
Cytoskeletal Responses and Aif-1 Expression in Caco-2 Monolayers Exposed to Phorbol-12-Myristate-13-Acetate and Carnosine


 ,
,  and
and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Cell Culture and Treatments
2.3. Fluorescence Imaging of Cytoskeleton/Nuclei and Quantitative Image Analyses
2.4. Immunocytoichemistry
2.5. Total RNA and Protein Extraction
2.6. Primer Design and Real Time PCR (qPCR) Assays
2.7. Western Blotting
2.8. Statistical Analysis
3. Results
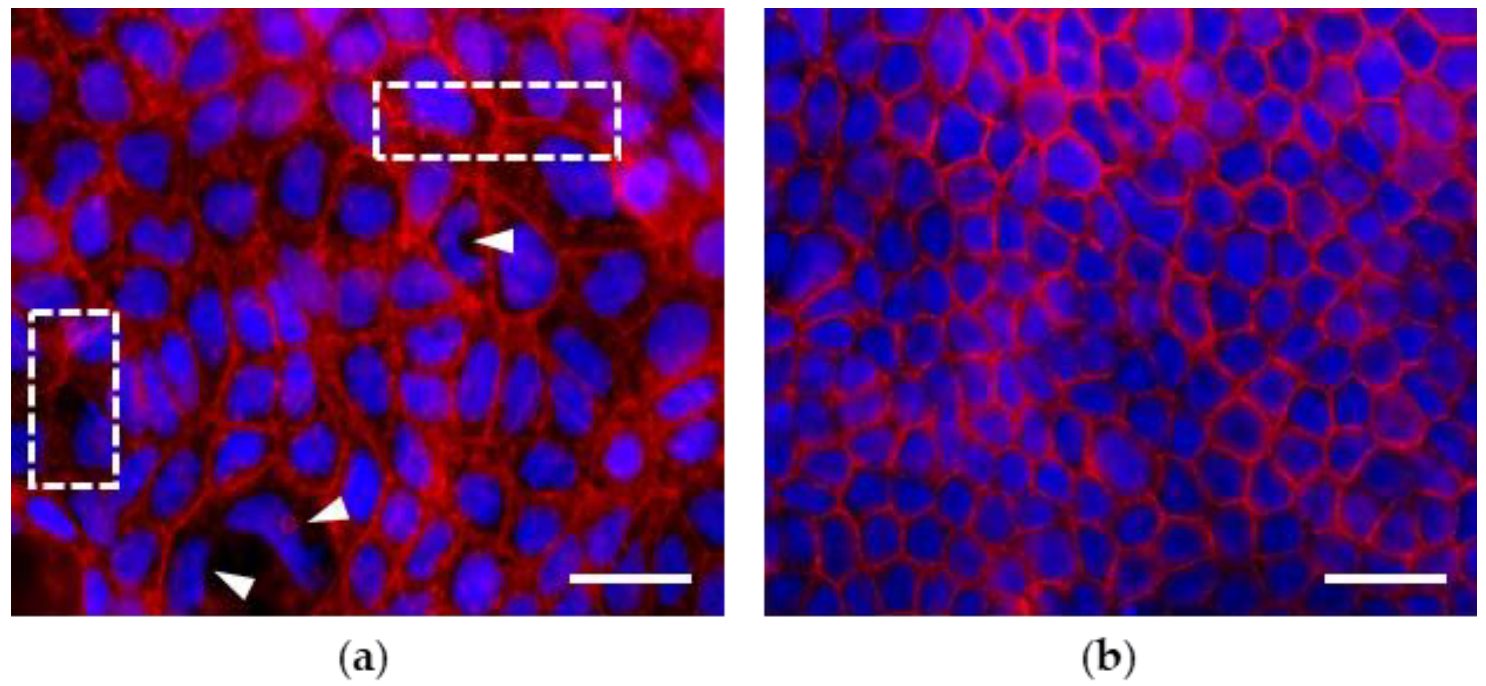
3.1. Morphological Features of Actin Cytoskeleton in Caco-2 Cell Monolayers Grown up to 7 and 21 Days Post Seeding (dps)
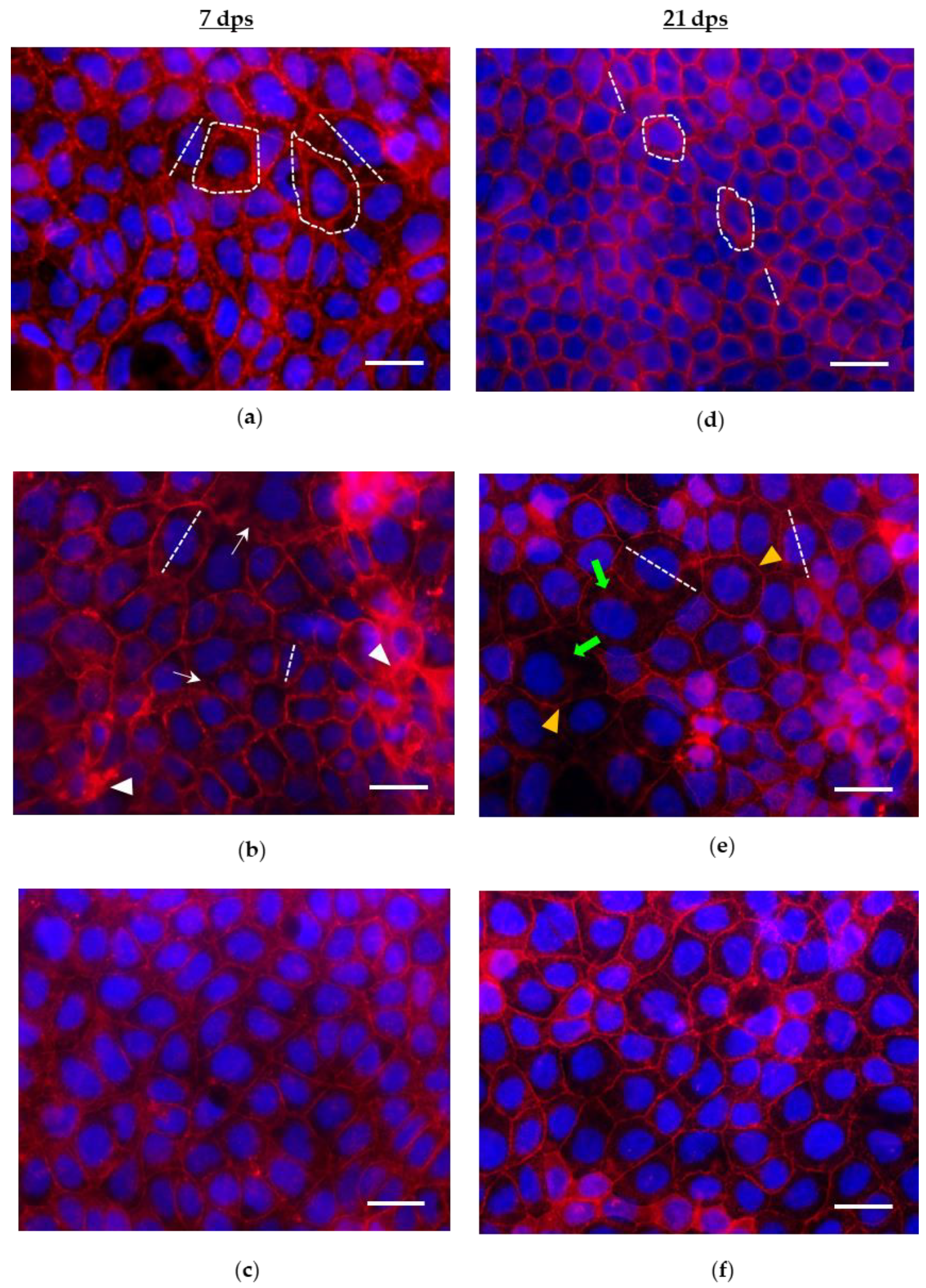
3.2. Effects of PMA Treatments on Morphology, Morphometry and Actin Cytoskeleton of Cells in Monolayers at 7 and 21 dps
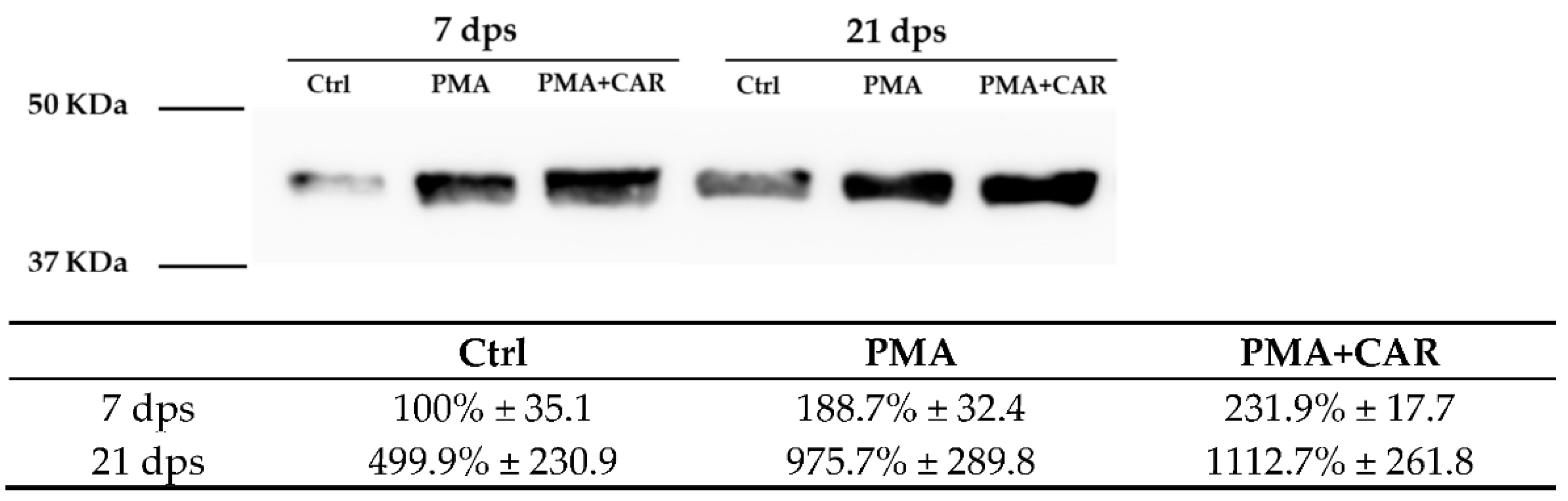
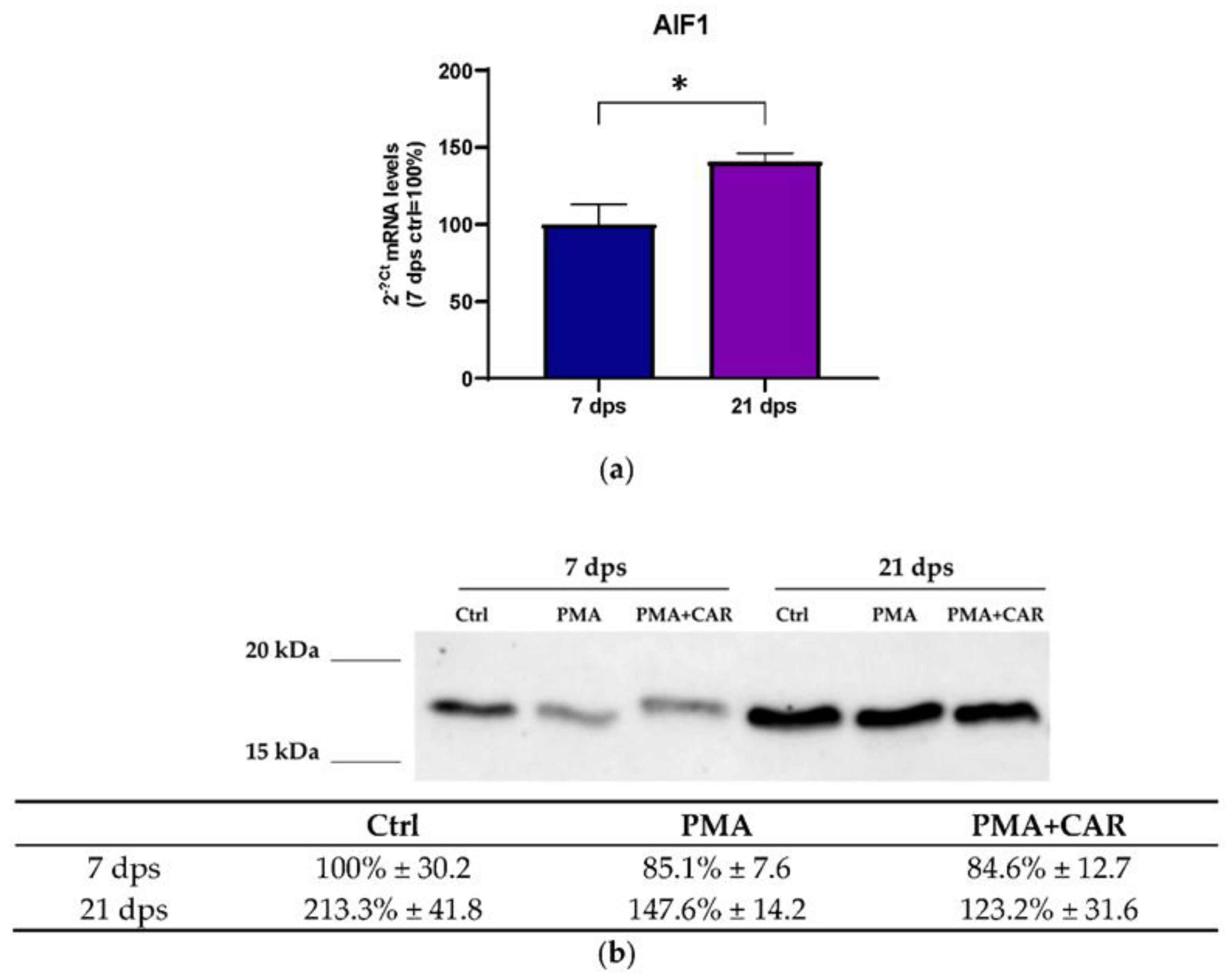
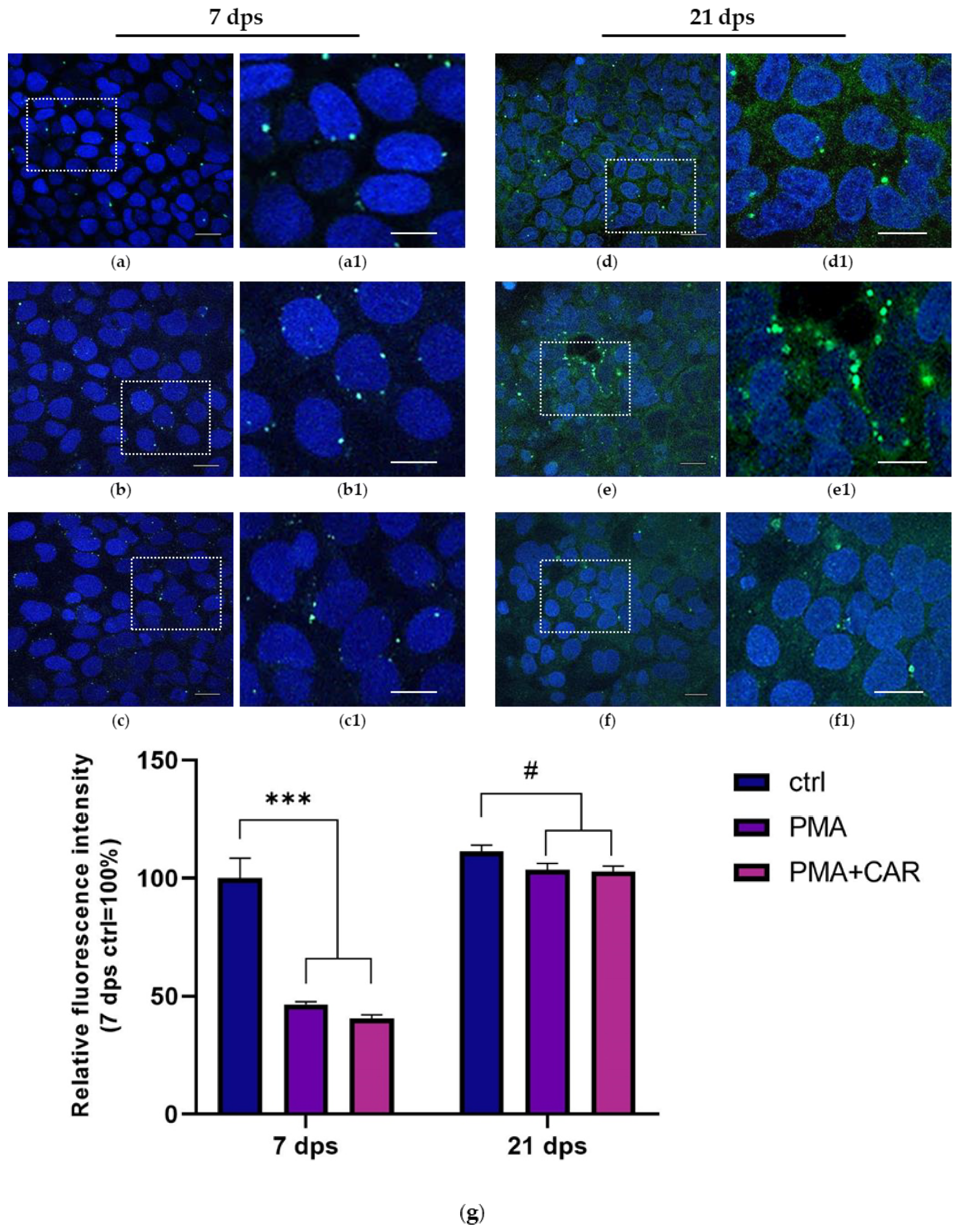
3.3. Identification of the Allograft Inflammatory Factor 1 (AIF-1) Gene Products in 7 and 21 dps Caco-2 Monolayers Exposed to PMA Challenge
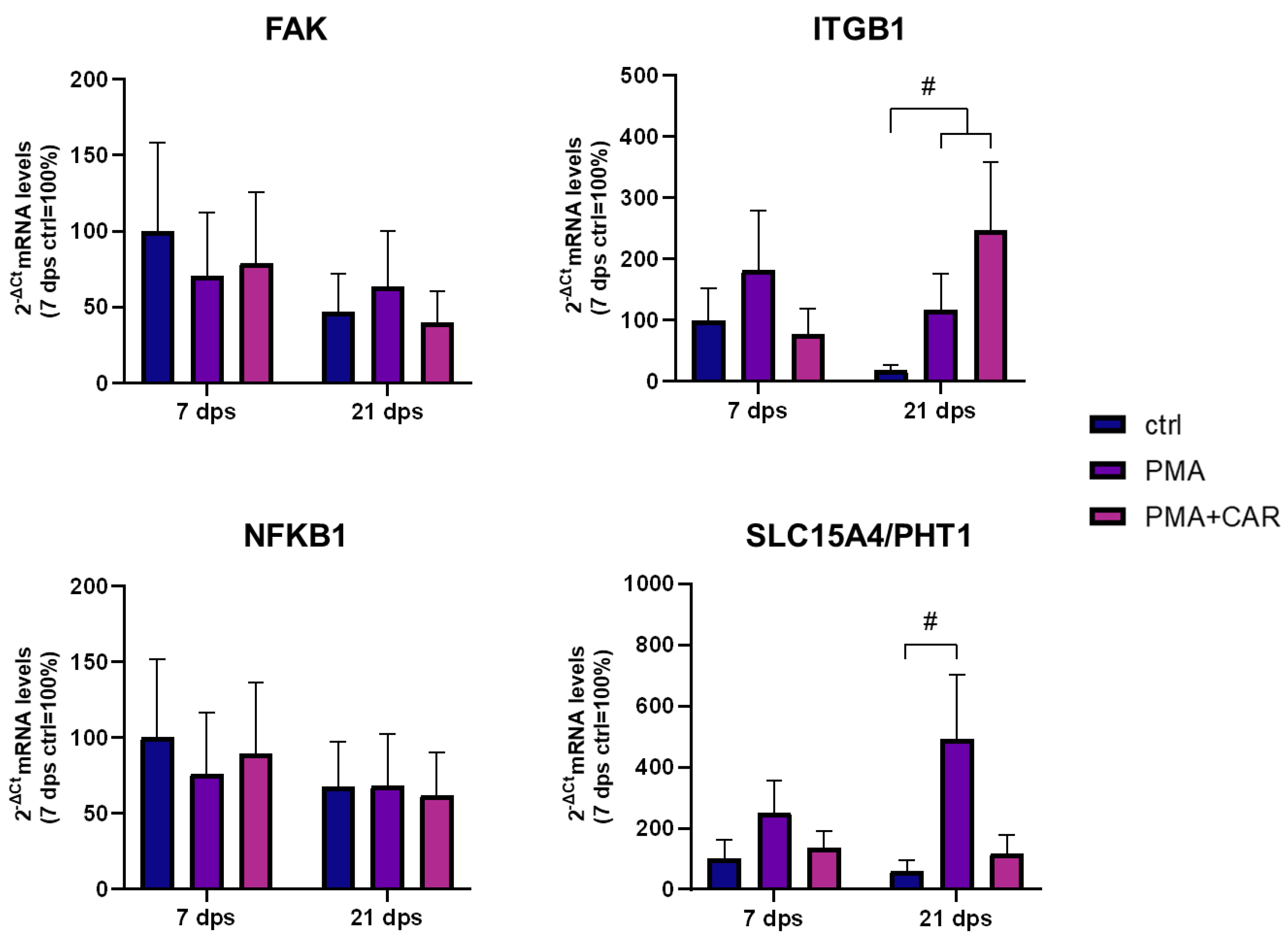
3.4. Cytoskeleton-/Inflammation-Related mRNA Expression Variations Induced by PMA Treatments in 7 and 21 dps Caco-2 Monolayers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M.; Madsen, K.; Spiller, R.; Greenwood-Van Meerveld, B.; Verne, G.N. Intestinal barrier function in health and gastrointestinal disease. Neurogastroenterol. Motil. 2012, 24, 503–512, Erratum in Neurogastroenterol. Motil. 2012, 24, 976. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.I.; Parkos, C.A.; Nusrat, A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am. J. Pathol. 2010, 177, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Esmaeilniakooshkghazi, A.; Patnaik, S.; Wang, Y.; George, S.P.; Ahrorov, A.; Hou, J.K.; Herron, A.J.; Sesaki, H.; Khurana, S. Villin-1 and Gelsolin Regulate Changes in Actin Dynamics That Affect Cell Survival Signaling Pathways and Intestinal Inflammation. Gastroenterology 2018, 154, 1405–1420.e2. [Google Scholar] [CrossRef]
