

Novel Covalent Modifier-Induced Local Conformational Changes within the Intrinsically Disordered Region of the Androgen Receptor

 ,
,  and
and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Systems and Method
2.1. Selection of Peptide Segments and Ligands
2.2. Simulation Setup and MD Procedure
2.3. Analysis of Conformations
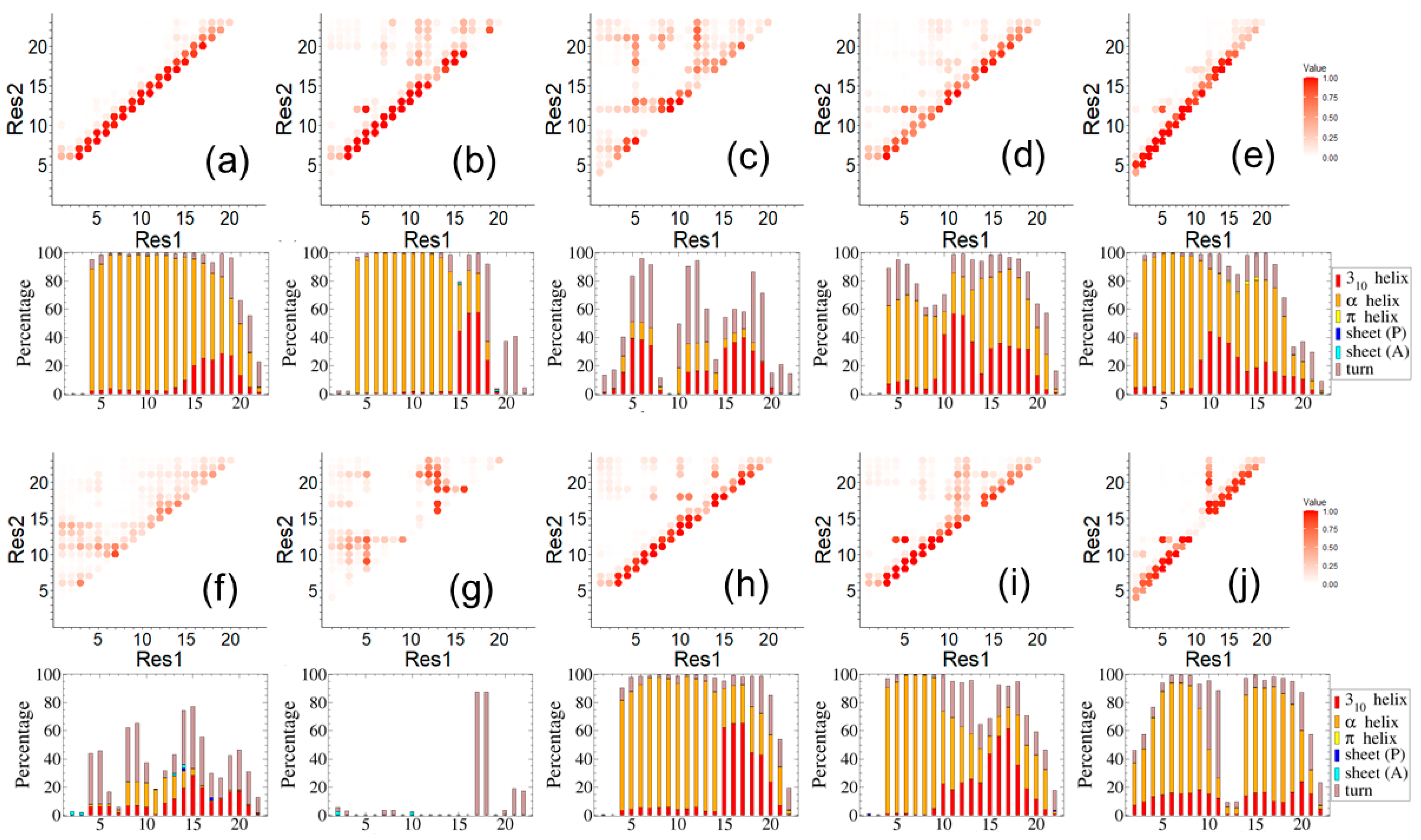
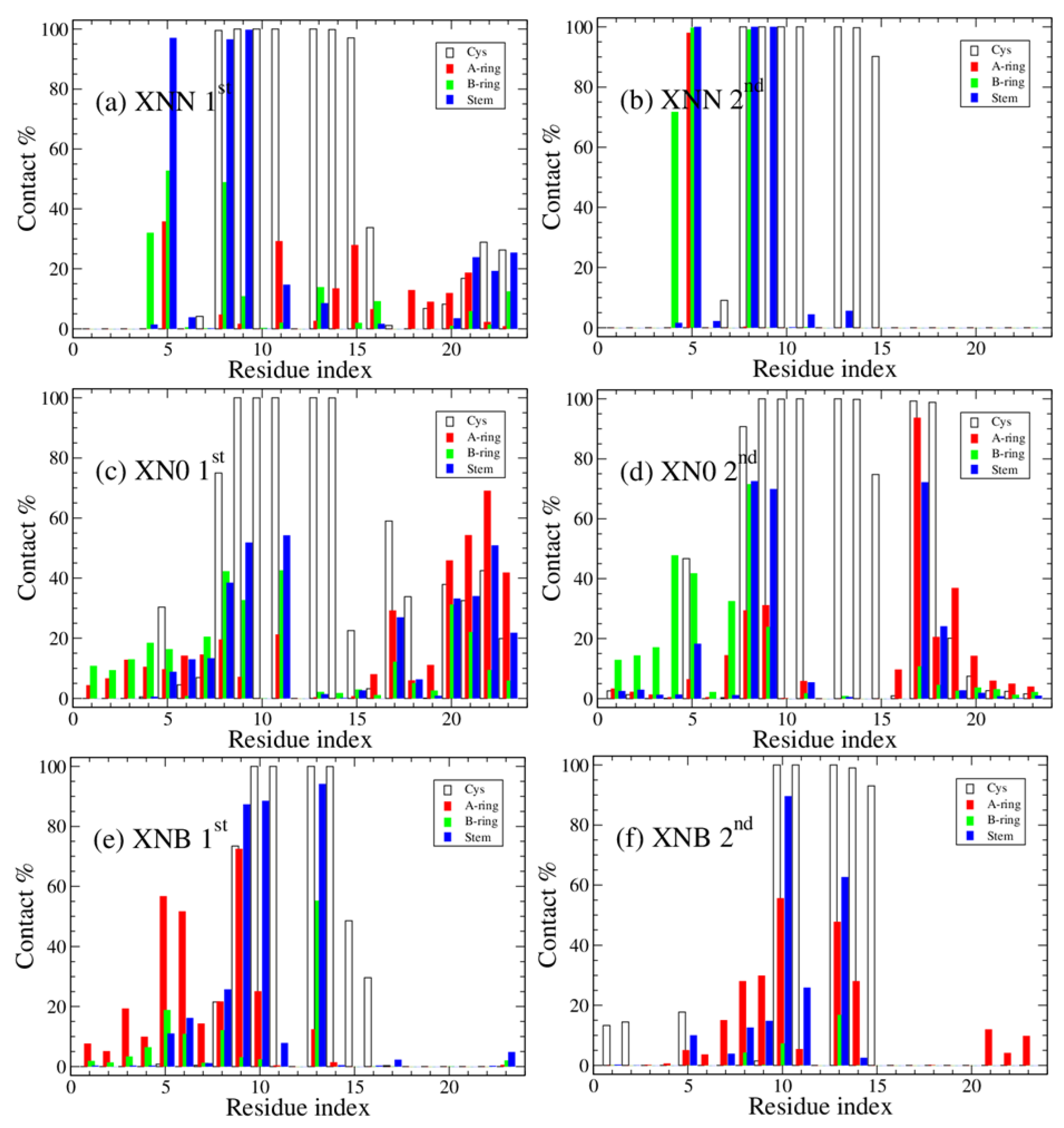
3. Results and Discussion
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically disordered proteins: A 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Basile, W.; Salvatore, M.; Bassot, C.; Elofsson, A. Why do eukaryotic proteins contain more intrinsically disordered regions? PLoS Comput. Biol. 2019, 15, e1007186. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Wang, J.; Choi, J.-M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 2018, 174, 688–699.e616. [Google Scholar] [CrossRef]
- Schuster, B.S.; Dignon, G.L.; Tang, W.S.; Kelley, F.M.; Ranganath, A.K.; Jahnke, C.N.; Simpkins, A.G.; Regy, R.M.; Hammer, D.A.; Good, M.C.; et al. Identifying sequence perturbations to an intrinsically disordered protein that determine its phase-separation behavior. Proc. Natl. Acad. Sci. USA 2020, 117, 11421–11431. [Google Scholar] [CrossRef]
- Alen, P.; Claessens, F.; Verhoeven, G.; Rombauts, W.; Peeters, B. The Androgen Receptor Amino-Terminal Domain Plays a Key Role in p160 Coactivator-Stimulated Gene Transcription. Mol. Cell. Biol. 1999, 19, 6085–6097. [Google Scholar] [CrossRef]
- Kumar, R.; Thompson, E.B. Transactivation functions of the N-terminal domains of nuclear hormone receptors: Protein folding and coactivator interactions. Mol. Endocrinol. 2003, 17, 1–10. [Google Scholar] [CrossRef]
- Kallio, H.M.L.; Hieta, R.; Latonen, L.; Brofeldt, A.; Annala, M.; Kivinummi, K.; Tammela, T.L.; Nykter, M.; Isaacs, W.B.; Lilja, H.G.; et al. Constitutively active androgen receptor splice variants AR-V3, AR-V7 and AR-V9 are co-expressed in castration-resistant prostate cancer metastases. Br. J. Cancer 2018, 119, 347–356. [Google Scholar] [CrossRef]
- Xie, J.; He, H.; Kong, W.; Li, Z.; Gao, Z.; Xie, D.; Sun, L.; Fan, X.; Jiang, X.; Zheng, Q.; et al. Targeting androgen receptor phase separation to overcome antiandrogen resistance. Nat. Chem. Biol. 2022, 18, 1341–1350. [Google Scholar] [CrossRef]
