-1 conjugation, activation, and senescence. Proc. Natl. Acad. Sci. USA 2015, 112, 14278–14283. [Google Scholar] [CrossRef]
- Xirodimas, D.P.; Saville, M.K.; Bourdon, J.C.; Hay, R.T.; Lane, D.P. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell 2004, 118, 83–97. [Google Scholar] [CrossRef]
- Basak, S.; Jacobs, S.B.; Krieg, A.J.; Pathak, N.; Zeng, Q.; Kaldis, P.; Giaccia, A.J.; Attardi, L.D. The metastasis-associated gene Prl-3 is a p53 target involved in cell-cycle regulation. Mol. Cell 2008, 30, 303–314. [Google Scholar] [CrossRef]
- Doumont, G.; Martoriati, A.; Marine, J.C. PTPRV is a key mediator of p53-induced cell cycle exit. Cell Cycle 2005, 4, 1703–1705. [Google Scholar] [CrossRef]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef]
- Hermeking, H.; Lengauer, C.; Polyak, K.; He, T.C.; Zhang, L.; Thiagalingam, S.; Kinzler, K.W.; Vogelstein, B. 14-3-3sigma is a p53-regulated inhibitor of G2/M progression. Mol. Cell 1997, 1, 3–11. [Google Scholar] [CrossRef]
- Chan, T.A.; Hermeking, H.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B. 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature 1999, 401, 616–620. [Google Scholar] [CrossRef]
- Wang, X.W.; Zhan, Q.; Coursen, J.D.; Khan, M.A.; Kontny, H.U.; Yu, L.; Hollander, M.C.; O’Connor, P.M.; Fornace, A.J., Jr.; Harris, C.C. GADD45 induction of a G2/M cell cycle checkpoint. Proc. Natl. Acad. Sci. USA 1999, 96, 3706–3711. [Google Scholar] [CrossRef]
- Ohki, R.; Nemoto, J.; Murasawa, H.; Oda, E.; Inazawa, J.; Tanaka, N.; Taniguchi, T. Reprimo, a new candidate mediator of the p53-mediated cell cycle arrest at the G2 phase. J. Biol. Chem. 2000, 275, 22627–22630. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T.W. Regulation of PTEN transcription by p53. Mol. Cell 2001, 8, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Arakawa, H.; Tanaka, T.; Matsuda, K.; Tanikawa, C.; Mori, T.; Nishimori, H.; Tamai, K.; Tokino, T.; Nakamura, Y.; et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 2000, 102, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Smeenk, L.; van Heeringen, S.J.; Koeppel, M.; Gilbert, B.; Janssen-Megens, E.; Stunnenberg, H.G.; Lohrum, M. Role of p53 serine 46 in p53 target gene regulation. PLoS ONE 2011, 6, e17574. [Google Scholar] [CrossRef] [PubMed]
- D’Orazi, G.; Cecchinelli, B.; Bruno, T.; Manni, I.; Higashimoto, Y.; Saito, S.; Gostissa, M.; Coen, S.; Marchetti, A.; Del Sal, G.; et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat. Cell Biol. 2002, 4, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Okamura, S.; Arakawa, H.; Tanaka, T.; Nakanishi, H.; Ng, C.C.; Taya, Y.; Monden, M.; Nakamura, Y. p53DINP1, a p53-inducible gene, regulates p53-dependent apoptosis. Mol. Cell 2001, 8, 85–94. [Google Scholar] [CrossRef]
- Yoshida, K.; Liu, H.; Miki, Y. Protein kinase C delta regulates Ser46 phosphorylation of p53 tumor suppressor in the apoptotic response to DNA damage. J. Biol. Chem. 2006, 281, 5734–5740. [Google Scholar] [CrossRef]
- Taira, N.; Nihira, K.; Yamaguchi, T.; Miki, Y.; Yoshida, K. DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol. Cell 2007, 25, 725–738. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef]
- Kaloni, D.; Diepstraten, S.T.; Strasser, A.; Kelly, G.L. BCL-2 protein family: Attractive targets for cancer therapy. Apoptosis 2023, 28, 20–38. [Google Scholar] [CrossRef]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, X.; Dantas Machado, A.C.; Ding, Y.; Chen, Z.; Qin, P.Z.; Rohs, R.; Chen, L. Structure of p53 binding to the BAX response element reveals DNA unwinding and compression to accommodate base-pair insertion. Nucleic Acids Res. 2013, 41, 8368–8376. [Google Scholar] [CrossRef]
- Moroni, M.C.; Hickman, E.S.; Lazzerini Denchi, E.; Caprara, G.; Colli, E.; Cecconi, F.; Muller, H.; Helin, K. Apaf-1 is a transcriptional target for E2F and p53. Nat. Cell Biol. 2001, 3, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Robles, A.I.; Bemmels, N.A.; Foraker, A.B.; Harris, C.C. APAF-1 is a transcriptional target of p53 in DNA damage-induced apoptosis. Cancer Res. 2001, 61, 6660–6664. [Google Scholar] [PubMed]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, V.; Jung, P.; Verdoodt, B.; Lodygin, D.; Epanchintsev, A.; Menssen, A.; Meister, G.; Hermeking, H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 2007, 6, 1586–1593. [Google Scholar] [CrossRef]
- Bommer, G.T.; Gerin, I.; Feng, Y.; Kaczorowski, A.J.; Kuick, R.; Love, R.E.; Zhai, Y.; Giordano, T.J.; Qin, Z.S.; Moore, B.B.; et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol. 2007, 17, 1298–1307. [Google Scholar] [CrossRef]
- Xia, L.; Zhang, D.; Du, R.; Pan, Y.; Zhao, L.; Sun, S.; Hong, L.; Liu, J.; Fan, D. miR-15b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int. J. Cancer 2008, 123, 372–379. [Google Scholar] [CrossRef]
- Xu, K.; Liang, X.; Cui, D.; Wu, Y.; Shi, W.; Liu, J. miR-1915 inhibits Bcl-2 to modulate multidrug resistance by increasing drug-sensitivity in human colorectal carcinoma cells. Mol. Carcinog. 2013, 52, 70–78. [Google Scholar] [CrossRef]
- Nakazawa, K.; Dashzeveg, N.; Yoshida, K. Tumor suppressor p53 induces miR-1915 processing to inhibit Bcl-2 in the apoptotic response to DNA damage. FEBS J. 2014, 281, 2937–2944. [Google Scholar] [CrossRef]
- Lin, X.; Guan, H.; Huang, Z.; Liu, J.; Li, H.; Wei, G.; Cao, X.; Li, Y. Downregulation of Bcl-2 expression by miR-34a mediates palmitate-induced Min6 cells apoptosis. J. Diabetes Res. 2014, 2014, 258695. [Google Scholar] [CrossRef]
- Li, W.J.; Wang, Y.; Liu, R.; Kasinski, A.L.; Shen, H.; Slack, F.J.; Tang, D.G. MicroRNA-34a: Potent Tumor Suppressor, Cancer Stem Cell Inhibitor, and Potential Anticancer Therapeutic. Front. Cell Dev. Biol. 2021, 9, 640587. [Google Scholar] [CrossRef] [PubMed]
- Taira, N.; Yamaguchi, T.; Kimura, J.; Lu, Z.G.; Fukuda, S.; Higashiyama, S.; Ono, M.; Yoshida, K. Induction of amphiregulin by p53 promotes apoptosis via control of microRNA biogenesis in response to DNA damage. Proc. Natl. Acad. Sci. USA 2014, 111, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Christophorou, M.A.; Ringshausen, I.; Finch, A.J.; Swigart, L.B.; Evan, G.I. The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature 2006, 443, 214–217. [Google Scholar] [CrossRef]
- Hinkal, G.; Parikh, N.; Donehower, L.A. Timed somatic deletion of p53 in mice reveals age-associated differences in tumor progression. PLoS ONE 2009, 4, e6654. [Google Scholar] [CrossRef]