- Werner, T.; Haller, D. Intestinal epithelial cell signalling and chronic inflammation: From the proteome to specific molecular mechanisms. Mutat. Res. 2007, 622, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.T.; Ghosh, C.; Hossain, M.; Linfield, D.; Rezaee, F.; Janigro, D.; Marchi, N.; van Boxel-Dezaire, A.H.H. IFN-γ, IL-17A, or zonulin rapidly increase the permeability of the blood-brain and small intestinal epithelial barriers: Relevance for neuro-inflammatory diseases. Biochem. Biophys. Res. Commun. 2018, 507, 274–279. [Google Scholar] [CrossRef]
- Utech, M.; Ivanov, A.I.; Samarin, S.N.; Bruewer, M.; Turner, J.R.; Mrsny, R.J.; Parkos, C.A.; Nusrat, A. Mechanism of IFN-gamma-induced endocytosis of tight junction proteins: Myosin II-dependent vacuolarization of the apical plasma membrane. Mol. Biol. Cell. 2005, 16, 5040–5052. [Google Scholar] [CrossRef]
- Musch, M.W.; Walsh-Reitz, M.M.; Chang, E.B. Roles of ZO-1, occludin, and actin in oxidant-induced barrier disruption. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G222–G231. [Google Scholar] [CrossRef]
- Lee, S.H.; Dominguez, R. Regulation of actin cytoskeleton dynamics in cells. Mol. Cells. 2010, 29, 311–325. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Zhang, L.; Yuan, X.; Hao, J.; Ni, J.; Hao, L. Upregulation of allograft inflammatory factor-1 expression and secretion by macrophages stimulated with aldosterone promotes renal fibroblasts to a profibrotic phenotype. Int. J. Mol. Med. 2018, 42, 861–872. [Google Scholar] [CrossRef]
- Kelemen, S.E.; Autieri, M.V. Expression of allograft inflammatory factor-1 in T lymphocytes: A role in T-lymphocyte activation and proliferative arteriopathies. Am. J. Pathol. 2005, 167, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Ohsawa, K.; Kanazawa, H.; Kohsaka, S.; Imai, Y. Iba1 is an actin-cross-linking protein in macrophages/microglia. Biochem. Biophys. Res. Commun. 