- Tan, M.H.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.-l. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef]
- Thiyagarajan, T.; Ponnusamy, S.; Hwang, D.-J.; He, Y.; Asemota, S.; Young, K.L.; Johnson, D.L.; Bocharova, V.; Zhou, W.; Jain, A.K.; et al. Inhibiting androgen receptor splice variants with cysteine-selective irreversible covalent inhibitors to treat prostate cancer. Proc. Natl. Acad. Sci. USA 2023, 120, e2211832120. [Google Scholar] [CrossRef] [PubMed]
- Reid, J.; Kelly, S.M.; Watt, K.; Price, N.C.; McEwan, I.J. Conformational analysis of the androgen receptor amino-terminal domain involved in transactivation. Influence of structure-stabilizing solutes and protein-protein interactions. J. Biol. Chem. 2002, 277, 20079–20086. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Dormann, D. Liquid-Liquid Phase Separation in Disease. Annu. Rev. Genet. 2019, 53, 171–194. [Google Scholar] [CrossRef]
- Tong, X.; Tang, R.; Xu, J.; Wang, W.; Zhao, Y.; Yu, X.; Shi, S. Liquid–liquid phase separation in tumor biology. Signal Transduct. Target. Ther. 2022, 7, 221. [Google Scholar] [CrossRef] [PubMed]
- McEwan, I.J. Breaking apart condensates. Nat. Chem. Biol. 2022, 18, 1292–1293. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; You, K.; Chen, T.; Hou, C.; Liang, Z.; Liu, M.; Wang, J.; Wei, T.; Qin, J.; Chen, Y.; et al. Quantifying the phase separation property of chromatin-associated proteins under physiological conditions using an anti-1,6-hexanediol index. Genome Biol. 2021, 22, 229. [Google Scholar] [CrossRef] [PubMed]
- Chemes, L.B.; Alonso, L.G.; Noval, M.G.; de Prat-Gay, G. Circular dichroism techniques for the analysis of intrinsically disordered proteins and domains. Methods Mol. Biol. 2012, 895, 387–404. [Google Scholar] [CrossRef]
- Micsonai, A.; Moussong, É.; Murvai, N.; Tantos, Á.; Tőke, O.; Réfrégiers, M.; Wien, F.; Kardos, J. Disordered–Ordered Protein Binary Classification by Circular Dichroism Spectroscopy. Front. Mol. Biosci. 2022, 9, 863141. [Google Scholar] [CrossRef]
- Nygaard, M.; Kragelund, B.B.; Papaleo, E.; Lindorff-Larsen, K. An Efficient Method for Estimating the Hydrodynamic Radius of Disordered Protein Conformations. Biophys. J. 2017, 113, 550–557. [Google Scholar] [CrossRef]