- Efeyan, A.; Garcia-Cao, I.; Herranz, D.; Velasco-Miguel, S.; Serrano, M. Tumour biology: Policing of oncogene activity by p53. Nature 2006, 443, 159. [Google Scholar] [CrossRef]
- Narasimha, A.M.; Kaulich, M.; Shapiro, G.S.; Choi, Y.J.; Sicinski, P.; Dowdy, S.F. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife 2014, 3, e02872. [Google Scholar] [CrossRef] [PubMed]
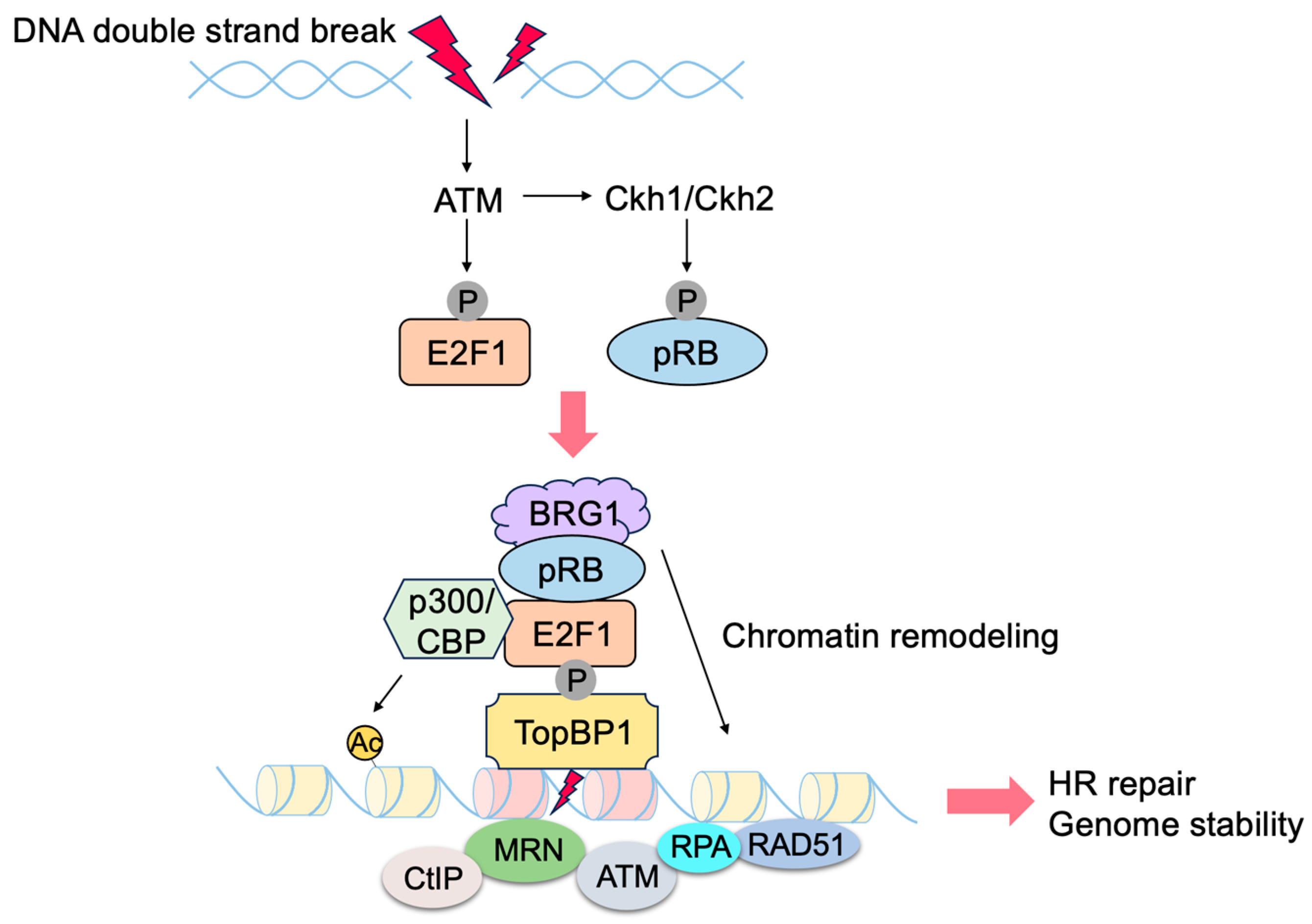
- Biswas, A.K.; Johnson, D.G. Transcriptional and nontranscriptional functions of E2F1 in response to DNA damage. Cancer Res. 2012, 72, 13–17. [Google Scholar] [CrossRef]
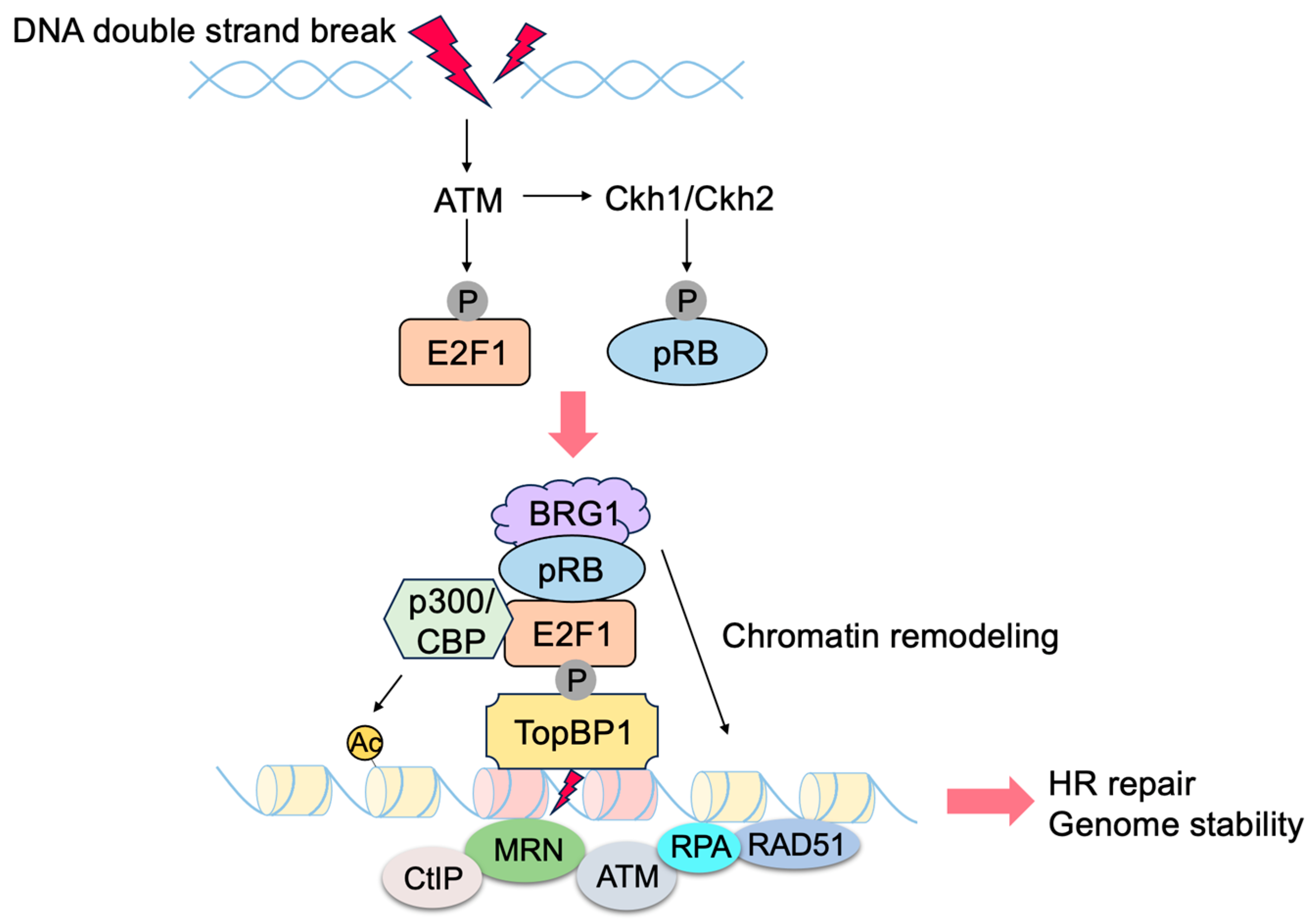
- Velez-Cruz, R.; Johnson, D.G. The Retinoblastoma (RB) Tumor Suppressor: Pushing Back against Genome Instability on Multiple Fronts. Int. J. Mol. Sci. 2017, 18, 1776. [Google Scholar] [CrossRef]
- Manickavinayaham, S.; Dennehey, B.K.; Johnson, D.G. Direct Regulation of DNA Repair by E2F and RB in Mammals and Plants: Core Function or Convergent Evolution? Cancers 2021, 13, 934. [Google Scholar] [CrossRef]
- Lin, W.C.; Lin, F.T.; Nevins, J.R. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 2001, 15, 1833–1844. [Google Scholar]
- Liu, K.; Lin, F.T.; Ruppert, J.M.; Lin, W.C. Regulation of E2F1 by BRCT domain-containing protein TopBP1. Mol. Cell. Biol. 2003, 23, 3287–3304. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Kitagawa, M.; Taya, Y. Phosphorylation of pRB at Ser612 by Chk1/2 leads to a complex between pRB and E2F-1 after DNA damage. EMBO J. 2007, 26, 2083–2093. [Google Scholar] [CrossRef]
- Velez-Cruz, R.; Manickavinayaham, S.; Biswas, A.K.; Clary, R.W.; Premkumar, T.; Cole, F.; Johnson, D.G. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev. 2016, 30, 2500–2512. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yam, J.C.; Tham, C.C.; Pang, C.P.; Chu, W.K. RB Regulates DNA Double Strand Break Repair Pathway Choice by Mediating CtIP Dependent End Resection. Int. J. Mol. Sci. 2020, 21, 9176. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhu, F.; Weaks, R.L.; Biswas, A.K.; Guo, R.; Li, Y.; Johnson, D.G. E2F1 promotes the recruitment of DNA repair factors to sites of DNA double-strand breaks. Cell Cycle 2011, 10, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Manickavinayaham, S.; Velez-Cruz, R.; Biswas, A.K.; Bedford, E.; Klein, B.J.; Kutateladze, T.G.; Liu, B.; Bedford, M.T.; Johnson, D.G. E2F1 acetylation directs p300/CBP-mediated histone acetylation at DNA double-strand breaks to facilitate repair. Nat. Commun. 2019, 10, 4951. [Google Scholar] [CrossRef]
- Choi, E.H.; Kim, K.P. E2F1 facilitates DNA break repair by localizing to break sites and enhancing the expression of homologous recombination factors. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Guo, R.; Chen, J.; Zhu, F.; Biswas, A.K.; Berton, T.R.; Mitchell, D.L.; Johnson, D.G. E2F1 localizes to sites of UV-induced DNA damage to enhance nucleotide excision repair. J. Biol. Chem. 2010, 285, 19308–19315. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.K.; Mitchell, D.L.; Johnson, D.G. E2F1 responds to ultraviolet radiation by directly stimulating DNA repair and suppressing carcinogenesis. Cancer Res. 2014, 74, 3369–3377. [Google Scholar] [CrossRef]
- Stevaux, O.; Dyson, N.J. A revised picture of the E2F transcriptional network and RB function. Curr. Opin. Cell Biol. 2002, 14, 684–691. [Google Scholar] [CrossRef]
- Manning, A.L.; Dyson, N.J. pRB, a tumor suppressor with a stabilizing presence. Trends Cell Biol. 2011, 21, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.L.; Dyson, N.J. RB: Mitotic implications of a tumour suppressor. Nat. Rev. Cancer 2012, 12, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Talluri, S.; Dick, F.A. Regulation of transcription and chromatin structure by pRB: Here, there and everywhere. Cell Cycle 2012, 11, 3189–3198. [Google Scholar] [CrossRef] [PubMed]
- Dick, F.A.; Rubin, S.M. Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell. Biol. 2013, 14, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Popov, B.; Petrov, N. pRb-E2F signaling in life of mesenchymal stem cells: Cell cycle, cell fate, and cell differentiation. Genes Dis. 2014, 1, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Indovina, P.; Pentimalli, F.; Casini, N.; Vocca, I.; Giordano, A. RB1 dual role in proliferation and apoptosis: Cell fate control and implications for cancer therapy. Oncotarget 2015, 6, 17873–17890. [Google Scholar] [CrossRef]
- Dyson, N.J. RB1: A prototype tumor suppressor and an enigma. Genes Dev. 2016, 30, 1492–1502. [Google Scholar] [CrossRef]
- Dick, F.A.; Goodrich, D.W.; Sage, J.; Dyson, N.J. Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 2018, 18, 442–451. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Pruitt, S.C.; Hershberger, P.A.; Witkiewicz, A.K.; Goodrich, D.W. Cell Cycle and Beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy. Trends Cancer 2019, 5, 308–324. [Google Scholar] [CrossRef]
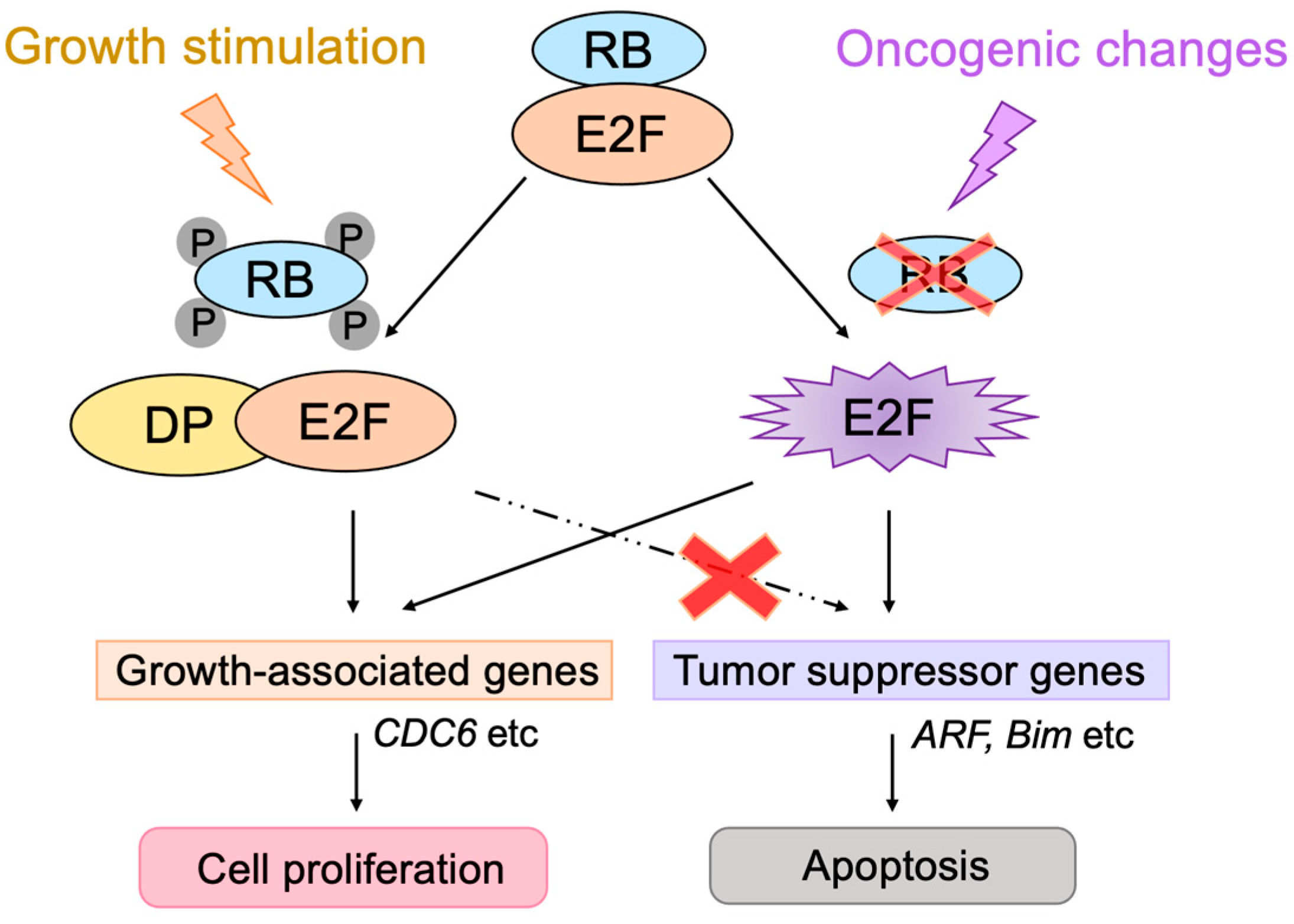
- Komori, H.; Enomoto, M.; Nakamura, M.; Iwanaga, R.; Ohtani, K. Distinct E2F-mediated transcriptional program regulates p14ARF gene expression. EMBO J. 2005, 24, 3724–3736. [Google Scholar] [CrossRef] [PubMed]
- Komori, H.; Goto, Y.; Kurayoshi, K.; Ozono, E.; Iwanaga, R.; Bradford, A.P.; Araki, K.; Ohtani, K. Differential requirement for dimerization partner DP between E2F-dependent activation of tumor suppressor and growth-related genes. Sci. Rep. 2018, 8, 8438. [Google Scholar] [CrossRef] [PubMed]
- Ozono, E.; Komori, H.; Iwanaga, R.; Ikeda, M.A.; Iseki, S.; Ohtani, K. E2F-like elements in p27(Kip1) promoter specifically sense deregulated E2F activity. Genes Cells 2009, 14, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Ozono, E.; Komori, H.; Iwanaga, R.; Tanaka, T.; Sakae, T.; Kitamura, H.; Yamaoka, S.; Ohtani, K. Tumor suppressor TAp73 gene specifically responds to deregulated E2F activity in human normal fibroblasts. Genes Cells 2012, 17, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Ozono, E.; Iwanaga, R.; Bradford, A.P.; Okuno, J.; Shimizu, E.; Kurayoshi, K.; Kugawa, K.; Toh, H.; Ohtani, K. Identification of novel target genes specifically activated by deregulated E2F in human normal fibroblasts. Genes Cells 2015, 20, 739–757. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.; Marin, M.C.; Phillips, A.C.; Seelan, R.S.; Smith, D.I.; Liu, W.; Flores, E.R.; Tsai, K.Y.; Jacks, T.; Vousden, K.H.; et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature 2000, 407, 645–648. [Google Scholar] [CrossRef]