2001, 286, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, H.; Ohsawa, K.; Sasaki, Y.; Kohsaka, S.; Imai, Y. Macrophage/microglia-specific protein Iba1 enhances membrane ruffling and Rac activation via phospholipase C-gamma -dependent pathway. J. Biol. Chem. 2002, 277, 20026–20032. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, K.; Imai, Y.; Kanazawa, H.; Sasaki, Y.; Kohsaka, S. Involvement of Iba1 in membrane ruffling and phagocytosis of macrophages/microglia. J. Cell. Sci. 2000, 113 Pt 17, 3073–3084. [Google Scholar] [CrossRef] [PubMed]
- Sikora, M.; Kopeć, B.; Piotrowska, K.; Pawlik, A. Role of allograft inflammatory factor-1 in pathogenesis of diseases. Immunol. Lett. 2020, 218, 1–4. [Google Scholar] [CrossRef]
- Zhao, Y.Y.; Yan, D.J.; Chen, Z.W. Role of AIF-1 in the regulation of inflammatory activation and diverse disease processes. Cell. Immunol. 2013, 284, 75–83. [Google Scholar] [CrossRef]
- Kishikawa, S.; Sato, S.; Kaneto, S.; Uchino, S.; Kohsaka, S.; Nakamura, S.; Kiyono, H. Allograft inflammatory factor 1 is a regulator of transcytosis in M cells. Nat. Commun. 2017, 8, 14509. [Google Scholar] [CrossRef]
- Jochems, P.G.M.; Garssen, J.; van Keulen, A.M.; Masereeuw, R.; Jeurink, P.V. Evaluating Human Intestinal Cell Lines for Studying Dietary Protein Absorption. Nutrients 2018, 10, 322. [Google Scholar] [CrossRef]
- Hidalgo, I.J.; Raub, T.J.; Borchardt, R.T. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology 1989, 96, 736–749. [Google Scholar] [CrossRef]
- Sambuy, Y.; De Angelis, I.; Ranaldi, G.; Scarino, M.L.; Stammati, A.; Zucco, F. The Caco-2 cell line as a model of the intestinal barrier: Influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell. Biol. Toxicol. 2005, 21, 1–26. [Google Scholar] [CrossRef]
- Olejnik, A.; Lewandowska, M.; Grajek, W.; Czaczyk, K. New rapid method of Caco-2 cell differentiation. Pol. J. Food Nutr. Sci. 2003, 12, 60–64. [Google Scholar]
- Kong, S.; Zhang, Y.H.; Zhang, W. Regulation of Intestinal Epithelial Cells Properties and Functions by Amino Acids. Biomed. Res. Int. 2018, 2018, 2819154. [Google Scholar] [CrossRef] [PubMed]
