
Alcohol and HIV-Derived Hepatocyte Apoptotic Bodies Induce Hepatic Stellate Cell Activation

 ,
,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. In Vitro Studies
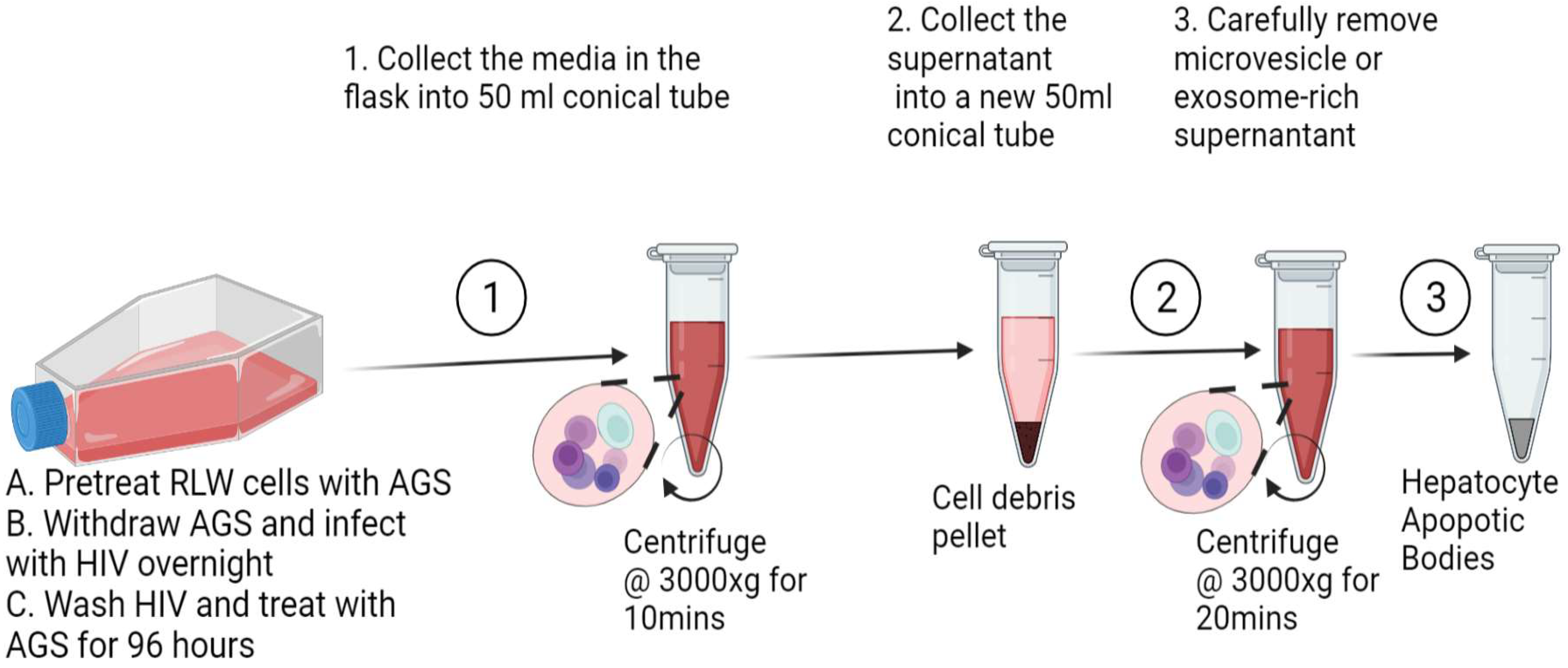
2.3. Isolation of ABs from RLW Cells
2.4. Generation of TAMRA-Labeled RLW ABs by Prolonged Incubation
2.5. siRNA Transfection of LX2 Cells
2.6. Imaging of ABs by Transmission Electron Microscopy
2.7. In Vivo Studies
- Control-fed mice exposed to (A) uninfected and (B) HIV-infected ABs generated from RLW cells.
- Ethanol-fed mice exposed to (A) uninfected and (B) HIV-infected ABs from RLW cells. Each subgroup contains 3 mice.
2.8. RNA Isolation and RT-PCR
2.9. Immunoblotting
2.10. Immunofluorescence
2.11. Statistical Analyses
3. Results
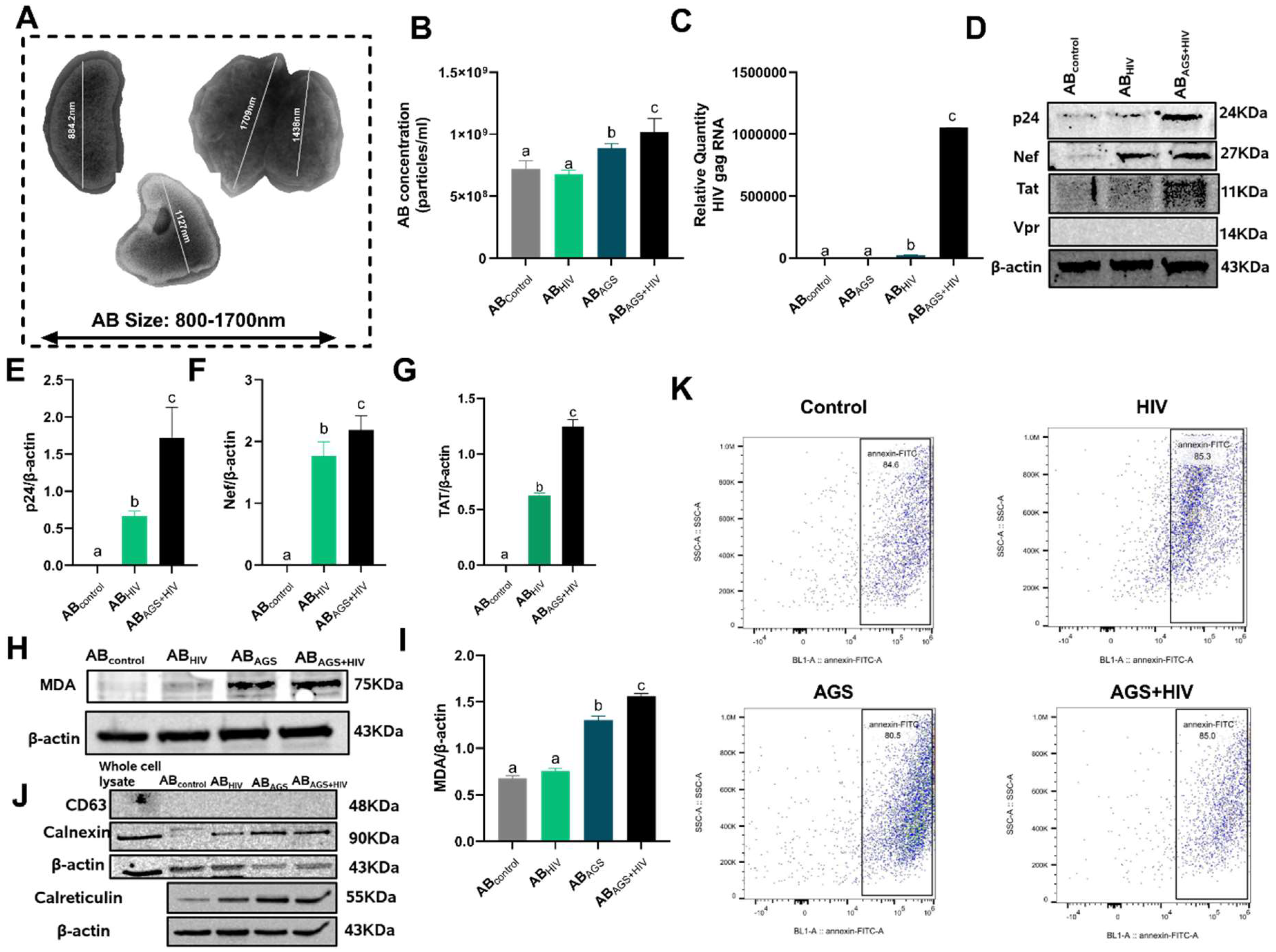
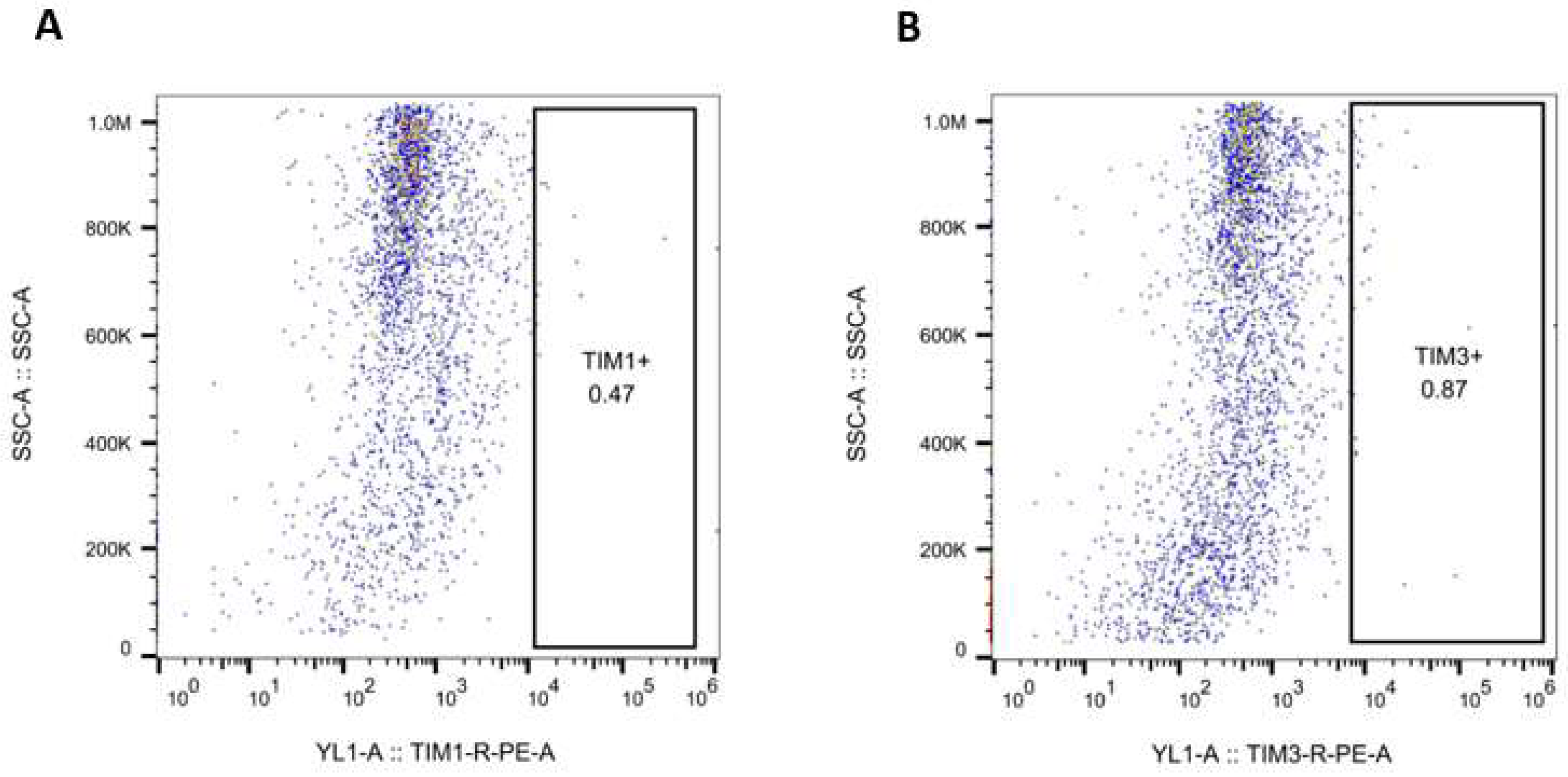
3.1. HIV RNA, HIV Proteins and Malondialdehyde Were Expressed by RLW ABAGS+HIV
3.2. Engulfment of RLW ABs by LX2 Cells
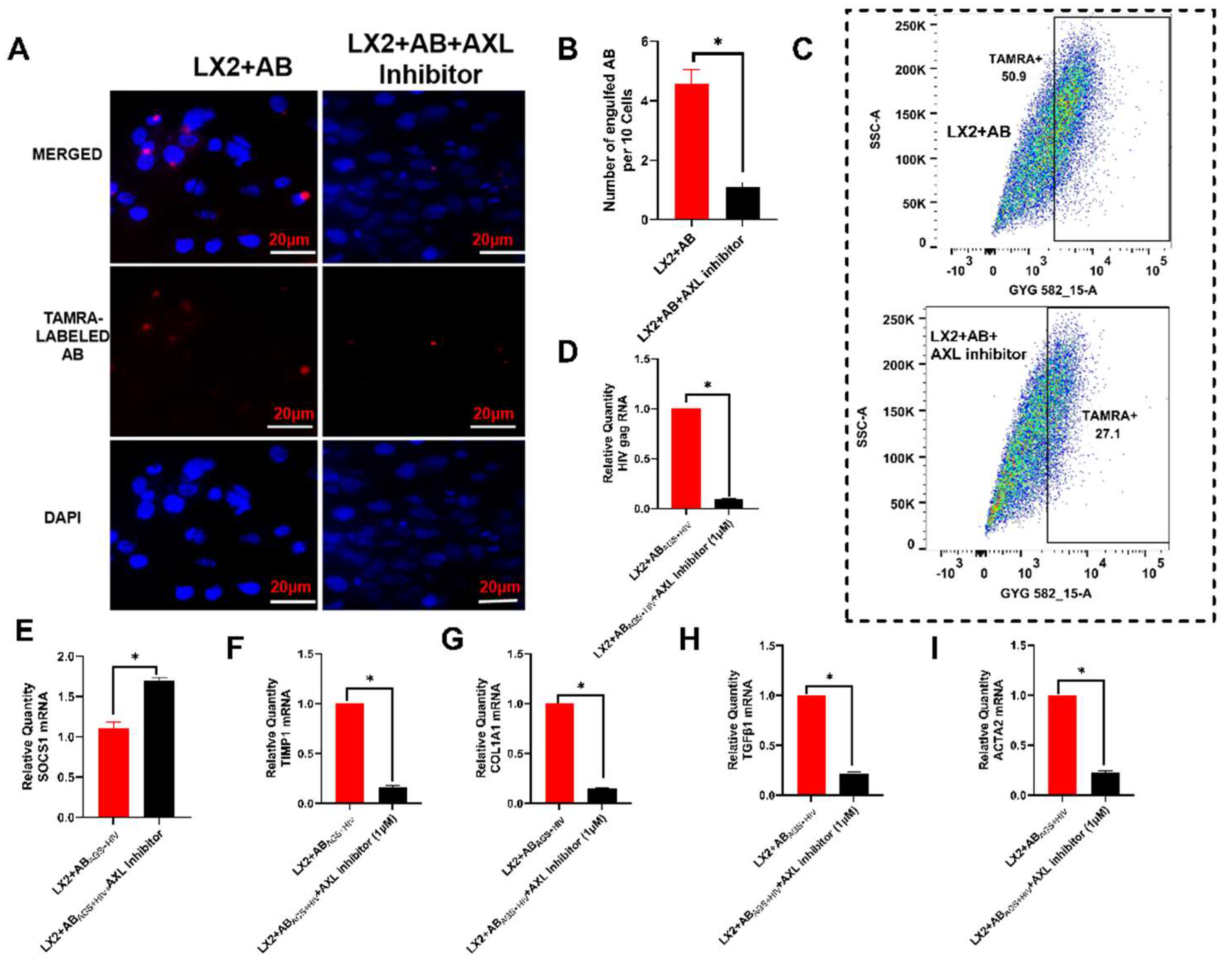