- Stiewe, T.; Putzer, B.M. Role of the p53-homologue p73 in E2F1-induced apoptosis. Nat. Genet. 2000, 26, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Wang, J.Y. Targeting the RB-pathway in cancer therapy. Clin. Cancer Res. 2010, 16, 1094–1099. [Google Scholar] [CrossRef]
- Laine, A.; Westermarck, J. Molecular pathways: Harnessing E2F1 regulation for prosenescence therapy in p53-defective cancer cells. Clin. Cancer Res. 2014, 20, 3644–3650. [Google Scholar] [CrossRef]
- Tsukuda, K.; Wiewrodt, R.; Molnar-Kimber, K.; Jovanovic, V.P.; Amin, K.M. An E2F-responsive replication-selective adenovirus targeted to the defective cell cycle in cancer cells: Potent antitumoral efficacy but no toxicity to normal cell. Cancer Res. 2002, 62, 3438–3447. [Google Scholar] [PubMed]
- Dubensky, T.W., Jr. (Re-)Engineering tumor cell-selective replicating adenoviruses: A step in the right direction toward systemic therapy for metastatic disease. Cancer Cell 2002, 1, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Jakubczak, J.L.; Ryan, P.; Gorziglia, M.; Clarke, L.; Hawkins, L.K.; Hay, C.; Huang, Y.; Kaloss, M.; Marinov, A.; Phipps, S.; et al. An oncolytic adenovirus selective for retinoblastoma tumor suppressor protein pathway-defective tumors: Dependence on E1A, the E2F-1 promoter, and viral replication for selectivity and efficacy. Cancer Res. 2003, 63, 1490–1499. [Google Scholar] [PubMed]
- Kurayoshi, K.; Ozono, E.; Iwanaga, R.; Bradford, A.P.; Komori, H.; Ohtani, K. Cancer cell specific cytotoxic gene expression mediated by ARF tumor suppressor promoter constructs. Biochem. Biophys. Res. Commun. 2014, 450, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, R.; Zhao, L.; Zhou, Y.; Shirasawa, M.; Uchida, A.; Murakawa, H.; Fikriyanti, M.; Iwanaga, R.; Bradford, A.P.; Araki, K.; et al. Deregulated E2F Activity as a Cancer-Cell Specific Therapeutic Tool. Genes 2023, 14, 393. [Google Scholar] [CrossRef]
- Hsu, J.; Sage, J. Novel functions for the transcription factor E2F4 in development and disease. Cell Cycle 2016, 15, 3183–3190. [Google Scholar] [CrossRef]
- Fontana, R.; Ranieri, M.; La Mantia, G.; Vivo, M. Dual Role of the Alternative Reading Frame ARF Protein in Cancer. Biomolecules 2019, 9, 87. [Google Scholar] [CrossRef]
- Kung, C.P.; Weber, J.D. It’s Getting Complicated—A Fresh Look at p53-MDM2-ARF Triangle in Tumorigenesis and Cancer Therapy. Front. Cell Dev. Biol. 2022, 10, 818744. [Google Scholar] [CrossRef]
- Weber, J.D.; Jeffers, J.R.; Rehg, J.E.; Randle, D.H.; Lozano, G.; Roussel, M.F.; Sherr, C.J.; Zambetti, G.P. p53-independent functions of the p19(ARF) tumor suppressor. Genes Dev. 2000, 14, 2358–2365. [Google Scholar] [CrossRef]
- Eymin, B.; Leduc, C.; Coll, J.L.; Brambilla, E.; Gazzeri, S. p14ARF induces G2 arrest and apoptosis independently of p53 leading to regression of tumours established in nude mice. Oncogene 2003, 22, 1822–1835. [Google Scholar] [CrossRef]
- Sandoval, R.; Xue, J.; Pilkinton, M.; Salvi, D.; Kiyokawa, H.; Colamonici, O.R. Different requirements for the cytostatic and apoptotic effects of type I interferons. Induction of apoptosis requires ARF but not p53 in osteosarcoma cell lines. J. Biol. Chem. 2004, 279, 32275–32280. [Google Scholar] [CrossRef]
- Muniz, V.P.; Barnes, J.M.; Paliwal, S.; Zhang, X.; Tang, X.; Chen, S.; Zamba, K.D.; Cullen, J.J.; Meyerholz, D.K.; Meyers, S.; et al. The ARF tumor suppressor inhibits tumor cell colonization independent of p53 in a novel mouse model of pancreatic ductal adenocarcinoma metastasis. Mol. Cancer Res. 2011, 9, 867–877. [Google Scholar] [CrossRef]
- Sugimoto, M.; Kuo, M.L.; Roussel, M.F.; Sherr, C.J. Nucleolar Arf tumor suppressor inhibits ribosomal RNA processing. Mol. Cell 2003, 11, 415–424. [Google Scholar] [CrossRef]
- Ayrault, O.; Andrique, L.; Larsen, C.J.; Seite, P. Human Arf tumor suppressor specifically interacts with chromatin containing the promoter of rRNA genes. Oncogene 2004, 23, 8097–8104. [Google Scholar] [CrossRef]
- Lessard, F.; Morin, F.; Ivanchuk, S.; Langlois, F.; Stefanovsky, V.; Rutka, J.; Moss, T. The ARF tumor suppressor controls ribosome biogenesis by regulating the RNA polymerase I transcription factor TTF-I. Mol. Cell 2010, 38, 539–550. [Google Scholar] [CrossRef]
- Brady, S.N.; Yu, Y.; Maggi, L.B., Jr.; Weber, J.D. ARF impedes NPM/B23 shuttling in an Mdm2-sensitive tumor suppressor pathway. Mol. Cell. Biol. 2004, 24, 9327–9338. [Google Scholar] [CrossRef]
- Bertwistle, D.; Sugimoto, M.; Sherr, C.J. Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Mol. Cell. Biol. 2004, 24, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.B., Jr.; Kuchenruether, M.; Dadey, D.Y.; Schwope, R.M.; Grisendi, S.; Townsend, R.R.; Pandolfi, P.P.; Weber, J.D. Nucleophosmin serves as a rate-limiting nuclear export chaperone for the Mammalian ribosome. Mol. Cell. Biol. 2008, 28, 7050–7065. [Google Scholar] [CrossRef] [PubMed]
- Moulin, S.; Llanos, S.; Kim, S.H.; Peters, G. Binding to nucleophosmin determines the localization of human and chicken ARF but not its impact on p53. Oncogene 2008, 27, 2382–2389. [Google Scholar] [CrossRef] [PubMed]
- Korgaonkar, C.; Hagen, J.; Tompkins, V.; Frazier, A.A.; Allamargot, C.; Quelle, F.W.; Quelle, D.E. Nucleophosmin (B23) targets ARF to nucleoli and inhibits its function. Mol. Cell. Biol. 2005, 25, 1258–1271. [Google Scholar] [CrossRef]
- Apicelli, A.J.; Maggi, L.B., Jr.; Hirbe, A.C.; Miceli, A.P.; Olanich, M.E.; Schulte-Winkeler, C.L.; Saporita, A.J.; Kuchenreuther, M.; Sanchez, J.; Weilbaecher, K.; et al. A non-tumor suppressor role for basal p19ARF in maintaining nucleolar structure and function. Mol. Cell. Biol. 2008, 28, 1068–1080. [Google Scholar] [CrossRef]
- Saporita, A.J.; Chang, H.C.; Winkeler, C.L.; Apicelli, A.J.; Kladney, R.D.; Wang, J.; Townsend, R.R.; Michel, L.S.; Weber, J.D. RNA helicase DDX5 is a p53-independent target of ARF that participates in ribosome biogenesis. Cancer Res. 2011, 71, 6708–6717. [Google Scholar] [CrossRef]
- Castelli, M.; Pieroni, S.; Brunacci, C.; Piobbico, D.; Bartoli, D.; Bellet, M.M.; Colombo, E.; Pelicci, P.G.; Della Fazia, M.A.; Servillo, G. Hepatocyte odd protein shuttling (HOPS) is a bridging protein in the nucleophosmin-p19 Arf network. Oncogene 2013, 32, 3350–3358. [Google Scholar] [CrossRef]