- Trexler, A.J.; Rhoades, E. Single Molecule Characterization of α-Synuclein in Aggregation-Prone States. Biophys. J. 2010, 99, 3048–3055. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Olsson, T.S.G.; Bowden, S.J.; Hall, R.J.; Verdonk, M.L.; Liebeschuetz, J.W.; Cole, J.C. Potential and Limitations of Ensemble Docking. J. Chem. Inf. Model. 2012, 52, 1262–1274. [Google Scholar] [CrossRef]
- Stone, L. UT-34: A promising new AR degrader. Nat. Rev. Urol. 2019, 16, 640. [Google Scholar] [CrossRef]
- Ponnusamy, S.; He, Y.; Hwang, D.-J.; Thiyagarajan, T.; Houtman, R.; Bocharova, V.; Sumpter, B.G.; Fernandez, E.; Johnson, D.; Du, Z.; et al. Orally Bioavailable Androgen Receptor Degrader, Potential Next-Generation Therapeutic for Enzalutamide-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 6764–6780. [Google Scholar] [CrossRef]
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The nuclear receptor superfamily: A structural perspective. Protein Sci. 2018, 27, 1876–1892. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The Nuclear Receptor Superfamily: The Second Decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Chandra, V.; Rastinejad, F. Structural Overview of the Nuclear Receptor Superfamily: Insights into Physiology and Therapeutics. Annu. Rev. Physiol. 2010, 72, 247–272. [Google Scholar] [CrossRef]
- Yu, X.; Yi, P.; Hamilton, R.A.; Shen, H.; Chen, M.; Foulds, C.E.; Mancini, M.A.; Ludtke, S.J.; Wang, Z.; O’Malley, B.W. Structural Insights of Transcriptionally Active, Full-Length Androgen Receptor Coactivator Complexes. Mol. Cell 2020, 79, 812–823.e4. [Google Scholar] [CrossRef]
- Porter, B.A.; Ortiz, M.A.; Bratslavsky, G.; Kotula, L. Structure and Function of the Nuclear Receptor Superfamily and Current Targeted Therapies of Prostate Cancer. Cancers 2019, 11, 1852. [Google Scholar] [CrossRef]
- Bohl, C.E.; Miller, D.D.; Chen, J.; Bell, C.E.; Dalton, J.T. Structural basis for accommodation of nonsteroidal ligands in the androgen receptor. J. Biol. Chem. 2005, 280, 37747–37754. [Google Scholar] [CrossRef] [PubMed]
- Lonergan, P.E.; Tindall, D.J. Androgen receptor signaling in prostate cancer development and progression. J. Carcinog. 2011, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- De Mol, E.; Szulc, E.; Di Sanza, C.; Martínez-Cristóbal, P.; Bertoncini, C.W.; Fenwick, R.B.; Frigolé-Vivas, M.; Masín, M.; Hunter, I.; Buzón, V.; et al. Regulation of Androgen Receptor Activity by Transient Interactions of Its Transactivation Domain with General Transcription Regulators. Structure 2018, 26, 145–152.e143. [Google Scholar] [CrossRef] [PubMed]