3.3. Pharmacological Inhibition of Axl Blocks LX2 Engulfment of RLW ABs and Attenuates Atcivation of Profibrotic Genes
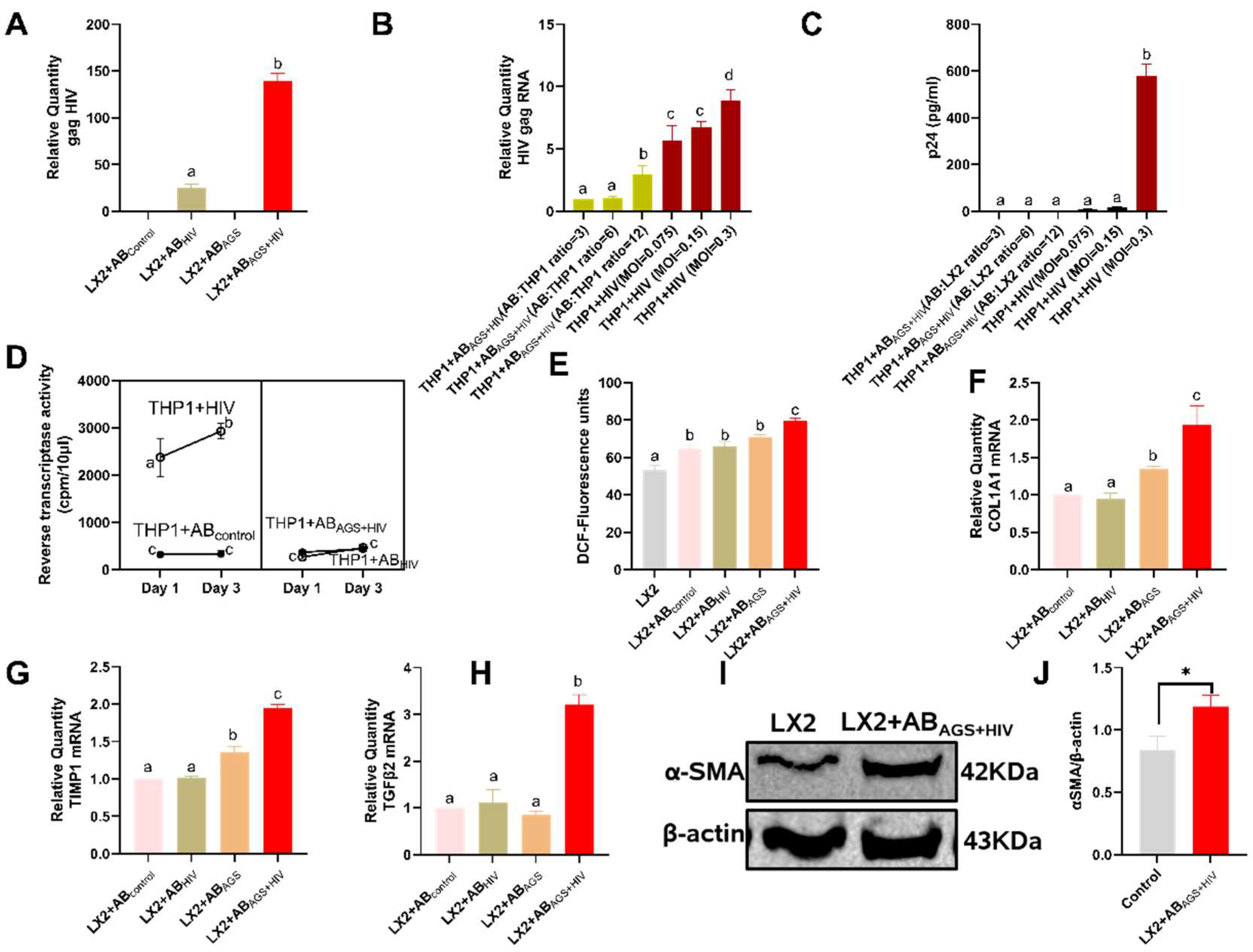
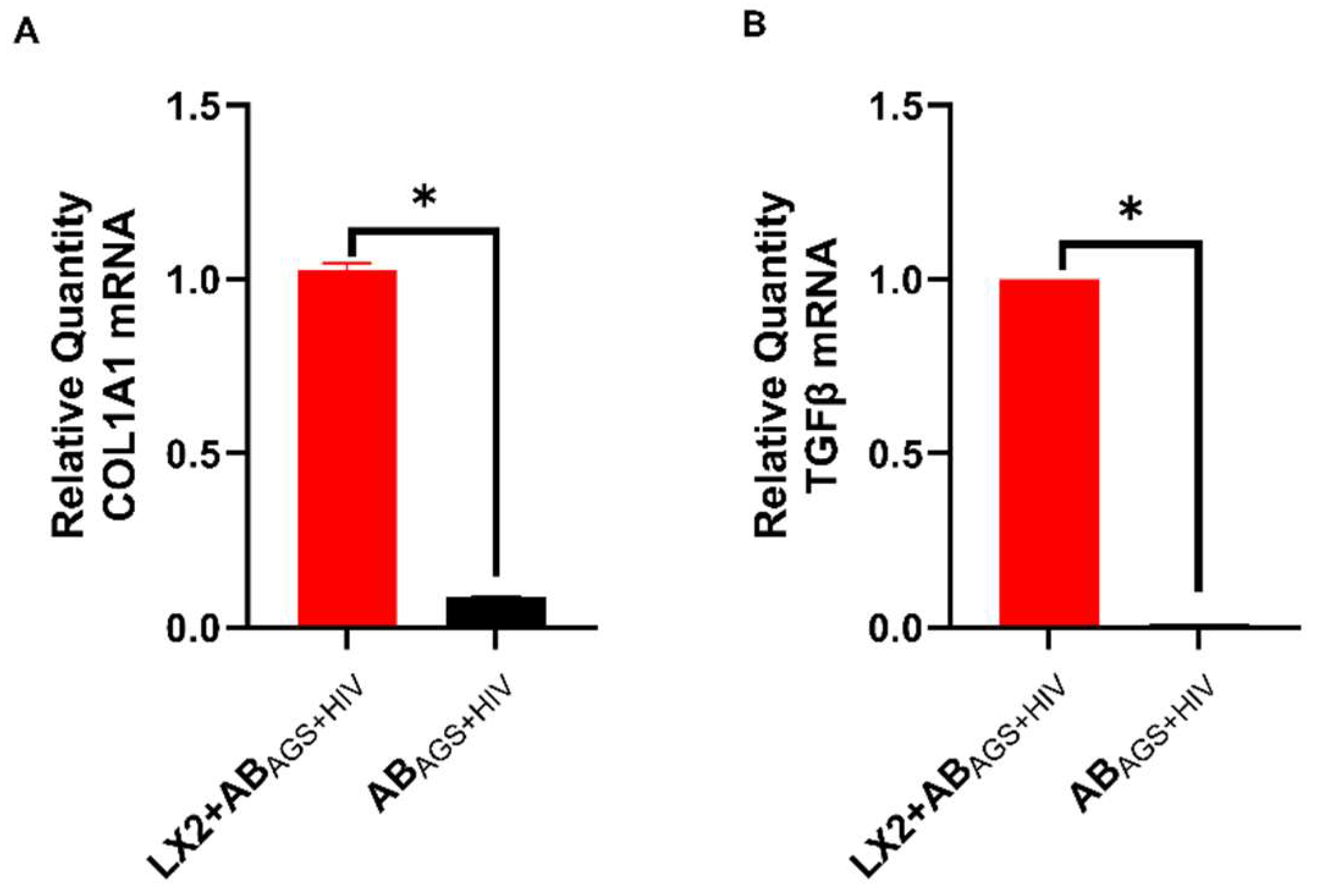
3.4. Engulfment of HIV- and MDA-Containing ABs Induces LX2 Profibrotic Activation
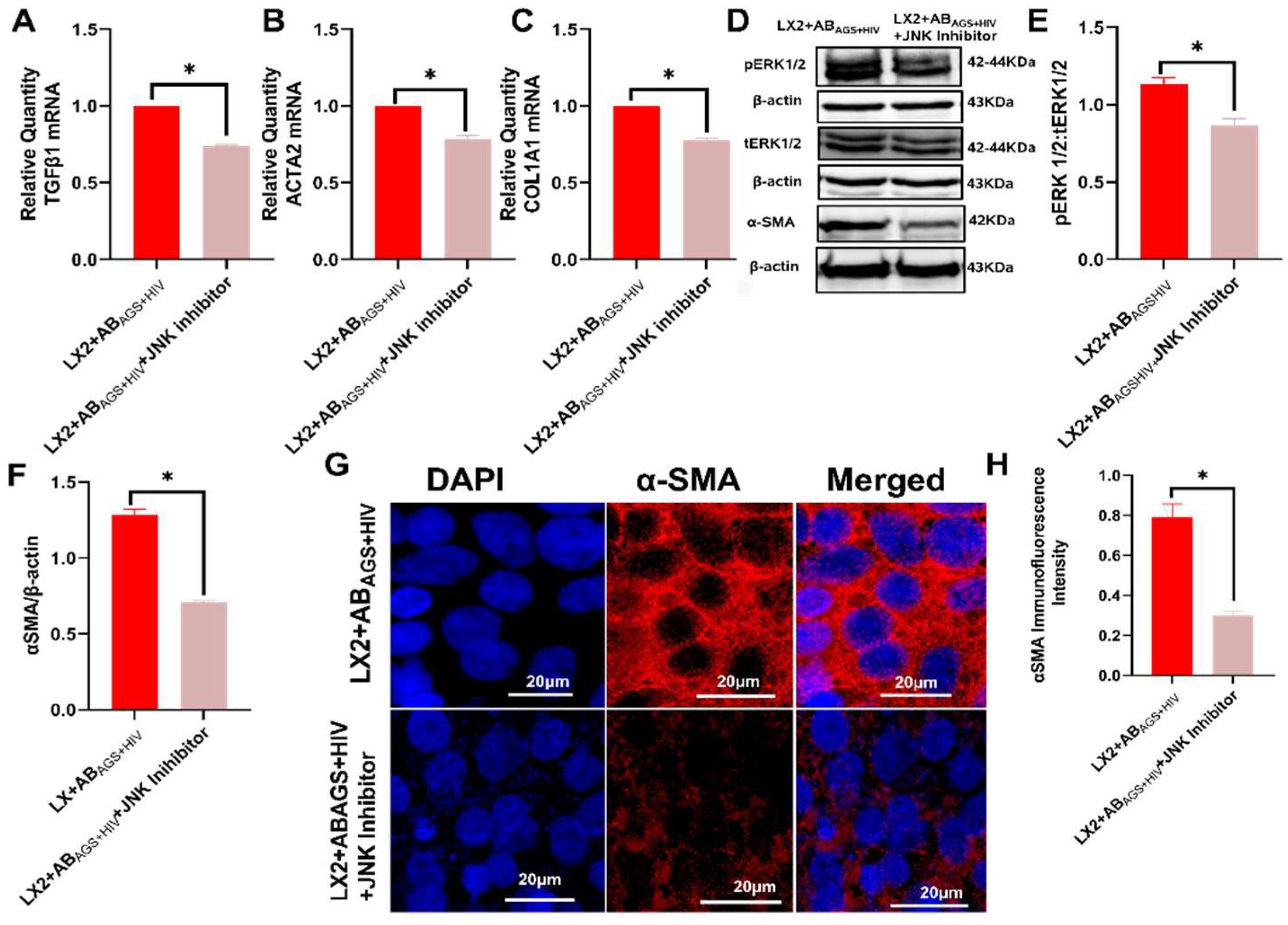
3.5. Pharmacological Inhibition of LX2 Cells Exposed to ABAGS+HIV Attenuates Profibrotic Activation via JNK and ERK1/2 Pathway
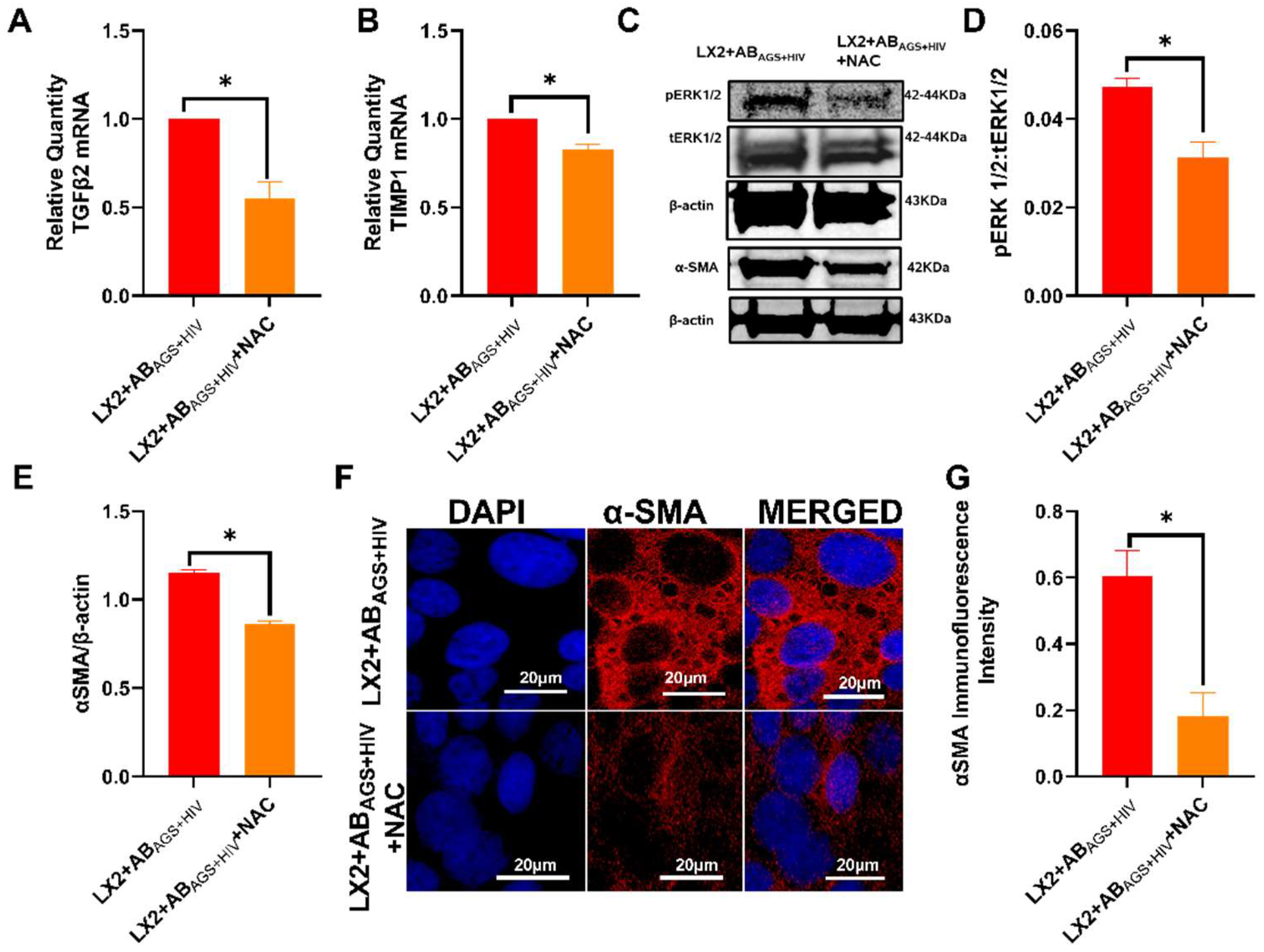
3.6. Oxidative Stress from ABAGS+HIV Activates JNK and ERK1/2 Pathway in LX2 Cells
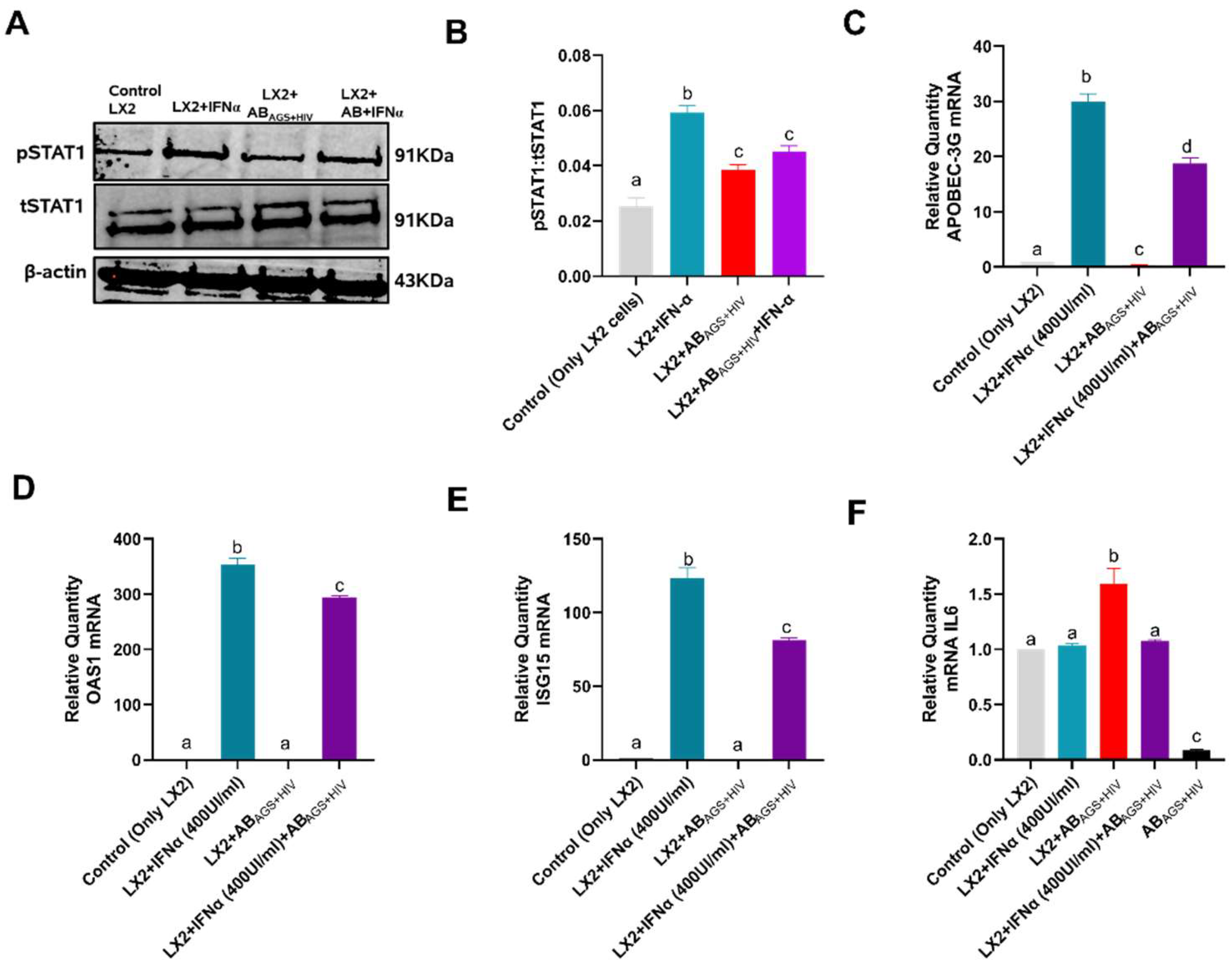
3.7. ABAGS+HIV Upregulates IL6 mRNA in LX2 Cells
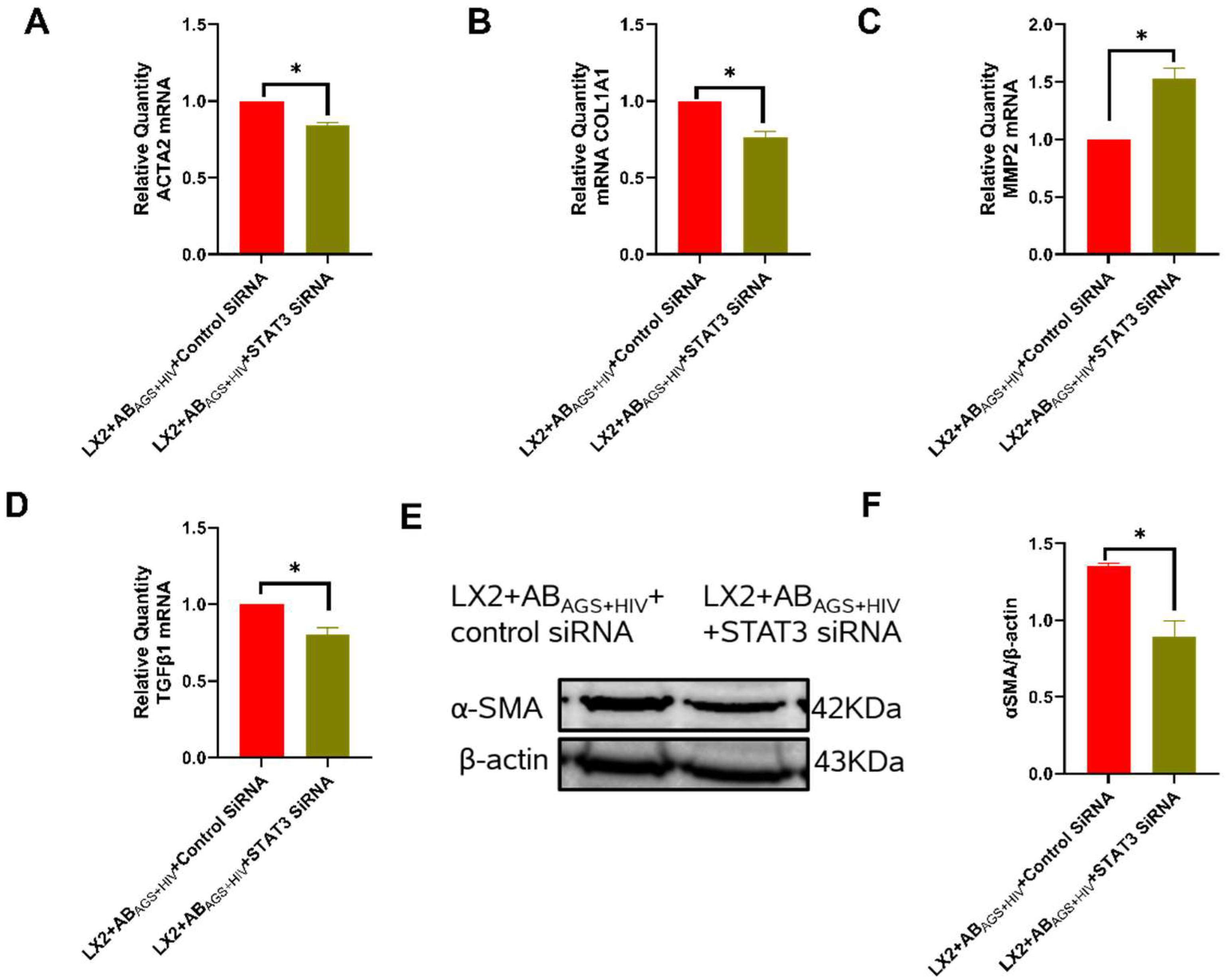
3.8. siRNA STAT3 Transfection Inhibits STAT3 Protein Expressions in LX2 Cells
3.9. Silencing STAT3 in LX2 Cells Attenuates ABAGS+HIV Induced Profibrotic Activation
3.10. In Vivo Effects of HIV Containing-Apoptotic Bodies on Ethanol-Fed Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A





References