- DeMarco, V.G.; Li, N.; Thomas, J.; West, C.M.; Neu, J. Glutamine and barrier function in cultured Caco-2 epithelial cell monolayers. J. Nutr. 2003, 133, 2176–2179. [Google Scholar] [CrossRef] [PubMed]
- Son, D.O.; Satsu, H.; Shimizu, M. Histidine inhibits oxidative stress- and TNF-alpha-induced interleukin-8 secretion in intestinal epithelial cells. FEBS Lett. 2005, 579, 4671–4677. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Sharma, A.K.; Shastri, V.; Madhu, M.K.; Sharma, V.K. Prediction of anti-inflammatory proteins/peptides: An in silico approach. J. Transl. Med. 2017, 15, 7. [Google Scholar] [CrossRef]
- de Medina, F.S.; Daddaoua, A.; Requena, P.; Capitán-Cañadas, F.; Zarzuelo, A.; Dolores Suárez, M.; Martínez-Augustin, O. New insights into the immunological effects of food bioactive peptides in animal models of intestinal inflammation. Proc. Nutr. Soc. 2010, 69, 454–462. [Google Scholar] [CrossRef]
- Boldyrev, A.A.; Aldini, G.; Derave, W. Physiology and pathophysiology of carnosine. Physiol. Rev. 2013, 93, 1803–1845. [Google Scholar] [CrossRef]
- Hipkiss, A.R. Aging, Proteotoxicity, Mitochondria, Glycation, NAD and Carnosine: Possible Inter-Relationships and Resolution of the Oxygen Paradox. Front. Aging Neurosci. 2010, 2, 10. [Google Scholar] [CrossRef]
- Barca, A.; Ippati, S.; Urso, E.; Vetrugno, C.; Storelli, C.; Maffia, M.; Romano, A.; Verri, T. Carnosine modulates the Sp1-Slc31a1/Ctr1 copper-sensing system and influences copper homeostasis in murine CNS-derived cells. Am. J. Physiol. Cell. Physiol. 2019, 316, C235–C245. [Google Scholar] [CrossRef]
- Aldini, G.; Facino, R.M.; Beretta, G.; Carini, M. Carnosine and related dipeptides as quenchers of reactive carbonyl species: From structural studies to therapeutic perspectives. Biofactors 2005, 24, 77–87. [Google Scholar] [CrossRef]