- Boike, L.; Henning, N.J.; Nomura, D.K. Advances in covalent drug discovery. Nat. Rev. Drug Discov. 2022, 21, 881–898. [Google Scholar] [CrossRef]
- Smith, A.J.T.; Zhang, X.; Leach, A.G.; Houk, K.N. Beyond Picomolar Affinities: Quantitative Aspects of Noncovalent and Covalent Binding of Drugs to Proteins. J. Med. Chem. 2009, 52, 225–233. [Google Scholar] [CrossRef]
- Li, J.; White, J.T.; Saavedra, H.; Wrabl, J.O.; Motlagh, H.N.; Liu, K.; Sowers, J.; Schroer, T.A.; Thompson, E.B.; Hilser, V.J. Genetically tunable frustration controls allostery in an intrinsically disordered transcription factor. eLife 2017, 6, e30688. [Google Scholar] [CrossRef]
- McCammon, J.A.; Harvey, S.C. Dynamics of Proteins and Nucleic Acids; Cambridge University Press: Cambridge, UK, 1988. [Google Scholar]
- Sethi, A.; Tian, J.; Vu, D.M.; Gnanakaran, S. Identification of Minimally Interacting Modules in an Intrinsically Disordered Protein. Biophys. J. 2012, 103, 748–757. [Google Scholar] [CrossRef]
- Lindsay, R.J.; Mansbach, R.A.; Gnanakaran, S.; Shen, T. Effects of pH on an IDP conformational ensemble explored by molecular dynamics simulation. Biophys. Chem. 2021, 271, 106552. [Google Scholar] [CrossRef]
- Papageorgiou, L.; Shalzi, L.; Efthimiadou, A.; Bacopoulou, F.; Chrousos, G.P.; Eliopoulos, E.; Vlachakis, D. Conserved functional motifs of the nuclear receptor superfamily as potential pharmacological targets. Int. J. Epigenetics 2021, 1, 3. [Google Scholar] [CrossRef]
- He, B.; Wang, K.; Liu, Y.; Xue, B.; Uversky, V.N.; Dunker, A.K. Predicting intrinsic disorder in proteins: An overview. Cell Res. 2009, 19, 929–949. [Google Scholar] [CrossRef]
- Lavery, D.N.; McEwan, I.J. Functional Characterization of the Native NH2-Terminal Transactivation Domain of the Human Androgen Receptor: Binding Kinetics for Interactions with TFIIF and SRC-1a. Biochemistry 2008, 47, 3352–3359. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves-Kulik, M.; Mier, P.; Kastano, K.; Cortés, J.; Bernadó, P.; Schmid, F.; Andrade-Navarro, M.A. Low Complexity Induces Structure in Protein Regions Predicted as Intrinsically Disordered. Biomolecules 2022, 12, 1098. [Google Scholar] [CrossRef] [PubMed]