- New-Aaron, M.; Kingi, H.; Meza, J.L.; Goedert, M.H.; Kibusi, S.M.; Mkhoi, M.L.; Mayengo, C.D.; Charles, J.; Shabani, S.; New-Aaron, T.O. Duration on ART, Alcohol Use and HIV Stage May Predict Risky Sexual Behavior in a Resource-limited Environment: A Cross-sectional Study. Curr. HIV Res. 2021, 19, 420–433. [Google Scholar] [CrossRef] [PubMed]
- New-Aaron, M.; Ganesan, M.; Dagur, R.S.; Kharbanda, K.K.; Poluektova, L.Y.; Osna, N.A. Pancreatogenic Diabetes: Triggering Effects of Alcohol and HIV. Biology 2021, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.E.; Nash, S.; Connor, N.; Kirwan, P.D.; Ogaz, D.; Croxford, S.; Angelis, D.; Delpech, V.C. Towards elimination of HIV transmission, AIDS and HIV-related deaths in the UK. HIV Med. 2018, 19, 505–512. [Google Scholar] [CrossRef]
- Granich, R.; Crowley, S.; Vitoria, M.; Smyth, C.; Kahn, J.G.; Bennett, R.; Lo, Y.R.; Souteyrand, Y.; Williams, B. Highly active antiretroviral treatment as prevention of HIV transmission: Review of scientific evidence and update. Curr. Opin. HIV AIDS 2010, 5, 298–304. [Google Scholar] [CrossRef]