- Fujii, K.; Abe, K.; Kadooka, K.; Matsumoto, T.; Katakura, Y. Carnosine activates the CREB pathway in Caco-2 cells. Cytotechnology 2017, 69, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, Y.; Onoue, S.; Tashiro, K.; Sato, M.; Hasegawa, T.; Katakura, Y. Carnosine induces intestinal cells to secrete exosomes that activate neuronal cells. PLoS ONE 2019, 14, e0217394. [Google Scholar] [CrossRef] [PubMed]
- Kadooka, K.; Fujii, K.; Matsumoto, T.; Sato, M.; Morimatsu, F.; Tashiro, K.; Kuhara, S.; Katakura, Y. Mechanisms and consequences of carnosine-induced activation of intestinal epithelial cells. J. Funct. Foods 2015, 13, 32–37. [Google Scholar] [CrossRef]
- Hewlings, S.; Kalman, D. A Review of Zinc-L-Carnosine and Its Positive Effects on Oral Mucositis, Taste Disorders, and Gastrointestinal Disorders. Nutrients 2020, 12, 665. [Google Scholar] [CrossRef] [PubMed]
- Son, D.O.; Satsu, H.; Kiso, Y.; Totsuka, M.; Shimizu, M. Inhibitory effect of carnosine on interleukin-8 production in intestinal epithelial cells through translational regulation. Cytokine 2008, 42, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.M.; Kitts, D.D. Determining conditions for nitric oxide synthesis in Caco-2 cells using Taguchi and factorial experimental designs. Anal. Biochem. 2008, 381, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Sumagin, R.; Robin, A.Z.; Nusrat, A.; Parkos, C.A. Activation of PKCβII by PMA facilitates enhanced epithelial wound repair through increased cell spreading and migration. PLoS ONE 2013, 8, e55775. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Tepperman, B.L.; Soper, B.D.; Chang, Q. Effect of protein kinase C activation on intracellular Ca2+ signaling and integrity of intestinal epithelial cells. Eur. J. Pharmacol. 2005, 518, 1–9. [Google Scholar] [CrossRef]
- Larsson, C. Protein kinase C and the regulation of the actin cytoskeleton. Cell. Signal. 2006, 18, 276–284. [Google Scholar] [CrossRef]
- Song, J.C.; Rangachari, P.K.; Matthews, J.B. Opposing effects of PKCalpha and PKCepsilon on basolateral membrane dynamics in intestinal epithelia. Am. J. Physiol. Cell. Physiol. 2002, 283, C1548–C1556. [Google Scholar] [CrossRef] [PubMed]