- Sheikhhassani, V.; Scalvini, B.; Ng, J.; Heling, L.; Ayache, Y.; Evers, T.M.J.; Estébanez-Perpiñá, E.; McEwan, I.J.; Mashaghi, A. Topological dynamics of an intrinsically disordered N-terminal domain of the human androgen receptor. Protein Sci. 2022, 31, e4334. [Google Scholar] [CrossRef]
- Skolnick, J.; Gao, M.; Zhou, H.; Singh, S. AlphaFold 2: Why It Works and Its Implications for Understanding the Relationships of Protein Sequence, Structure, and Function. J. Chem. Inf. Model. 2021, 61, 4827–4831. [Google Scholar] [CrossRef] [PubMed]
- Tien, A.H.; Sadar, M.D. Order within a Disordered Structure. Structure 2018, 26, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham III, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Johnson, Q.R.; Lindsay, R.J.; Shen, T. CAMERRA: An analysis tool for the computation of conformational dynamics by evaluating residue–residue associations. J. Comput. Chem. 2018, 39, 1568–1578. [Google Scholar] [CrossRef]
- Kolinski, A.; Godzik, A.; Skolnick, J. A general method for the prediction of the three dimensional structure and folding pathway of globular proteins: Application to designed helical proteins. J. Chem. Phys. 1993, 98, 7420–7433. [Google Scholar] [CrossRef]
- Foutch, D.; Pham, B.; Shen, T. Protein conformational switch discerned via network centrality properties. Comput. Struct. Biotechnol. J. 2021, 19, 3599–3608. [Google Scholar] [CrossRef]
- Lindsay, R.J.; Pham, B.; Shen, T.; McCord, R.P. Characterizing the 3D structure and dynamics of chromosomes and proteins in a common contact matrix framework. Nucleic Acids Res. 2018, 46, 8143–8152. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Golloshi, R.; McCord, R.P.; Shen, T. Using contact statistics to characterize structure transformation of biopolymer ensembles. Phys. Rev. E 2020, 101, 012419. [Google Scholar] [CrossRef] [PubMed]
- Brunton, S.L.; Kutz, J.N. Data-Driven Science and Engineering: Machine Learning, Dynamical Systems, and Control; Cambridge University Press: Cambridge, UK, 2019. [Google Scholar]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2020, arXiv:1802.03426. [Google Scholar] [CrossRef]
- Jolliffe, I.T. Principal Component Analysis for Special Types of Data; Springer: Berlin/Heidelberg, Germany, 2002. [Google Scholar]