- Kuchenreuther, M.J.; Weber, J.D. The ARF tumor-suppressor controls Drosha translation to prevent Ras-driven transformation. Oncogene 2014, 33, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.B., Jr.; Winkeler, C.L.; Miceli, A.P.; Apicelli, A.J.; Brady, S.N.; Kuchenreuther, M.J.; Weber, J.D. ARF tumor suppression in the nucleolus. Biochim. Biophys. Acta 2014, 1842, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Eymin, B.; Karayan, L.; Seite, P.; Brambilla, C.; Brambilla, E.; Larsen, C.J.; Gazzeri, S. Human ARF binds E2F1 and inhibits its transcriptional activity. Oncogene 2001, 20, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.L.; Loughran, O.; La Thangue, N.B. p14(ARF) regulates E2F activity. Oncogene 2002, 21, 4220–4230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.J.; Li, W.J.; Gu, Y.Y.; Li, S.Y.; An, G.S.; Ni, J.H.; Jia, H.T. p14ARF interacts with E2F factors to form p14ARF-E2F/partner-DNA complexes repressing E2F-dependent transcription. J. Cell Biochem. 2010, 109, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Nag, A.; Raychaudhuri, P. Differential regulation of E2F1, DP1, and the E2F1/DP1 complex by ARF. Mol. Cell. Biol. 2002, 22, 8398–8408. [Google Scholar] [CrossRef]
- Datta, A.; Sen, J.; Hagen, J.; Korgaonkar, C.K.; Caffrey, M.; Quelle, D.E.; Hughes, D.E.; Ackerson, T.J.; Costa, R.H.; Raychaudhuri, P. ARF directly binds DP1: Interaction with DP1 coincides with the G1 arrest function of ARF. Mol. Cell. Biol. 2005, 25, 8024–8036. [Google Scholar] [CrossRef] [PubMed]
- Martelli, F.; Hamilton, T.; Silver, D.P.; Sharpless, N.E.; Bardeesy, N.; Rokas, M.; DePinho, R.A.; Livingston, D.M.; Grossman, S.R. p19ARF targets certain E2F species for degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 4455–4460. [Google Scholar] [CrossRef]
- Datta, A.; Nag, A.; Pan, W.; Hay, N.; Gartel, A.L.; Colamonici, O.; Mori, Y.; Raychaudhuri, P. Myc-ARF (alternate reading frame) interaction inhibits the functions of Myc. J. Biol. Chem. 2004, 279, 36698–36707. [Google Scholar] [CrossRef]
- Tago, K.; Funakoshi-Tago, M.; Itoh, H.; Furukawa, Y.; Kikuchi, J.; Kato, T.; Suzuki, K.; Yanagisawa, K. Arf tumor suppressor disrupts the oncogenic positive feedback loop including c-Myc and DDX5. Oncogene 2015, 34, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Fatyol, K.; Szalay, A.A. The p14ARF tumor suppressor protein facilitates nucleolar sequestration of hypoxia-inducible factor-1alpha (HIF-1alpha) and inhibits HIF-1-mediated transcription. J. Biol. Chem. 2001, 276, 28421–28429. [Google Scholar] [CrossRef]
- Rocha, S.; Campbell, K.J.; Perkins, N.D. p53- and Mdm2-independent repression of NF-kappa B transactivation by the ARF tumor suppressor. Mol. Cell 2003, 12, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.; Garrett, M.D.; Campbell, K.J.; Schumm, K.; Perkins, N.D. Regulation of NF-kappaB and p53 through activation of ATR and Chk1 by the ARF tumour suppressor. EMBO J. 2005, 24, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Hyder, U.; McCann, J.L.; Wang, J.; Fung, V.; Bayo, J.; D’Orso, I. The ARF tumor suppressor targets PPM1G/PP2Cgamma to counteract NF-kappaB transcription tuning cell survival and the inflammatory response. Proc. Natl. Acad. Sci. USA 2020, 117, 32594–32605. [Google Scholar] [CrossRef] [PubMed]
- Kawagishi, H.; Nakamura, H.; Maruyama, M.; Mizutani, S.; Sugimoto, K.; Takagi, M.; Sugimoto, M. ARF suppresses tumor angiogenesis through translational control of VEGFA mRNA. Cancer Res. 2010, 70, 4749–4758. [Google Scholar] [CrossRef]
- Paliwal, S.; Pande, S.; Kovi, R.C.; Sharpless, N.E.; Bardeesy, N.; Grossman, S.R. Targeting of C-terminal binding protein (CtBP) by ARF results in p53-independent apoptosis. Mol. Cell. Biol. 2006, 26, 2360–2372. [Google Scholar] [CrossRef]
- Kovi, R.C.; Paliwal, S.; Pande, S.; Grossman, S.R. An ARF/CtBP2 complex regulates BH3-only gene expression and p53-independent apoptosis. Cell Death Differ. 2010, 17, 513–521. [Google Scholar] [CrossRef]
- Lu, W.; Xie, Y.; Ma, Y.; Matusik, R.J.; Chen, Z. ARF represses androgen receptor transactivation in prostate cancer. Mol. Endocrinol. 2013, 27, 635–648. [Google Scholar] [CrossRef]
- Herkert, B.; Dwertmann, A.; Herold, S.; Abed, M.; Naud, J.F.; Finkernagel, F.; Harms, G.S.; Orian, A.; Wanzel, M.; Eilers, M. The Arf tumor suppressor protein inhibits Miz1 to suppress cell adhesion and induce apoptosis. J. Cell Biol. 2010, 188, 905–918. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Tavana, O.; Chu, B.; Erber, L.; Chen, Y.; Baer, R.; Gu, W. NRF2 Is a Major Target of ARF in p53-Independent Tumor Suppression. Mol. Cell 2017, 68, 224–232.e224. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Zhang, Y. Mitochondrial p32 is a critical mediator of ARF-induced apoptosis. Cancer Cell 2008, 13, 542–553. [Google Scholar] [CrossRef]
- Irvine, M.; Philipsz, S.; Frausto, M.; Mijatov, B.; Gallagher, S.J.; Fung, C.; Becker, T.M.; Kefford, R.F.; Rizos, H. Amino terminal hydrophobic import signals target the p14(ARF) tumor suppressor to the mitochondria. Cell Cycle 2010, 9, 829–839. [Google Scholar] [CrossRef]
- Muer, A.; Overkamp, T.; Gillissen, B.; Richter, A.; Pretzsch, T.; Milojkovic, A.; Dorken, B.; Daniel, P.T.; Hemmati, P. p14(ARF)-induced apoptosis in p53 protein-deficient cells is mediated by BH3-only protein-independent derepression of Bak protein through down-regulation of Mcl-1 and Bcl-xL proteins. J. Biol. Chem. 2012, 287, 17343–17352. [Google Scholar] [CrossRef]
- Repenning, A.; Happel, D.; Bouchard, C.; Meixner, M.; Verel-Yilmaz, Y.; Raifer, H.; Holembowski, L.; Krause, E.; Kremmer, E.; Feederle, R.; et al. PRMT1 promotes the tumor suppressor function of p14(ARF) and is indicative for pancreatic cancer prognosis. EMBO J. 2021, 40, e106777. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Abida, W.M.; Gu, W. p53-Dependent and p53-independent activation of autophagy by ARF. Cancer Res. 2008, 68, 352–357. [Google Scholar] [CrossRef]
- Pimkina, J.; Humbey, O.; Zilfou, J.T.; Jarnik, M.; Murphy, M.E. ARF induces autophagy by virtue of interaction with Bcl-xl. J. Biol. Chem. 2009, 284, 2803–2810. [Google Scholar] [CrossRef]
- Pimkina, J.; Murphy, M.E. Interaction of the ARF tumor suppressor with cytosolic HSP70 contributes to its autophagy function. Cancer Biol. Ther. 2011, 12, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Budina-Kolomets, A.; Hontz, R.D.; Pimkina, J.; Murphy, M.E. A conserved domain in exon 2 coding for the human and murine ARF tumor suppressor protein is required for autophagy induction. Autophagy 2013, 9, 1553–1565. [Google Scholar] [CrossRef]