- Sterne, J.A.C.; Hernán, M.A.; Ledergerber, B.; Tilling, K.; Weber, R.; Sendi, P.; Rickenbach, M.; Robins, J.M.; Egger, M. Long-term effectiveness of potent antiretroviral therapy in preventing AIDS and death: A prospective cohort study. Lancet 2005, 366, 378–384. [Google Scholar] [CrossRef]
- Debes, J.D.; Bohjanen, P.R.; Boonstra, A. Mechanisms of Accelerated Liver Fibrosis Progression during HIV Infection. J. Clin. Transl. Hepatol. 2016, 4, 328–335. [Google Scholar] [CrossRef]
- Rein, S.M.; Lampe, F.C.; Chaloner, C.; Stafford, A.; Rodger, A.J.; Johnson, M.A.; McDonnell, J.; Burns, F.; Madge, S.; Miners, A.; et al. Causes of hospitalisation among a cohort of people with HIV from a London centre followed from 2011 to 2018. BMC Infect. Dis. 2021, 21, 395. [Google Scholar] [CrossRef]
- Althoff, K.N.; Gebo, K.A.; Moore, R.D.; Boyd, C.M.; Justice, A.C.; Wong, C.; Lucas, G.M.; Klein, M.B.; Kitahata, M.M.; Crane, H.; et al. Contributions of