- Balsera, M.A.; Wriggers, W.; Oono, Y.; Schulten, K. Principal Component Analysis and Long Time Protein Dynamics. J. Phys. Chem. 1996, 100, 2567–2572. [Google Scholar] [CrossRef]
- García, A.E. Large-amplitude nonlinear motions in proteins. Phys. Rev. Lett. 1992, 68, 2696–2699. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, O.F.; Jas, G.S.; Lin, M.M.; Zewail, A.H. Primary Peptide Folding Dynamics Observed with Ultrafast Temperature Jump. Angew. Chem. Int. Ed. 2009, 48, 5628–5632. [Google Scholar] [CrossRef]
- Lin, M.M.; Mohammed, O.F.; Jas, G.S.; Zewail, A.H. Speed limit of protein folding evidenced in secondary structure dynamics. Proc. Natl. Acad. Sci. USA 2011, 108, 16622–16627. [Google Scholar] [CrossRef]
- De Sancho, D.; Best, R.B. What Is the Time Scale for α-Helix Nucleation? J. Am. Chem. Soc. 2011, 133, 6809–6816. [Google Scholar] [CrossRef]
- Daggett, V.; Levitt, M. Molecular dynamics simulations of helix denaturation. J. Mol. Biol. 1992, 223, 1121–1138. [Google Scholar] [CrossRef]
- Chavez, L.L.; Onuchic, J.N.; Clementi, C. Quantifying the Roughness on the Free Energy Landscape: Entropic Bottlenecks and Protein Folding Rates. J. Am. Chem. Soc. 2004, 126, 8426–8432. [Google Scholar] [CrossRef] [PubMed]
- Johnson, Q.; Doshi, U.; Shen, T.; Hamelberg, D. Water’s Contribution to the Energetic Roughness from Peptide Dynamics. J. Chem. Theory Comput. 2010, 6, 2591–2597. [Google Scholar] [CrossRef] [PubMed]
- Farahi, N.; Lazar, T.; Wodak, S.J.; Tompa, P.; Pancsa, R. Integration of Data from Liquid-Liquid Phase Separation Databases Highlights Concentration and Dosage Sensitivity of LLPS Drivers. Int. J. Mol. Sci. 2021, 22, 3017. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, A.K.; Nutter-Upham, A.; Lindquist, S.; King, O.D. PLAAC: A web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics 2014, 30, 2501–2502. [Google Scholar] [CrossRef]
- Mollica, L.; Bessa, L.M.; Hanoulle, X.; Jensen, M.R.; Blackledge, M.; Schneider, R. Binding Mechanisms of Intrinsically Disordered Proteins: Theory, Simulation, and Experiment. Front. Mol. Biosci. 2016, 3, 52. [Google Scholar] [CrossRef]
- Shoemaker, B.A.; Portman, J.J.; Wolynes, P.G. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Natl. Acad. Sci. USA 2000, 97, 8868–8873. [Google Scholar] [CrossRef]