- Fontana, R.; Guidone, D.; Angrisano, T.; Calabro, V.; Pollice, A.; La Mantia, G.; Vivo, M. Mutation of the Conserved Threonine 8 within the Human ARF Tumour Suppressor Protein Regulates Autophagy. Biomolecules 2022, 12, 126. [Google Scholar] [CrossRef] [PubMed]
- Reef, S.; Zalckvar, E.; Shifman, O.; Bialik, S.; Sabanay, H.; Oren, M.; Kimchi, A. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol. Cell 2006, 22, 463–475. [Google Scholar] [CrossRef]
- Reef, S.; Shifman, O.; Oren, M.; Kimchi, A. The autophagic inducer smARF interacts with and is stabilized by the mitochondrial p32 protein. Oncogene 2007, 26, 6677–6683. [Google Scholar] [CrossRef]
- Tompkins, V.S.; Hagen, J.; Frazier, A.A.; Lushnikova, T.; Fitzgerald, M.P.; di Tommaso, A.; Ladeveze, V.; Domann, F.E.; Eischen, C.M.; Quelle, D.E. A novel nuclear interactor of ARF and MDM2 (NIAM) that maintains chromosomal stability. J. Biol. Chem. 2007, 282, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Britigan, E.M.; Wan, J.; Zasadil, L.M.; Ryan, S.D.; Weaver, B.A. The ARF tumor suppressor prevents chromosomal instability and ensures mitotic checkpoint fidelity through regulation of Aurora B. Mol. Biol. Cell 2014, 25, 2761–2773. [Google Scholar] [CrossRef]
- Tago, K.; Chiocca, S.; Sherr, C.J. Sumoylation induced by the Arf tumor suppressor: A p53-independent function. Proc. Natl. Acad. Sci. USA 2005, 102, 7689–7694. [Google Scholar] [CrossRef]
- Calabro, V.; Mansueto, G.; Santoro, R.; Gentilella, A.; Pollice, A.; Ghioni, P.; Guerrini, L.; La Mantia, G. Inhibition of p63 transcriptional activity by p14ARF: Functional and physical link between human ARF tumor suppressor and a member of the p53 family. Mol. Cell. Biol. 2004, 24, 8529–8540. [Google Scholar] [CrossRef]
- Vivo, M.; Di Costanzo, A.; Fortugno, P.; Pollice, A.; Calabro, V.; La Mantia, G. Downregulation of DeltaNp63alpha in keratinocytes by p14ARF-mediated SUMO-conjugation and degradation. Cell Cycle 2009, 8, 3545–3551. [Google Scholar] [CrossRef]
- Ranieri, M.; Vivo, M.; De Simone, M.; Guerrini, L.; Pollice, A.; La Mantia, G.; Calabro, V. Sumoylation and ubiquitylation crosstalk in the control of DeltaNp63alpha protein stability. Gene 2018, 645, 34–40. [Google Scholar] [CrossRef]
- Haindl, M.; Harasim, T.; Eick, D.; Muller, S. The nucleolar SUMO-specific protease SENP3 reverses SUMO modification of nucleophosmin and is required for rRNA processing. EMBO Rep. 2008, 9, 273–279. [Google Scholar] [CrossRef]
- Kuo, M.L.; den Besten, W.; Thomas, M.C.; Sherr, C.J. Arf-induced turnover of the nucleolar nucleophosmin-associated SUMO-2/3 protease Senp3. Cell Cycle 2008, 7, 3378–3387. [Google Scholar] [CrossRef]
- Xiao, Z.X.; Chen, J.; Levine, A.J.; Modjtahedi, N.; Xing, J.; Sellers, W.R.; Livingston, D.M. Interaction between the retinoblastoma protein and the oncoprotein MDM2. Nature 1995, 375, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.K.; Chan, F.S.; O’Connor, D.J.; Mittnacht, S.; Zhong, S.; Lu, X. RB regulates the stability and the apoptotic function of p53 via MDM2. Mol. Cell 1999, 3, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.; Trouche, D.; Hagemeier, C.; Sorensen, T.S.; La Thangue, N.B.; Kouzarides, T. Stimulation of E2F1/DP1 transcriptional activity by MDM2 oncoprotein. Nature 1995, 375, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Loughran, O.; La Thangue, N.B. Apoptotic and growth-promoting activity of E2F modulated by MDM2. Mol. Cell. Biol. 2000, 20, 2186–2197. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, K.; Montes de Oca Luna, R.; McNeill, Y.B.; Emerick, E.P.; Spencer, B.; Barfield, C.R.; Lozano, G.; Rosenberg, M.P.; Finlay, C.A. Targeted expression of MDM2 uncouples S phase from mitosis and inhibits mammary gland development independent of p53. Genes Dev. 1997, 11, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Candeias, M.M.; Malbert-Colas, L.; Powell, D.J.; Daskalogianni, C.; Maslon, M.M.; Naski, N.; Bourougaa, K.; Calvo, F.; Fahraeus, R. P53 mRNA controls p53 activity by managing Mdm2 functions. Nat. Cell Biol. 2008, 10, 1098–1105. [Google Scholar] [CrossRef]
- Gajjar, M.; Candeias, M.M.; Malbert-Colas, L.; Mazars, A.; Fujita, J.; Olivares-Illana, V.; Fahraeus, R. The p53 mRNA-Mdm2 interaction controls Mdm2 nuclear trafficking and is required for p53 activation following DNA damage. Cancer Cell 2012, 21, 25–35. [Google Scholar] [CrossRef]
- Takagi, M.; Absalon, M.J.; McLure, K.G.; Kastan, M.B. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005, 123, 49–63. [Google Scholar] [CrossRef]
- Ofir-Rosenfeld, Y.; Boggs, K.; Michael, D.; Kastan, M.B.; Oren, M. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol. Cell 2008, 32, 180–189. [Google Scholar] [CrossRef]
- Bouska, A.; Eischen, C.M. Mdm2 affects genome stability independent of p53. Cancer Res. 2009, 69, 1697–1701. [Google Scholar] [CrossRef]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Wei, Y.; Ali-Seyed, M.; Li, Z.; Broglio, K.; Berry, D.A.; Hung, M.C. MDM2 promotes cell motility and invasiveness by regulating E-cadherin degradation. Mol. Cell. Biol. 2006, 26, 7269–7282. [Google Scholar] [CrossRef]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef]
- Jung, C.H.; Kim, J.; Park, J.K.; Hwang, S.G.; Moon, S.K.; Kim, W.J.; Um, H.D. Mdm2 increases cellular invasiveness by binding to and stabilizing the Slug mRNA. Cancer Lett. 2013, 335, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.P.; Wang, W.L.; Chang, Y.L.; Wu, C.T.; Chao, Y.C.; Kao, S.H.; Yuan, A.; Lin, C.W.; Yang, S.C.; Chan, W.K.; et al. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat. Cell Biol. 2009, 11, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Chen, L.; Jost, C.A.; Maya, R.; Keller, D.; Wang, X.; Kaelin, W.G., Jr.; Oren, M.; Chen, J.; Lu, H. MDM2 suppresses p73 function without promoting p73 degradation. Mol. Cell. Biol. 1999, 19, 3257–3266. [Google Scholar] [CrossRef] [PubMed]
- Balint, E.; Bates, S.; Vousden, K.H. Mdm2 binds p73 alpha without targeting degradation. Oncogene 1999, 18, 3923–3929. [Google Scholar] [CrossRef]
- Ongkeko, W.M.; Wang, X.Q.; Siu, W.Y.; Lau, A.W.; Yamashita, K.; Harris, A.L.; Cox, L.S.; Poon, R.Y. MDM2 and MDMX bind and stabilize the p53-related protein p73. Curr. Biol. 1999, 9, 829–832. [Google Scholar] [CrossRef]