traditional and HIV-related risk factors on non-AIDS-defining cancer, myocardial infarction, and end-stage liver and renal diseases in adults with HIV in the USA and Canada: A collaboration of cohort studies. Lancet HIV 2019, 6, e93–e104. [Google Scholar] [CrossRef]
- Madhombiro, M.; Dube, B.; Dube, M.; Zunza, M.; Chibanda, D.; Rusakaniko, S.; Seedat, S. Intervention for alcohol use disorders at an HIV care clinic in Harare: A pilot and feasibility study. Addict. Sci. Clin. Pract. 2019, 14, 16. [Google Scholar] [CrossRef]
- Cook, J.A.; Burke-Miller, J.K.; Steigman, P.J.; Schwartz, R.M.; Hessol, N.A.; Milam, J.; Merenstein, D.J.; Anastos, K.; Golub, E.T.; Cohen, M.H. Prevalence, Comorbidity, and Correlates of Psychiatric and Substance Use Disorders and Associations with HIV Risk Behaviors in a Multisite Cohort of Women Living with HIV. AIDS Behav. 2018, 22, 3141–3154. [Google Scholar] [CrossRef]
- Petoumenos, K.; Law, M.G. Smoking, alcohol and illicit drug use effects on survival in HIV-positive persons. Curr. Opin. HIV AIDS 2016, 11, 514–520. [Google Scholar] [CrossRef]