- Ilie, I.M.; Caflisch, A. Simulation Studies of Amyloidogenic Polypeptides and Their Aggregates. Chem. Rev. 2019, 119, 6956–6993. [Google Scholar] [CrossRef]
- Gui, X.; Feng, S.; Li, Z.; Li, Y.; Reif, B.; Shi, B.; Niu, Z. Liquid-liquid phase separation of amyloid-beta oligomers modulates amyloid fibrils formation. J. Biol. Chem. 2023, 299, 102926. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Notation | Center | S.C. | Ligand | Run Time | Ions | H2O# |
|---|---|---|---|---|---|---|
| XNN_4H | 406 | H | XNN | 100 ns | 1 Cl− | 2701 |
| XNN_4E | 406 | E | XNN | 100 ns | 1 Cl− | 6898 |
| NTD_4H | 406 | H | - | 100 ns | 1 Cl− | 2731 |
| NTD_4E | 406 | E | - | 100 ns | 1 Cl− | 5570 |
| XN0_4H | 406 | H | XN0 | 100 ns | 1 Cl− | 2687 |
| XNB_4H | 406 | H | XNB | 100 ns | 1 Cl− | 2886 |
| XNN_3H | 327 | H | XNN | 100 ns | 1 Na+ | 2701 |
| XNN_3E | 327 | E | XNN | 50 ns | 1 Na+ | 6880 |
| NTD_3H | 327 | H | - | 100 ns | 1 Na+ | 2522 |
| NTD_3E | 327 | E | - | 100 ns | 1 Na+ | 5460 |
| XNN_2H | 240 | H | XNN | 50 ns | - | 2980 |
| NTD_2H | 240 | H | - | 100 ns | - | 2774 |
| NTD_2E | 240 | E | - | 100 ns | - | 3415 |
| XEN_4H | 406 | H | XEN | 50 ns | 1 Cl− | 2695 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harnish, M.T.; Lopez, D.; Morrison, C.T.; Narayanan, R.; Fernandez, E.J.; Shen, T. Novel Covalent Modifier-Induced Local Conformational Changes within the Intrinsically Disordered Region of the Androgen Receptor. Biology 2023, 12, 1442. https://doi.org/10.3390/biology12111442
Harnish MT, Lopez D, Morrison CT, Narayanan R, Fernandez EJ, Shen T. Novel Covalent Modifier-Induced Local Conformational Changes within the Intrinsically Disordered Region of the Androgen Receptor. Biology. 2023; 12(11):1442. https://doi.org/10.3390/biology12111442
Chicago/Turabian StyleHarnish, Michael T., Daniel Lopez, Corbin T. Morrison, Ramesh Narayanan, Elias J. Fernandez, and Tongye Shen. 2023. "Novel Covalent Modifier-Induced Local Conformational Changes within the Intrinsically Disordered Region of the Androgen Receptor" Biology 12, no. 11: 1442. https://doi.org/10.3390/biology12111442
APA StyleHarnish, M. T., Lopez, D., Morrison, C. T., Narayanan, R., Fernandez, E. J., & Shen, T. (2023). Novel Covalent Modifier-Induced Local Conformational Changes within the Intrinsically Disordered Region of the Androgen Receptor. Biology, 12(11), 1442. https://doi.org/10.3390/biology12111442