- Zdzalik, M.; Pustelny, K.; Kedracka-Krok, S.; Huben, K.; Pecak, A.; Wladyka, B.; Jankowski, S.; Dubin, A.; Potempa, J.; Dubin, G. Interaction of regulators Mdm2 and Mdmx with transcription factors p53, p63 and p73. Cell Cycle 2010, 9, 4584–4591. [Google Scholar] [CrossRef]
- Klein, A.M.; Biderman, L.; Tong, D.; Alaghebandan, B.; Plumber, S.A.; Mueller, H.S.; van Vlimmeren, A.; Katz, C.; Prives, C. MDM2, MDMX, and p73 regulate cell-cycle progression in the absence of wild-type p53. Proc. Natl. Acad. Sci. USA 2021, 118, e2102420118. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.M.; Mitra, R.; Xiao, Y.; Michener, P.; Palazzo, J.; Chao, A.; Gour, J.; Cassel, J.; Salvino, J.M.; Eischen, C.M. Targeted MDM2 Degradation Reveals a New Vulnerability for p53-Inactivated Triple-Negative Breast Cancer. Cancer Discov. 2023, 13, 1210–1229. [Google Scholar] [CrossRef] [PubMed]
- Valente, L.J.; Gray, D.H.; Michalak, E.M.; Pinon-Hofbauer, J.; Egle, A.; Scott, C.L.; Janic, A.; Strasser, A. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep. 2013, 3, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Janic, A.; Valente, L.J.; Wakefield, M.J.; Di Stefano, L.; Milla, L.; Wilcox, S.; Yang, H.; Tai, L.; Vandenberg, C.J.; Kueh, A.J.; et al. DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat. Med. 2018, 24, 947–953. [Google Scholar] [CrossRef]
- Mello, S.S.; Valente, L.J.; Raj, N.; Seoane, J.A.; Flowers, B.M.; McClendon, J.; Bieging-Rolett, K.T.; Lee, J.; Ivanochko, D.; Kozak, M.M.; et al. A p53 Super-tumor Suppressor Reveals a Tumor Suppressive p53-Ptpn14-Yap Axis in Pancreatic Cancer. Cancer Cell 2017, 32, 460–473.e466. [Google Scholar] [CrossRef]
- Moon, S.H.; Huang, C.H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P.t.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019, 176, 564–580.e519. [Google Scholar] [CrossRef]
- Jiang, D.; Brady, C.A.; Johnson, T.M.; Lee, E.Y.; Park, E.J.; Scott, M.P.; Attardi, L.D. Full p53 transcriptional activation potential is dispensable for tumor suppression in diverse lineages. Proc. Natl. Acad. Sci. USA 2011, 108, 17123–17128. [Google Scholar] [CrossRef]
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Kenzelmann Broz, D.; Basak, S.; Park, E.J.; McLaughlin, M.E.; et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011, 145, 571–583. [Google Scholar] [CrossRef]
- Bieging-Rolett, K.T.; Kaiser, A.M.; Morgens, D.W.; Boutelle, A.M.; Seoane, J.A.; Van Nostrand, E.L.; Zhu, C.; Houlihan, S.L.; Mello, S.S.; Yee, B.A.; et al. Zmat3 Is a Key Splicing Regulator in the p53 Tumor Suppression Program. Mol. Cell 2020, 80, 452–469.e459. [Google Scholar] [CrossRef] [PubMed]
- Muys, B.R.; Anastasakis, D.G.; Claypool, D.; Pongor, L.; Li, X.L.; Grammatikakis, I.; Liu, M.; Wang, X.; Prasanth, K.V.; Aladjem, M.I.; et al. The p53-induced RNA-binding protein ZMAT3 is a splicing regulator that inhibits the splicing of oncogenic CD44 variants in colorectal carcinoma. Genes Dev. 2021, 35, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Gnanapradeepan, K.; Indeglia, A.; Stieg, D.C.; Clarke, N.; Shao, C.; Dougherty, J.F.; Murali, N.; Murphy, M.E. PLTP is a p53 target gene with roles in cancer growth suppression and ferroptosis. J. Biol. Chem. 2022, 298, 102637. [Google Scholar] [CrossRef]
- Indeglia, A.; Leung, J.C.; Miller, S.A.; Leu, J.I.; Dougherty, J.F.; Clarke, N.L.; Kirven, N.A.; Shao, C.; Ke, L.; Lovell, S.; et al. An African-Specific Variant of TP53 Reveals PADI4 as a Regulator of p53-Mediated Tumor Suppression. Cancer Discov. 2023, 13, 1696–1719. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, Y.; Wu, X.; Li, J.; Liu, K.; Fang, D.; Li, B.; Shan, G.; Mei, X.; Wang, F.; et al. Reciprocal modulation of long noncoding RNA EMS and p53 regulates tumorigenesis. Proc. Natl. Acad. Sci. USA 2022, 119, e2111409119. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Marchenko, N.D.; Moll, U.M. Mitochondrial death functions of p53. Mol. Cell Oncol. 2014, 1, e955995. [Google Scholar] [CrossRef]
- Marchenko, N.D.; Zaika, A.; Moll, U.M. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J. Biol. Chem. 2000, 275, 16202–16212. [Google Scholar] [CrossRef]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef]
- Tomita, Y.; Marchenko, N.; Erster, S.; Nemajerova, A.; Dehner, A.; Klein, C.; Pan, H.; Kessler, H.; Pancoska, P.; Moll, U.M. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J. Biol. Chem. 2006, 281, 8600–8606. [Google Scholar] [CrossRef]
- Hagn, F.; Klein, C.; Demmer, O.; Marchenko, N.; Vaseva, A.; Moll, U.M.; Kessler, H. BclxL changes conformation upon binding to wild-type but not mutant p53 DNA binding domain. J. Biol. Chem. 2010, 285, 3439–3450. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Qu, L.; Dai, S.; Li, Y.; Wang, H.; Feng, Y.; Chen, X.; Jiang, L.; Guo, M.; Li, J.; et al. Structural insight into the molecular mechanism of p53-mediated mitochondrial apoptosis. Nat. Commun. 2021, 12, 2280. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D.L. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 2004, 6, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Marchenko, N.D.; Wolff, S.; Erster, S.; Becker, K.; Moll, U.M. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J. 2007, 26, 923–934. [Google Scholar] [CrossRef]
- Castelli, M.; Piobbico, D.; Chiacchiaretta, M.; Brunacci, C.; Pieroni, S.; Bartoli, D.; Gargaro, M.; Fallarino, F.; Puccetti, P.; Soddu, S.; et al. HOPS/TMUB1 retains p53 in the cytoplasm and sustains p53-dependent mitochondrial apoptosis. EMBO Rep. 2020, 21, e48073. [Google Scholar] [CrossRef]
- Ferracchiato, S.; Di-Iacovo, N.; Scopetti, D.; Piobbico, D.; Castelli, M.; Pieroni, S.; Gargaro, M.; Manni, G.; Brancorsini, S.; Della-Fazia, M.A.; et al. Hops/Tmub1 Heterozygous Mouse Shows Haploinsufficiency Effect in Influencing p53-Mediated Apoptosis. Int. J. Mol. Sci. 2021, 22, 7186. [Google Scholar] [CrossRef]
- Sorrentino, G.; Mioni, M.; Giorgi, C.; Ruggeri, N.; Pinton, P.; Moll, U.; Mantovani, F.; Del Sal, G. The prolyl-isomerase Pin1 activates the mitochondrial death program of p53. Cell Death Differ. 2013, 20, 198–208. [Google Scholar] [CrossRef]
- Follis, A.V.; Llambi, F.; Merritt, P.; Chipuk, J.E.; Green, D.R.; Kriwacki, R.W. Pin1-Induced Proline Isomerization in Cytosolic p53 Mediates BAX Activation and Apoptosis. Mol. Cell 2015, 59, 677–684. [Google Scholar] [CrossRef]