- Caviston, J.P.; Cohen, L.A.; Donaldson, J.G. Arf1 and Arf6 promote ventral actin structures formed by acute activation of protein kinase C and Src. Cytoskeleton 2014, 71, 380–394. [Google Scholar] [CrossRef] [PubMed]
- Davison, G.; Marchbank, T.; March, D.S.; Thatcher, R.; Playford, R.J. Zinc carnosine works with bovine colostrum in truncating heavy exercise-induced increase in gut permeability in healthy volunteers. Am. J. Clin. Nutr. 2016, 104, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Miao, L.; Wu, X.; Liu, G.; Peng, Y.; Xin, X.; Jiao, B.; Kong, X. Carnosine Inhibits the Proliferation of Human Gastric Carcinoma Cells by Retarding Akt/mTOR/p70S6K Signaling. J. Cancer 2014, 5, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef] [PubMed]
- Moujaber, O.; Stochaj, U. The Cytoskeleton as Regulator of Cell Signaling Pathways. Trends Biochem. Sci. 2020, 45, 96–107. [Google Scholar] [CrossRef]
- Furukawa, K.T.; Yamashita, K.; Sakurai, N.; Ohno, S. The Epithelial Circumferential Actin Belt Regulates YAP/TAZ through Nucleocytoplasmic Shuttling of Merlin. Cell Rep. 2017, 20, 1435–1447. [Google Scholar] [CrossRef]
- Chen, G.; Han, Y.; Feng, Y.; Wang, A.; Li, X.; Deng, S.; Zhang, L.; Xiao, J.; Li, Y.; Li, N. Extract of Ilex rotunda Thunb alleviates experimental colitis-associated cancer via suppressing inflammation-induced miR-31-5p/YAP overexpression. Phytomedicine 2019, 62, 152941. [Google Scholar] [CrossRef]
- Hageman, J.H.; Heinz, M.C.; Kretzschmar, K.; van der Vaart, J.; Clevers, H.; Snippert, H.J.G. Intestinal Regeneration: Regulation by the Microenvironment. Dev. Cell. 2020, 54, 435–446. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, Y.; Wu, H.; Barry, E.; Yin, Y.; Lawrence, E.; Dawson, D.; Willis, J.E.; Markowitz, S.D.; Camargo, F.D.; et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc. Natl. Acad. Sci. USA 2011, 108, E1312–E1320. [Google Scholar] [CrossRef]