- Li, L.; Su, Z.; Zou, Z.; Tan, H.; Cai, D.; Su, L.; Gu, Z. Ser46 phosphorylation of p53 is an essential event in prolyl-isomerase Pin1-mediated p53-independent apoptosis in response to heat stress. Cell Death Dis. 2019, 10, 96. [Google Scholar] [CrossRef]
- Sykes, S.M.; Stanek, T.J.; Frank, A.; Murphy, M.E.; McMahon, S.B. Acetylation of the DNA binding domain regulates transcription-independent apoptosis by p53. J. Biol. Chem. 2009, 284, 20197–20205. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Yamamoto, K.; Mak, T.W. Regulation of transactivation-independent proapoptotic activity of p53 by FOXO3a. Proc. Natl. Acad. Sci. USA 2006, 103, 9051–9056. [Google Scholar] [CrossRef] [PubMed]
- Follis, A.V.; Chipuk, J.E.; Fisher, J.C.; Yun, M.K.; Grace, C.R.; Nourse, A.; Baran, K.; Ou, L.; Min, L.; White, S.W.; et al. PUMA binding induces partial unfolding within BCL-xL to disrupt p53 binding and promote apoptosis. Nat. Chem. Biol. 2013, 9, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Galindo Ramirez, F.; Rizzuto, R.; Di Virgilio, F.; Zito, E.; et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 2015, 112, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.J.; Kao, T.Y.; Kuo, C.L.; Fan, C.C.; Cheng, A.N.; Fang, W.C.; Chou, H.Y.; Lo, Y.K.; Chen, C.H.; Jiang, S.S.; et al. Mitochondrial Lon sequesters and stabilizes p53 in the matrix to restrain apoptosis under oxidative stress via its chaperone activity. Cell Death Dis. 2018, 9, 697. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Mah, L.Y.; O’Prey, J.; Baudot, A.D.; Hoekstra, A.; Ryan, K.M. DRAM-1 encodes multiple isoforms that regulate autophagy. Autophagy 2012, 8, 18–28. [Google Scholar] [CrossRef]
- Mrschtik, M.; Ryan, K.M. Another DRAM involved in autophagy and cell death. Autophagy 2016, 12, 603–605. [Google Scholar] [CrossRef]
- Kenzelmann Broz, D.; Spano Mello, S.; Bieging, K.T.; Jiang, D.; Dusek, R.L.; Brady, C.A.; Sidow, A.; Attardi, L.D. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013, 27, 1016–1031. [Google Scholar] [CrossRef]
- Seillier, M.; Peuget, S.; Gayet, O.; Gauthier, C.; N’Guessan, P.; Monte, M.; Carrier, A.; Iovanna, J.L.; Dusetti, N.J. TP53INP1, a tumor suppressor, interacts with LC3 and ATG8-family proteins through the LC3-interacting region (LIR) and promotes autophagy-dependent cell death. Cell Death Differ. 2012, 19, 1525–1535. [Google Scholar] [CrossRef]
- Wu, G.S.; Saftig, P.; Peters, C.; El-Deiry, W.S. Potential role for cathepsin D in p53-dependent tumor suppression and chemosensitivity. Oncogene 1998, 16, 2177–2183. [Google Scholar] [CrossRef]
- Ikeguchi, M.; Sakatani, T.; Ueta, T.; Fukuda, K.; Oka, S.; Hisamitsu, K.; Yamaguchi, K.; Tsujitani, S.; Kaibara, N. Correlation between cathepsin D expression and p53 protein nuclear accumulation in oesophageal squamous cell carcinoma. J. Clin. Pathol. 2002, 55, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Chen, Q.; Wang, C.; Yao, D.; Zhu, L.; Pan, Y.; Zhang, J.; Bai, Y.; Shao, C. Inhibition of Cathepsin D (CTSD) enhances radiosensitivity of glioblastoma cells by attenuating autophagy. Mol. Carcinog. 2020, 59, 651–660. [Google Scholar] [CrossRef]
- Yeo, S.Y.; Itahana, Y.; Guo, A.K.; Han, R.; Iwamoto, K.; Nguyen, H.T.; Bao, Y.; Kleiber, K.; Wu, Y.J.; Bay, B.H.; et al. Transglutaminase 2 contributes to a TP53-induced autophagy program to prevent oncogenic transformation. eLife 2016, 5, e07101. [Google Scholar] [CrossRef]
- Zhang, X.D.; Wang, Y.; Wang, Y.; Zhang, X.; Han, R.; Wu, J.C.; Liang, Z.Q.; Gu, Z.L.; Han, F.; Fukunaga, K.; et al. p53 mediates mitochondria dysfunction-triggered autophagy activation and cell death in rat striatum. Autophagy 2009, 5, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.Y.; Gang, H.; Aviv, Y.; Dhingra, R.; Margulets, V.; Kirshenbaum, L.A. p53 mediates autophagy and cell death by a mechanism contingent on Bnip3. Hypertension 2013, 62, 70–77. [Google Scholar] [CrossRef]
- Chang, H.W.; Kim, M.R.; Lee, H.J.; Lee, H.M.; Kim, G.C.; Lee, Y.S.; Nam, H.Y.; Lee, M.; Jang, H.J.; Lee, K.E.; et al. p53/BNIP3-dependent mitophagy limits glycolytic shift in radioresistant cancer. Oncogene 2019, 38, 3729–3742. [Google Scholar] [CrossRef]
- Feng, W.; Huang, S.; Wu, H.; Zhang, M. Molecular basis of Bcl-xL’s target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J. Mol. Biol. 2007, 372, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.C.; et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018, 558, 136–140. [Google Scholar] [CrossRef]
- Lee, E.F.; Smith, N.A.; Soares da Costa, T.P.; Meftahi, N.; Yao, S.; Harris, T.J.; Tran, S.; Pettikiriarachchi, A.; Perugini, M.A.; Keizer, D.W.; et al. Structural insights into BCL2 pro-survival protein interactions with the key autophagy regulator BECN1 following phosphorylation by STK4/MST1. Autophagy 2019, 15, 785–795. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Kon, N.; Ou, Y.; Wang, S.J.; Li, H.; Rustgi, A.K.; Gu, W. mTOR inhibition acts as an unexpected checkpoint in p53-mediated tumor suppression. Genes Dev. 2021, 35, 59–64. [Google Scholar] [CrossRef]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Wang, J.; Hu, W.; Feng, Z. The Regulation of Ferroptosis by Tumor Suppressor p53 and its Pathway. Int. J. Mol. Sci. 2020, 21, 8387. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gu, W. p53 in ferroptosis regulation: The new weapon for the old guardian. Cell Death Differ. 2022, 29, 895–910. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wang, W.; Zhang, W. Ferroptosis and the bidirectional regulatory factor p53. Cell Death Discov. 2023, 9, 197. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [PubMed]
- Jennis, M.; Kung, C.P.; Basu, S.; Budina-Kolomets, A.; Leu, J.I.; Khaku, S.; Scott, J.P.; Cai, K.Q.; Campbell, M.R.; Porter, D.K.; et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 2016, 30, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.; Murphy, M.E.; George, D.L. Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc. Natl. Acad. Sci. USA 2019, 116, 8390–8396. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Lin, M.; Zhu, W.; Liang, Y.; Hong, X.; Zhao, Y.; Young, K.H.; Hu, W.; Feng, Z. Glutaminase 2 negatively regulates the PI3K/AKT signaling and shows tumor suppression activity in human hepatocellular carcinoma. Oncotarget 2014, 5, 2635–2647. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qian, Y.; Zhang, J.; Yan, W.; Jung, Y.S.; Chen, M.; Huang, E.; Lloyd, K.; Duan, Y.; Wang, J.; et al. Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes Dev. 2017, 31, 1243–1256. [Google Scholar] [CrossRef]