- Kaur, H.; Moreau, R. Role of mTORC1 in intestinal epithelial repair and tumorigenesis. Cell Mol. Life Sci. 2019, 76, 2525–2546. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.; Ghazi, P.C.; Starchenko, A.; Tovaglieri, A.; Baldwin, K.R.; Poulin, E.J.; Gierut, J.J.; Genetti, C.; Yajnik, V.; Breault, D.T.; et al. The colonic epithelium plays an active role in promoting colitis by shaping the tissue cytokine profile. PLoS Biol. 2018, 16, e2002417. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, L.G.; Cano-Martínez, D.; Toledo-Lobo, M.V.; Ruiz-Llorente, L.; Chaparro, M.; Guerra, I.; Iborra, M.; Cabriada, J.L.; Bujanda, L.; Taxonera, C.; et al. Evaluation of AIF-1 (Allograft Inflammatory Factor-1) as a Biomarker of Crohn’s Disease Severity. Biomedicines 2022, 10, 727. [Google Scholar] [CrossRef] [PubMed]
- Román, I.D.; Cano-Martínez, D.; Lobo, M.V.; Fernández-Moreno, M.D.; Hernández-Breijo, B.; Sacristán, S.; Sanmartín-Salinas, P.; Monserrat, J.; Gisbert, J.P.; Guijarro, L.G. Infliximab therapy reverses the increase of allograft inflammatory factor-1 in serum and colonic mucosa of rats with inflammatory bowel disease. Biomarkers 2017, 22, 133–144. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE | RefSeq mRNA Acc. No. | Sense Primer 5′-3′ (Tm) | Antisense Primer 5′-3′ (Tm) | PCR Size (bp) |
|---|---|---|---|---|
| ACTB | NM_001101.5 | ATCGTGCGTGACATTAAGGAGA (56 °C) | TCCTCCCCTGGAGAAGAGCT (55 °C) | 102 |
| FAK | NM_001352694.2 | ATTAAATGGATGGCTCCA (54 °C) | CTCCCACATACACACACC (58 °C) | 121 |
| AIF1 | NM_001623.5 | GAAGCGAATGCTGGAGAA (54 °C) | ATCTCTTGCCCAGCATCA (54 °C) | 98 |
| ITGB1 | NM_002211.3 | CAAATGCCAAATCATGTGGA (51 °C) | TTCTCTGCTGTTCCTTTGCT (54 °C) | 226 |
| NFKB1b | NM_003998.4 | AATGCCTTCCGGCTGAGTC (59 °C) | AGGCTGCCTGGATCACTTCA (60 °C) | 140 |
| SLC15A4 | NM_145648.4 | TGAAGGCATTGGAGTCTTT (51 °C) | TGGAAATACACTGTCCAGTAA (51 °C) | 168 |
| GAPDH | NM_002046.7 | AAACCTGCCAAGTATGATGA (51 °C) | TACTCCTTGGAGGCCATGT (54 °C) | 217 |
| Ctrl | PMA | PMA+CAR | |
|---|---|---|---|
| Cell count (%) | |||
| 7 dps | 100 ± 6.1 (n = 24 ± 1) | 86.5 ± 13.9 (n = 21 ± 3) | 87.5 ± 4.5 (n = 21 ± 1) |
| 21 dps | 244.8 ± 20.2 ## (n = 59 ± 5) (100 ± 8.3) | 87.5 ± 19.2 (n = 21 ± 5) (35.7 ± 7.8 *) | 109.4 ± 10.3 (n = 26 ± 2) (44.7 ± 4.2 *) |
| Diameter max length (%) | |||
| 7 dps | 100 ± 5.9 | 94.5 ± 4.8 | 100.7 ± 3.7 |
| 21 dps | 48.8 ± 1.3 #### (100 ± 2.7) | 112.3 ± 6.0 (230.3 ± 12.3 ****) | 104.6 ± 6.3 (214.4 ± 13.0 ****) |
| Area (%) | |||
| 7 dps | 100 ± 8.8 | 115.9 ± 17.5 | 108.5 ± 8.9 |
| 21 dps | 41.7 ± 1.8 ## (100 ± 4.3) | 161.0 ± 26.2 (386.5 ± 62.9 *) | 94.8 ± 11.5 (227.64 ± 27.5 *) |
| Fluorescence intensity (%) | |||
| 7 dps | 100 ± 11.5 | 115.4 ± 19.6 | 81.2 ± 4.5 |
| 21 dps | 55.6 ± 4.6 # (100 ± 8.2) | 129.2 ± 10.6 (232.4 ± 19.1 **) | 40.6 ± 3.4 (73.1 ± 6.1 *) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzei, A.; Pagliara, P.; Del Vecchio, G.; Giampetruzzi, L.; Croce, F.; Schiavone, R.; Verri, T.; Barca, A. Cytoskeletal Responses and Aif-1 Expression in Caco-2 Monolayers Exposed to Phorbol-12-Myristate-13-Acetate and Carnosine. Biology 2023, 12, 36. https://doi.org/10.3390/biology12010036
Mazzei A, Pagliara P, Del Vecchio G, Giampetruzzi L, Croce F, Schiavone R, Verri T, Barca A. Cytoskeletal Responses and Aif-1 Expression in Caco-2 Monolayers Exposed to Phorbol-12-Myristate-13-Acetate and Carnosine. Biology. 2023; 12(1):36. https://doi.org/10.3390/biology12010036
Chicago/Turabian StyleMazzei, Aurora, Patrizia Pagliara, Gianmarco Del Vecchio, Lucia Giampetruzzi, Francesca Croce, Roberta Schiavone, Tiziano Verri, and Amilcare Barca. 2023. "Cytoskeletal Responses and Aif-1 Expression in Caco-2 Monolayers Exposed to Phorbol-12-Myristate-13-Acetate and Carnosine" Biology 12, no. 1: 36. https://doi.org/10.3390/biology12010036
APA StyleMazzei, A., Pagliara, P., Del Vecchio, G., Giampetruzzi, L., Croce, F., Schiavone, R., Verri, T., & Barca, A. (2023). Cytoskeletal Responses and Aif-1 Expression in Caco-2 Monolayers Exposed to Phorbol-12-Myristate-13-Acetate and Carnosine. Biology, 12(1), 36. https://doi.org/10.3390/biology12010036