- Zhang, Z.; Guo, M.; Shen, M.; Kong, D.; Zhang, F.; Shao, J.; Tan, S.; Wang, S.; Chen, A.; Cao, P.; et al. The BRD7-P53-SLC25A28 axis regulates ferroptosis in hepatic stellate cells. Redox Biol. 2020, 36, 101619. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Wang, S.J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, L.; Zhang, X.; Cui, W.; Liu, Y.; Sun, Q.R.; He, Q.; Zhao, S.; Zhang, G.A.; Wang, Y.; et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 2019, 20, e47563. [Google Scholar] [CrossRef]
- Wang, M.; Mao, C.; Ouyang, L.; Liu, Y.; Lai, W.; Liu, N.; Shi, Y.; Chen, L.; Xiao, D.; Yu, F.; et al. Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ. 2019, 26, 2329–2343. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; Wang, S.J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, C.; Xu, Z.; Scuoppo, C.; Rillahan, C.D.; Gao, J.; Spitzer, B.; Bosbach, B.; Kastenhuber, E.R.; Baslan, T.; et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature 2016, 531, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef]
- Yang, X.; Liu, J.; Wang, C.; Cheng, K.K.; Xu, H.; Li, Q.; Hua, T.; Jiang, X.; Sheng, L.; Mao, J.; et al. miR-18a promotes glioblastoma development by down-regulating ALOXE3-mediated ferroptotic and anti-migration activities. Oncogenesis 2021, 10, 15. [Google Scholar] [CrossRef]
- Chen, D.; Chu, B.; Yang, X.; Liu, Z.; Jin, Y.; Kon, N.; Rabadan, R.; Jiang, X.; Stockwell, B.R.; Gu, W. iPLA2beta-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat. Commun. 2021, 12, 3644. [Google Scholar] [CrossRef] [PubMed]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell. Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Riscal, R.; Arena, G.; Linares, L.K.; Le Cam, L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol. Metab. 2020, 33, 2–22. [Google Scholar] [CrossRef]
- Humpton, T.; Vousden, K.H. Taking up the reins of power: Metabolic functions of p53. J. Mol. Cell. Biol. 2019, 11, 610–614. [Google Scholar] [CrossRef]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Zawacka-Pankau, J.; Grinkevich, V.V.; Hunten, S.; Nikulenkov, F.; Gluch, A.; Li, H.; Enge, M.; Kel, A.; Selivanova, G. Inhibition of glycolytic enzymes mediated by pharmacologically activated p53: Targeting Warburg effect to fight cancer. J. Biol. Chem. 2011, 286, 41600–41615. [Google Scholar] [CrossRef] [PubMed]
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 2005, 202, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Contractor, T.; Harris, C.R. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012, 72, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Barnoud, T.; Parris, J.L.D.; Murphy, M.E. Tumor cells containing the African-Centric S47 variant of TP53 show increased Warburg metabolism. Oncotarget 2019, 10, 1217–1223. [Google Scholar] [CrossRef]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef]
- Zhu, Y.; Gu, L.; Lin, X.; Zhou, X.; Lu, B.; Liu, C.; Li, Y.; Prochownik, E.V.; Karin, M.; Wang, F.; et al. P53 deficiency affects cholesterol esterification to exacerbate hepatocarcinogenesis. Hepatology 2023, 77, 1499–1511. [Google Scholar] [CrossRef]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef]
- McConnell, A.M.; Yao, C.; Yeckes, A.R.; Wang, Y.; Selvaggio, A.S.; Tang, J.; Kirsch, D.G.; Stripp, B.R. p53 Regulates Progenitor Cell Quiescence and Differentiation in the Airway. Cell Rep. 2016, 17, 2173–2182. [Google Scholar] [CrossRef]
- Spike, B.T.; Wahl, G.M. p53, Stem Cells, and Reprogramming: Tumor Suppression beyond Guarding the Genome. Genes Cancer 2011, 2, 404–419. [Google Scholar] [CrossRef]
- Hong, H.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Kanagawa, O.; Nakagawa, M.; Okita, K.; Yamanaka, S. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 2009, 460, 1132–1135. [Google Scholar] [CrossRef]
- Jain, A.K.; Allton, K.; Iacovino, M.; Mahen, E.; Milczarek, R.J.; Zwaka, T.P.; Kyba, M.; Barton, M.C. p53 regulates cell cycle and microRNAs to promote differentiation of human embryonic stem cells. PLoS Biol. 2012, 10, e1001268. [Google Scholar] [CrossRef]
- Shi, L.; Jackstadt, R.; Siemens, H.; Li, H.; Kirchner, T.; Hermeking, H. p53-induced miR-15a/16-1 and AP4 form a double-negative feedback loop to regulate epithelial-mesenchymal transition and metastasis in colorectal cancer. Cancer Res. 2014, 74, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Ouadah, Y.; Rojas, E.R.; Riordan, D.P.; Capostagno, S.; Kuo, C.S.; Krasnow, M.A. Rare Pulmonary Neuroendocrine Cells Are Stem Cells Regulated by Rb, p53, and Notch. Cell 2019, 179, 403–416.e423. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.P.t.; Yashinskie, J.J.; Koche, R.; Chandwani, R.; Tian, S.; Chen, C.C.; Baslan, T.; Marinkovic, Z.S.; Sanchez-Rivera, F.J.; Leach, S.D.; et al. alpha-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019, 573, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Cubuk, C.; Loucera, C.; Pena-Chilet, M.; Dopazo, J. Crosstalk between Metabolite Production and Signaling Activity in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 7450. [Google Scholar] [CrossRef] [PubMed]
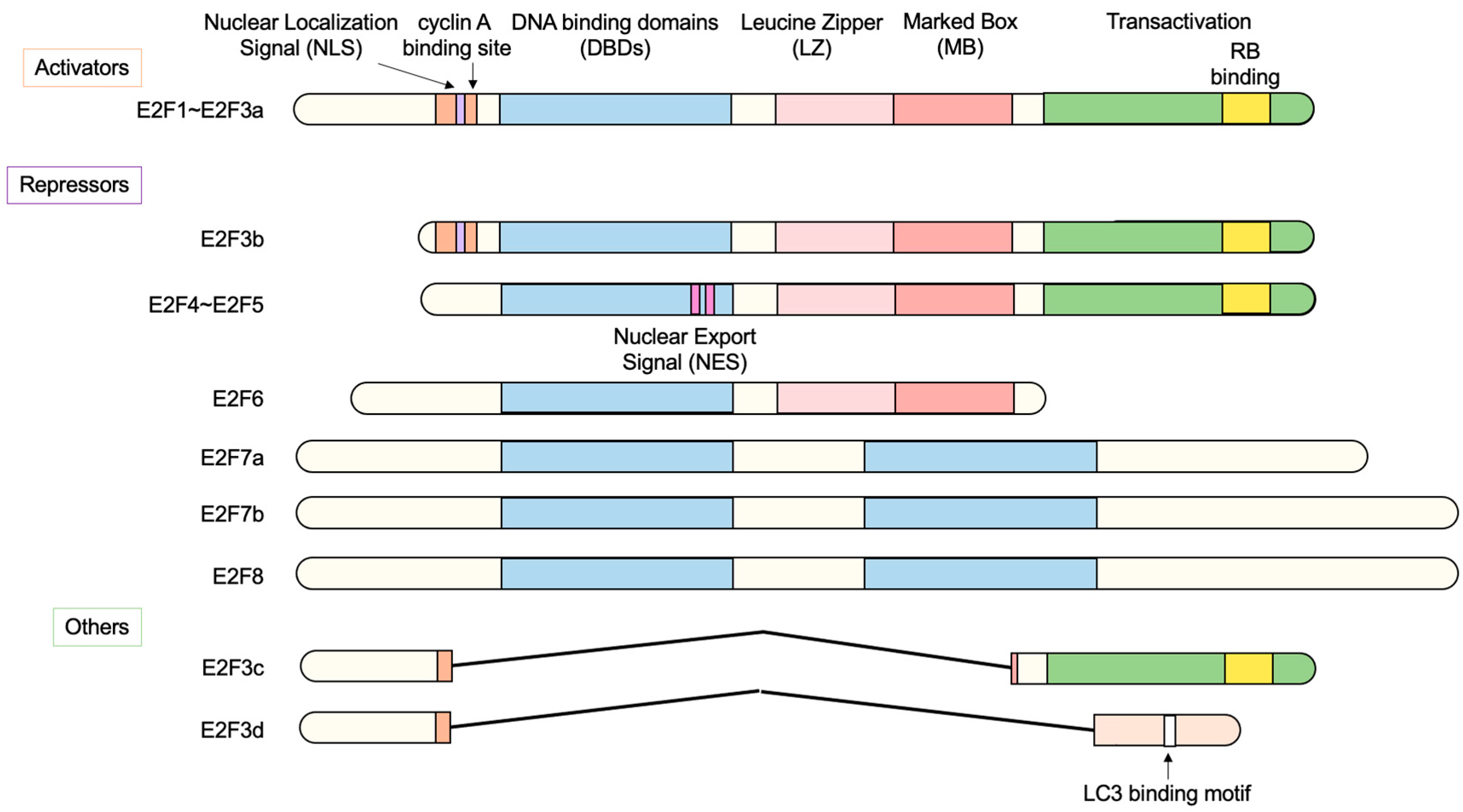
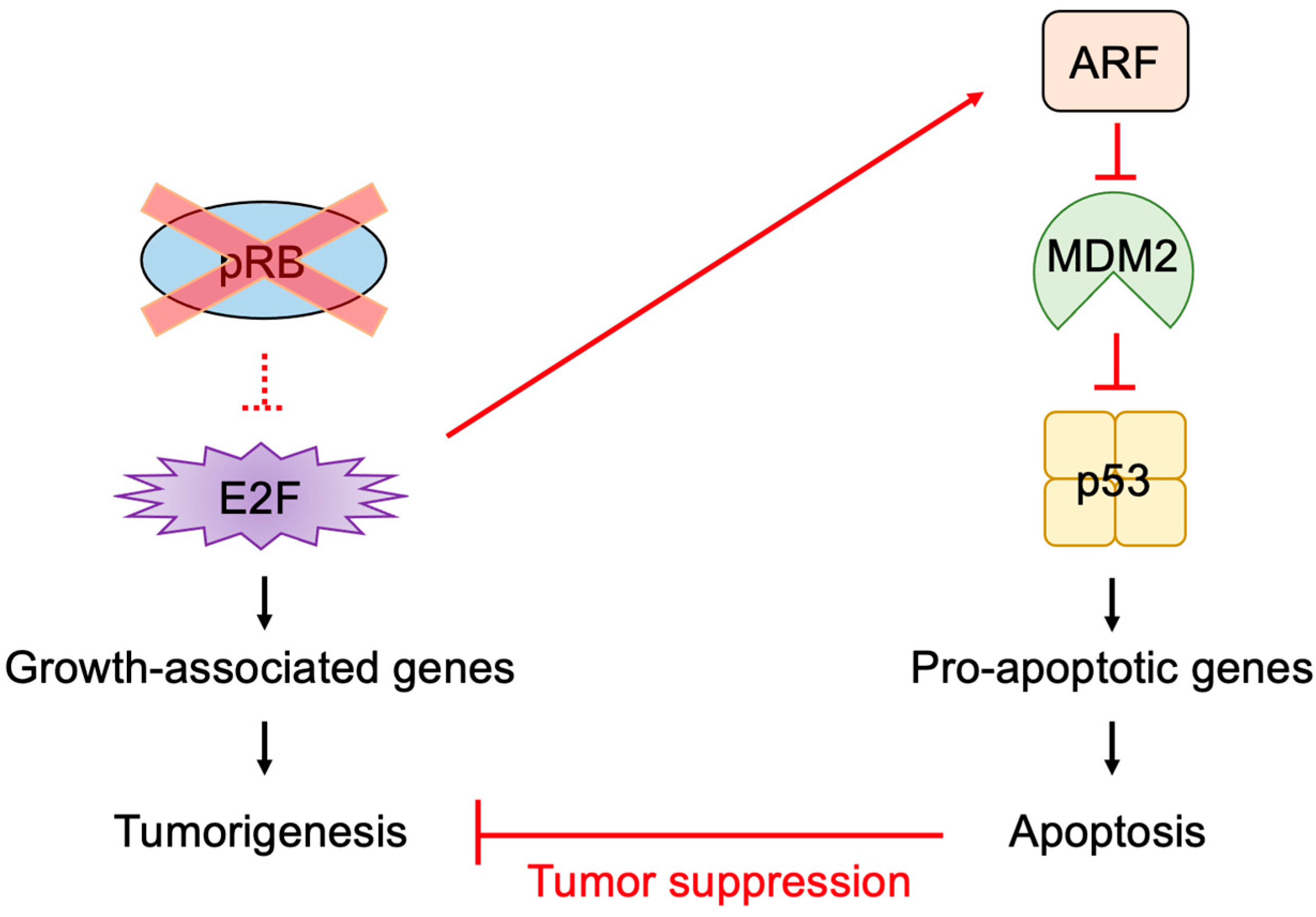




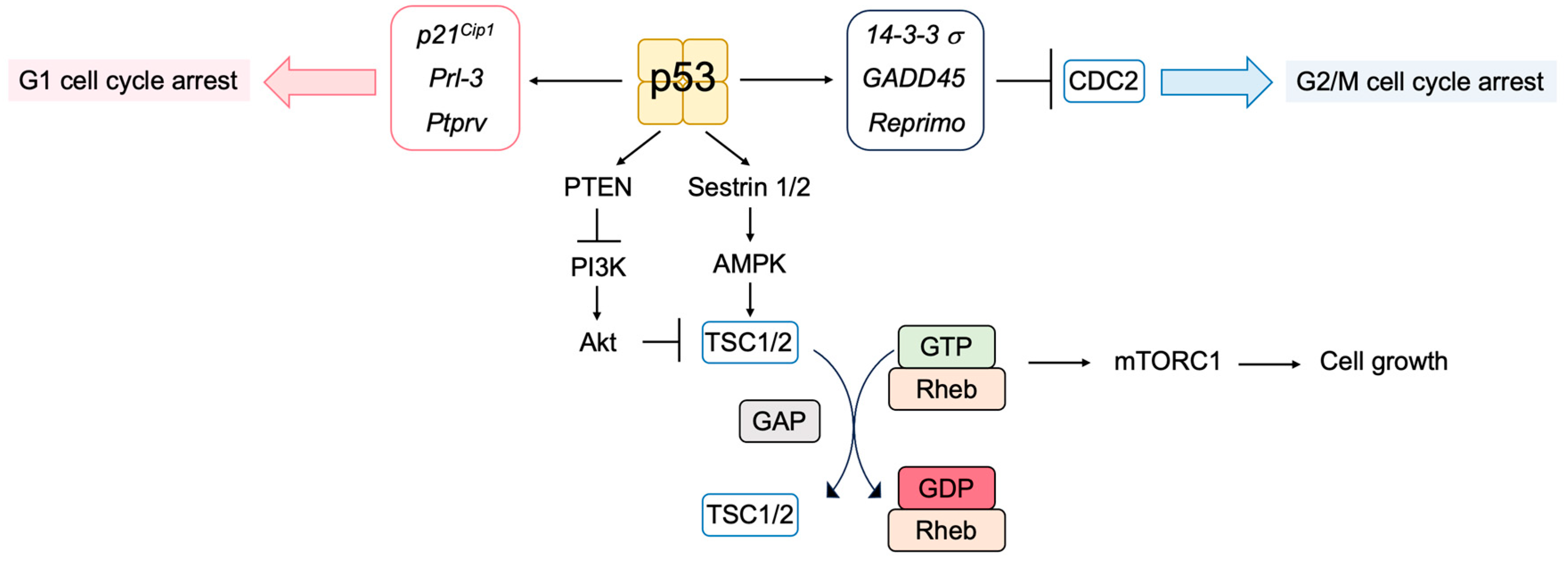
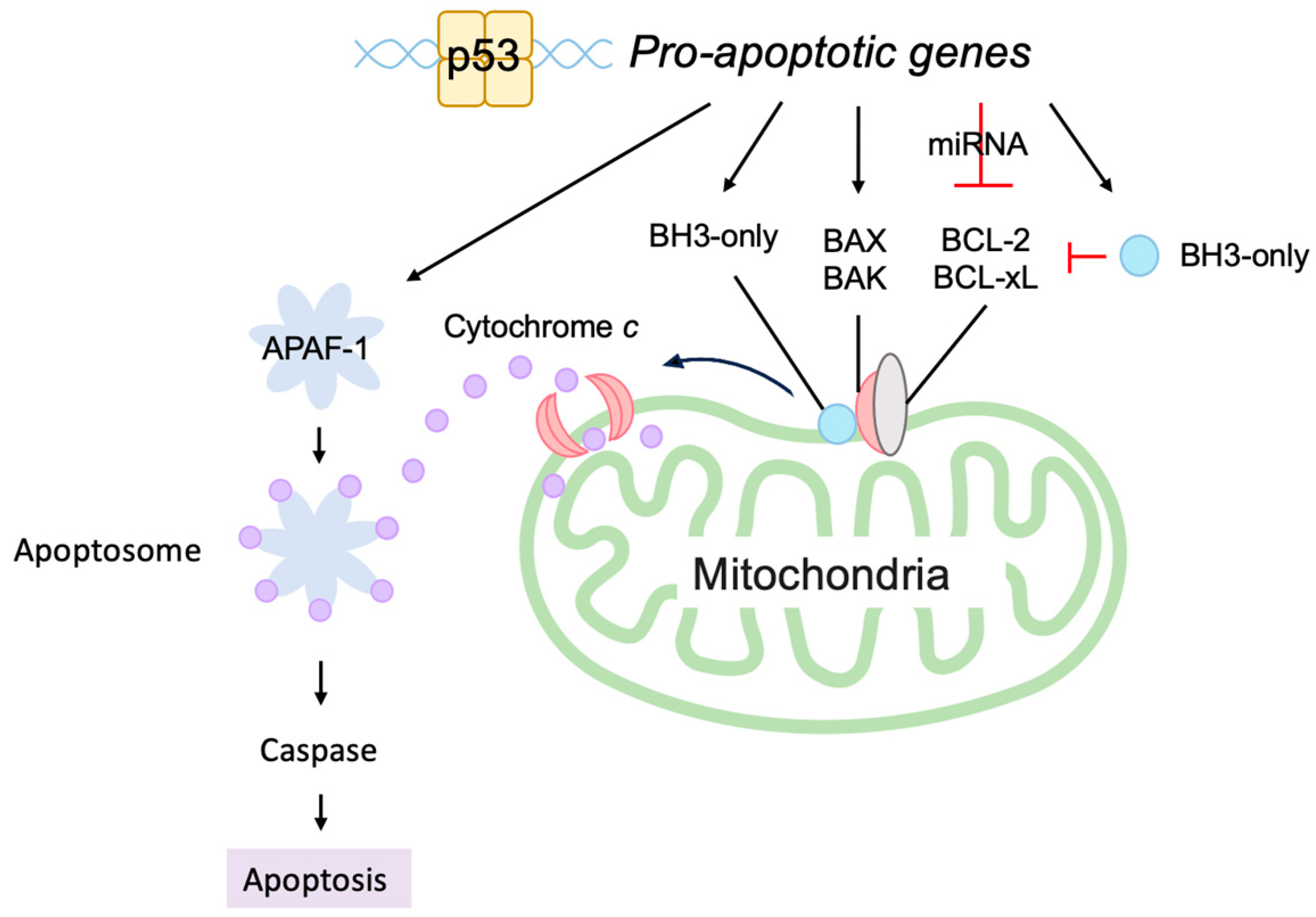

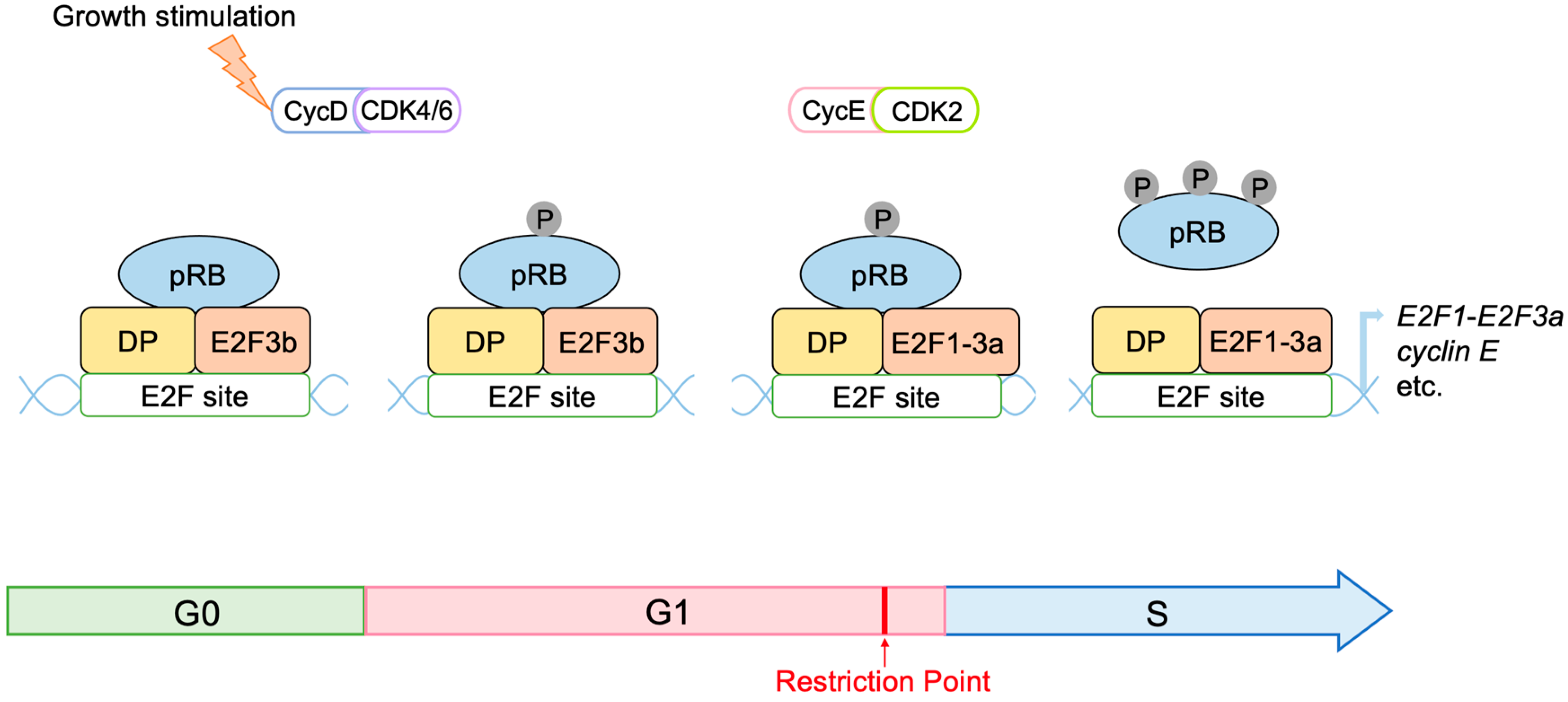
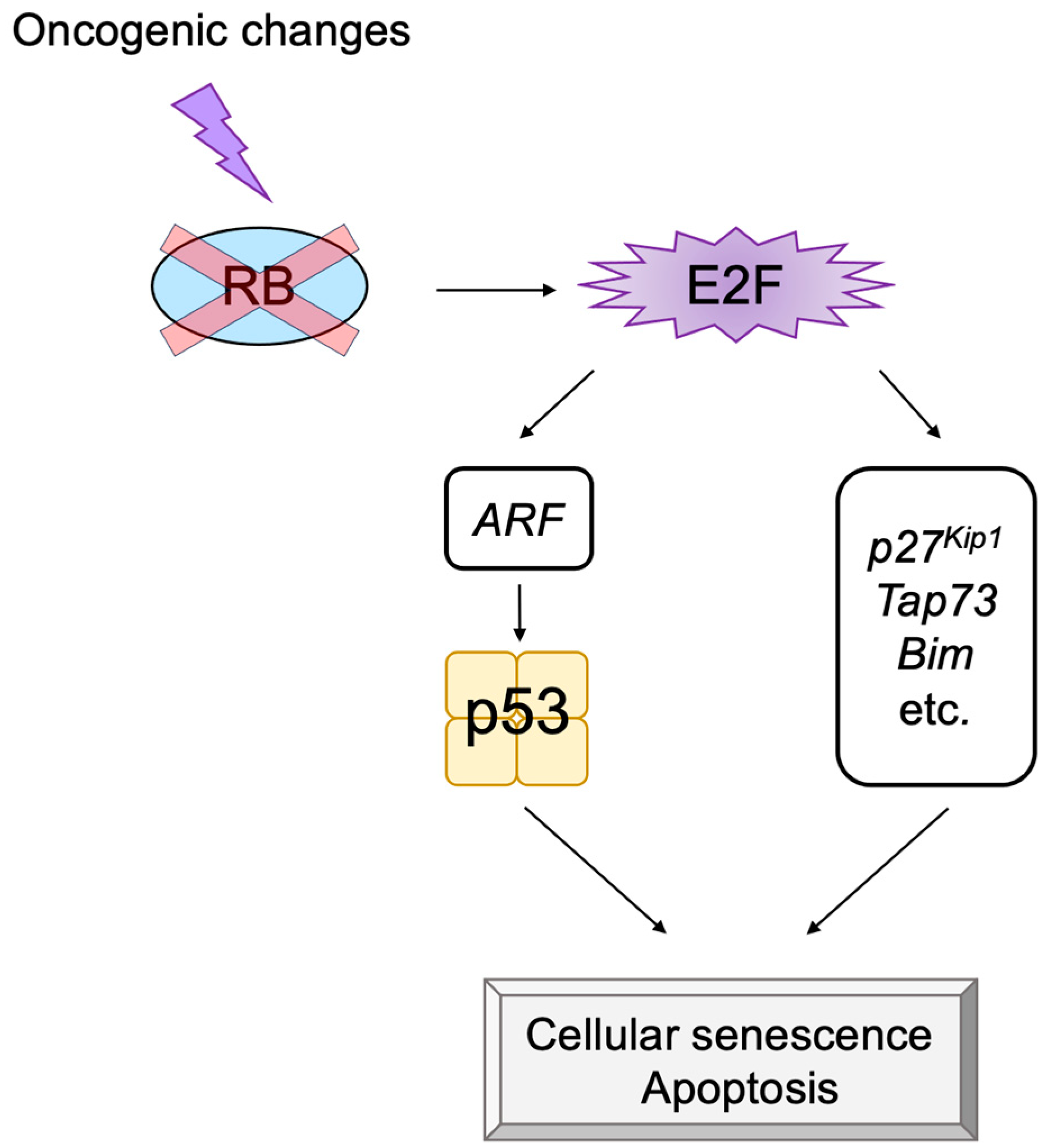
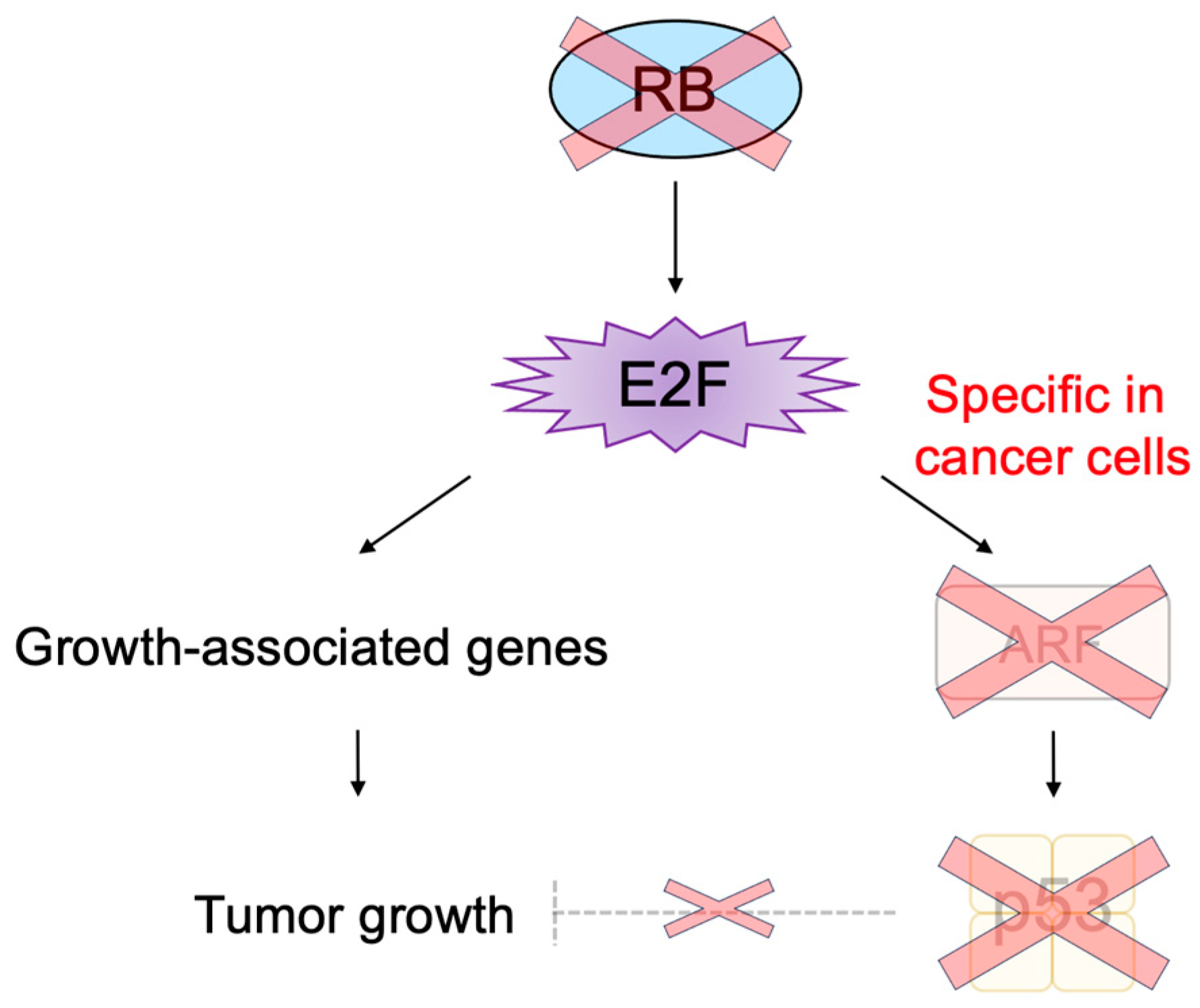
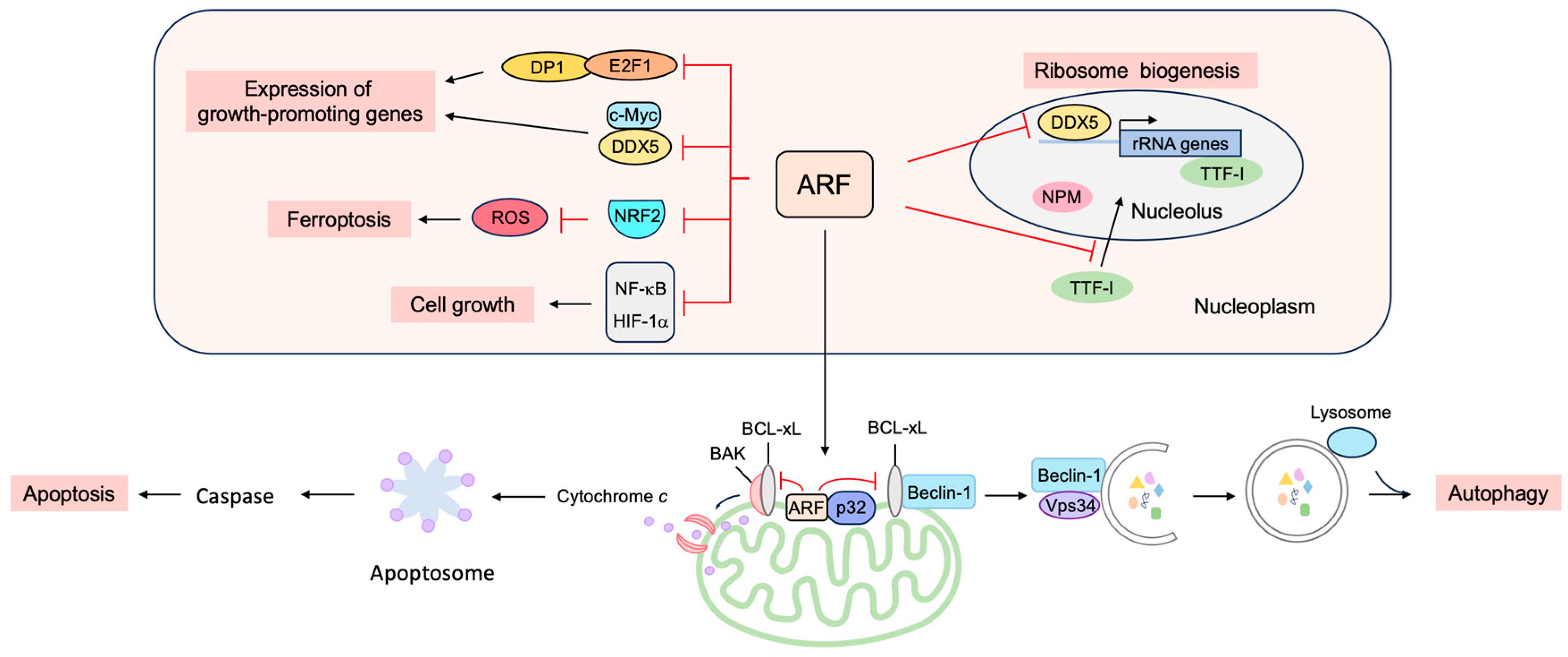
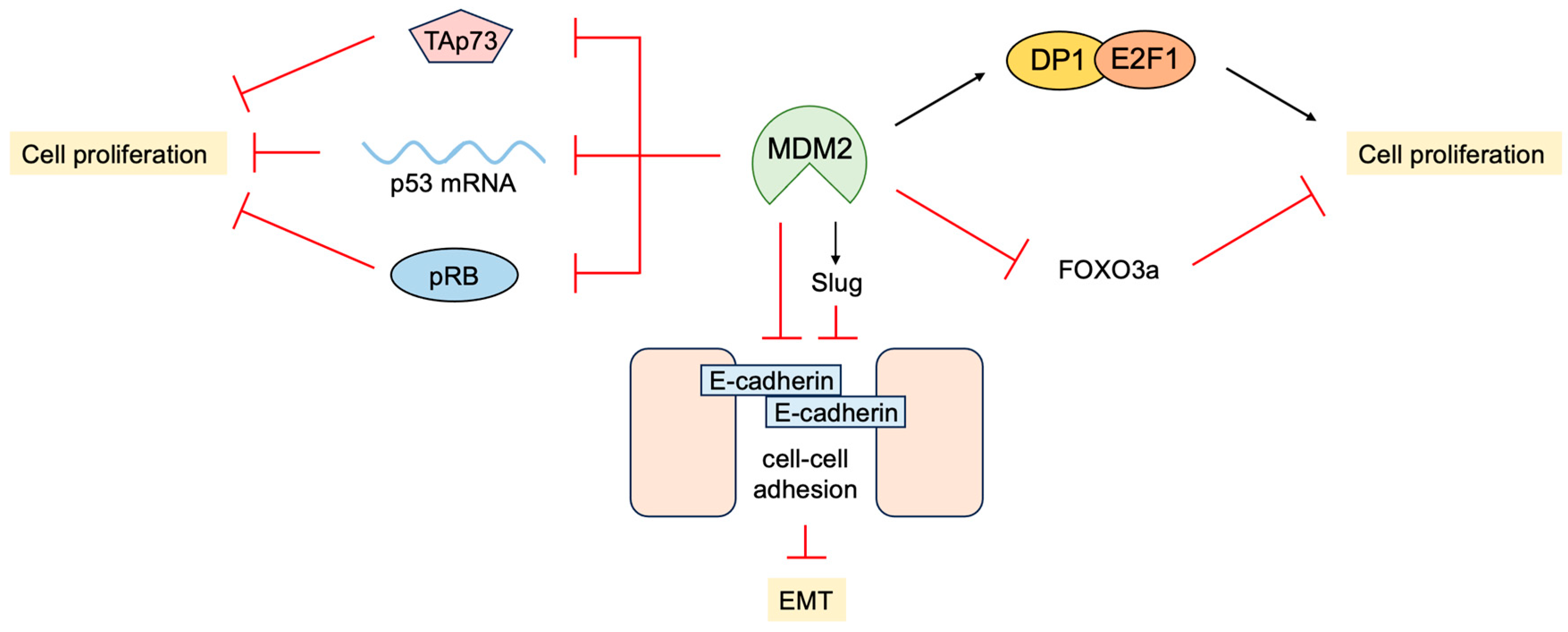
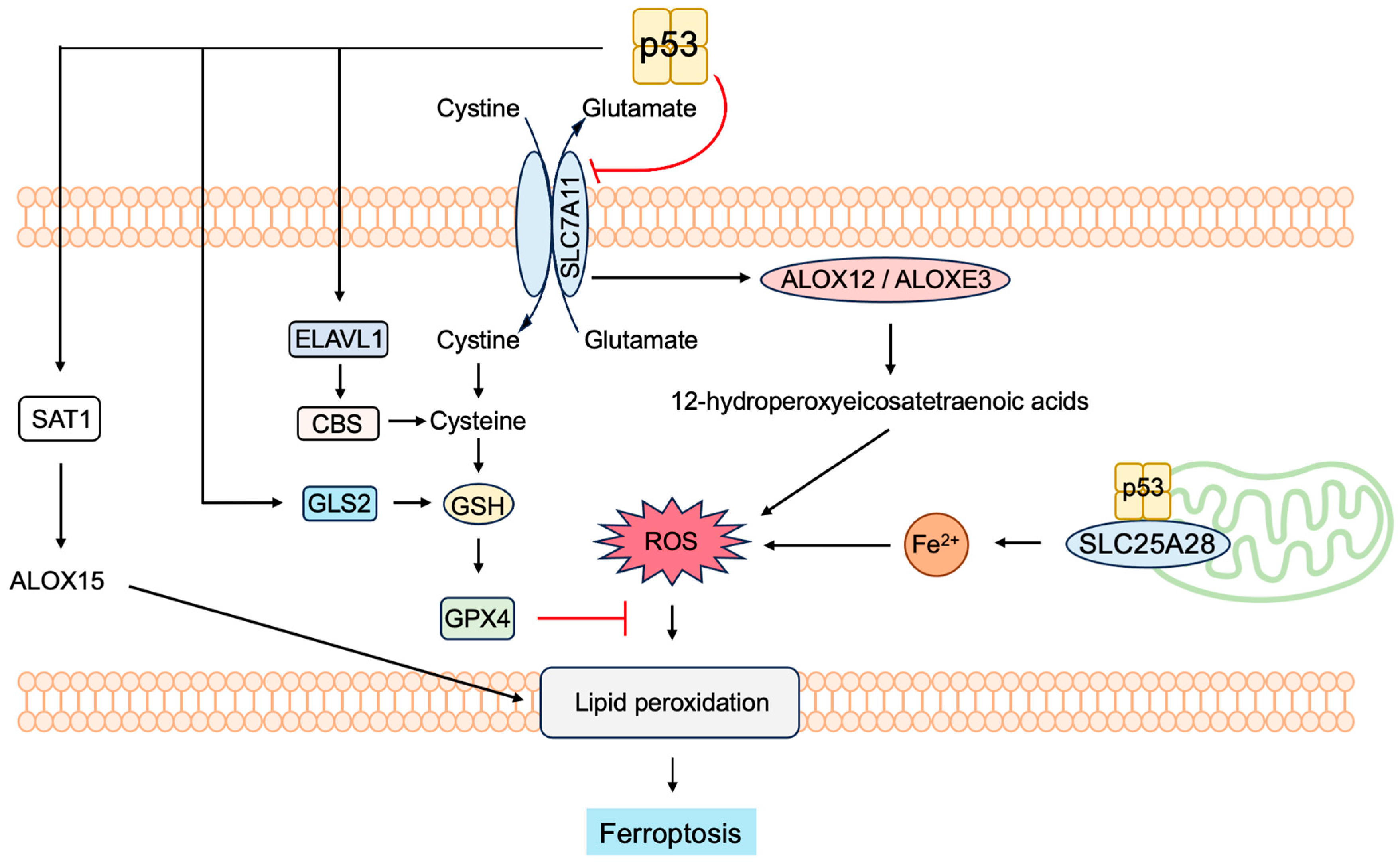
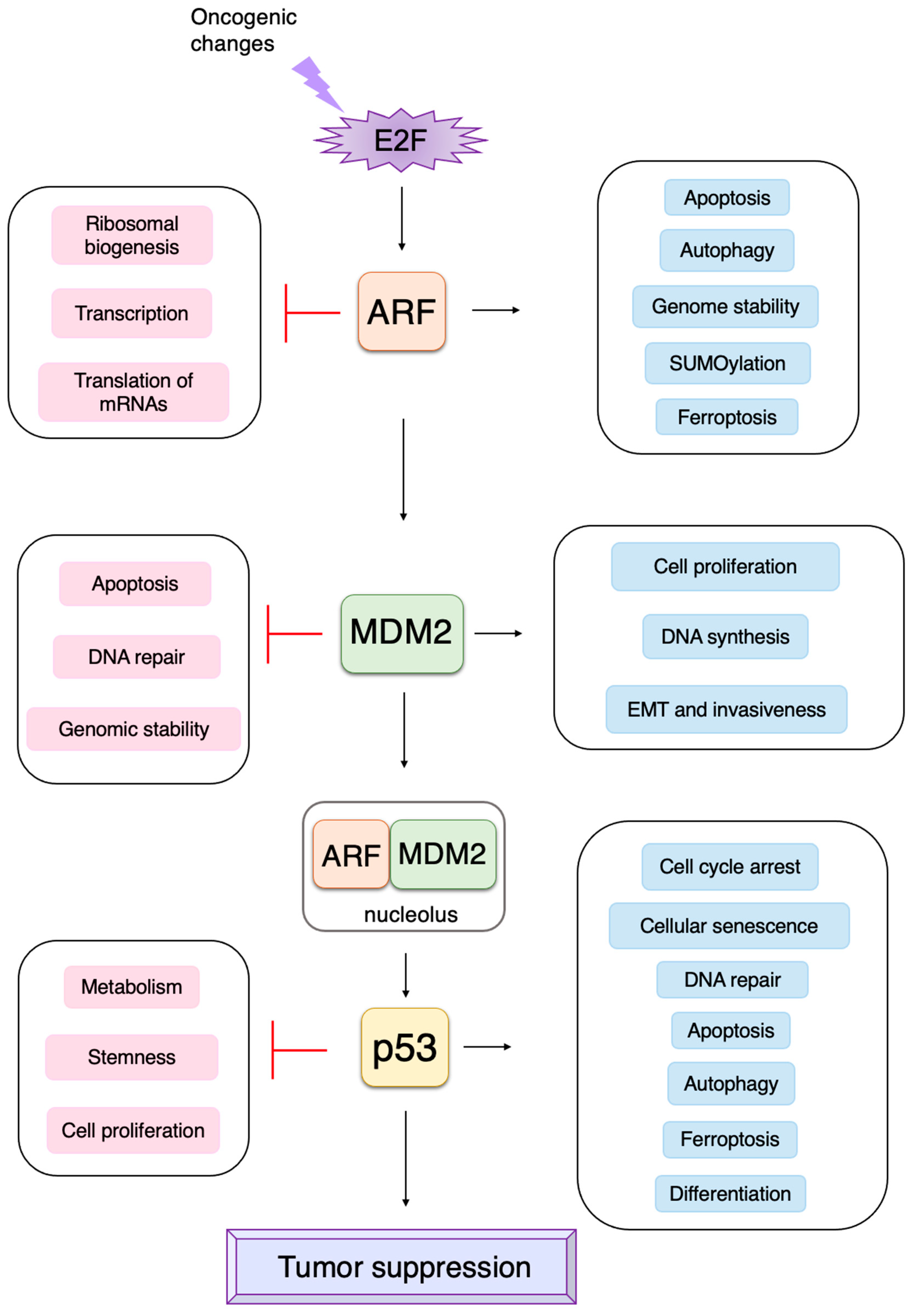
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Nakajima, R.; Shirasawa, M.; Fikriyanti, M.; Zhao, L.; Iwanaga, R.; Bradford, A.P.; Kurayoshi, K.; Araki, K.; Ohtani, K. Expanding Roles of the E2F-RB-p53 Pathway in Tumor Suppression. Biology 2023, 12, 1511. https://doi.org/10.3390/biology12121511
Zhou Y, Nakajima R, Shirasawa M, Fikriyanti M, Zhao L, Iwanaga R, Bradford AP, Kurayoshi K, Araki K, Ohtani K. Expanding Roles of the E2F-RB-p53 Pathway in Tumor Suppression. Biology. 2023; 12(12):1511. https://doi.org/10.3390/biology12121511
Chicago/Turabian StyleZhou, Yaxuan, Rinka Nakajima, Mashiro Shirasawa, Mariana Fikriyanti, Lin Zhao, Ritsuko Iwanaga, Andrew P. Bradford, Kenta Kurayoshi, Keigo Araki, and Kiyoshi Ohtani. 2023. "Expanding Roles of the E2F-RB-p53 Pathway in Tumor Suppression" Biology 12, no. 12: 1511. https://doi.org/10.3390/biology12121511