- Galvan, F.H.; Bing, E.G.; Fleishman, J.A.; London, A.S.; Caetano, R.; Burnam, M.A.; Longshore, D.; Morton, S.C.; Orlando, M.; Shapiro, M. The prevalence of alcohol consumption and heavy drinking among people with HIV in the United States: Results from the HIV Cost and Services Utilization Study. J. Stud. Alcohol 2002, 63, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, M.; New-Aaron, M.; Dagur, R.S.; Makarov, E.; Wang, W.; Kharbanda, K.K.; Kidambi, S.; Poluektova, L.Y.; Osna, N.A. Alcohol Metabolism Potentiates HIV-Induced Hepatotoxicity: Contribution to End-Stage Liver Disease. Biomolecules 2019, 9, 851. [Google Scholar] [CrossRef] [PubMed]
- New-Aaron, M.; Thomes, P.G.; Ganesan, M.; Dagur, R.S.; Donohue, T.M.; Kusum, K.K.; Poluektova, L.Y.; Osna, N.A. Alcohol-Induced Lysosomal Damage and Suppression of Lysosome Biogenesis Contribute to Hepatotoxicity in HIV-Exposed Liver Cells. Biomolecules 2021, 11, 1497. [Google Scholar] [CrossRef]
- Chen, M.; Liu, J.; Yang, W.; Ling, W. Lipopolysaccharide mediates hepatic stellate cell activation by regulating autophagy and retinoic acid signaling. Autophagy 2017, 13, 1813–1827. [Google Scholar] [CrossRef]
- Ramos-Tovar, E.; Muriel, P. Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants 2020, 9, 1279. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.-C.; Tseng, C.-P.; Liao, M.-H.; Peng, C.-Y.; Yu, J.-S.; Chuang, P.-H.; Huang, J.-T.; Chen, J.J.W. Activation of hepatic stellate cells by the ubiquitin C-terminal hydrolase 1 protein secreted from hepatitis C virus-infected hepatocytes. Sci. Rep. 2017, 7, 4448. [Google Scholar] [CrossRef]
- Gäbele, E.; Mühlbauer, M.; Dorn, C.; Weiss, T.S.; Froh, M.; Schnabl, B.; Wiest, R.; Schölmerich, J.; Obermeier, F.; Hellerbrand, C. Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem. Biophys. Res. Commun. 2008, 376, 271–276. [Google Scholar] [CrossRef]
- Kiziltas, S. Toll-like receptors in pathophysiology of liver diseases. World J. Hepatol. 2016, 8, 1354. [Google Scholar] [CrossRef]
- Nakamoto, N.; Kanai, T. Role of Toll-Like Receptors in Immune Activation and Tolerance in the Liver. Front. Immunol. 2014, 5, 221. [Google Scholar] [CrossRef]
- Watanabe, A.; Hashmi, A.; Gomes, D.A.; Town, T.; Badou, A.; Flavell, R.A.; Mehal, W.Z. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology 2007, 46, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; Li, S.P.; Westermarck, J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008, 22, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal. Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef]
- Ichijo, H. From receptors to stress-activated MAP kinases. Oncogene 1999, 18, 6087–6093. [Google Scholar] [CrossRef]
- Simon, A.R.; Rai, U.; Fanburg, B.L.; Cochran, B.H. Activation of the JAK-STAT pathway by reactive oxygen species. Am. J. Physiol.-Cell Physiol. 1998, 275, C1640–C1652. [Google Scholar] [CrossRef]
- Butturini, E.; Carcereri de Prati, A.; Mariotto, S. Redox Regulation of STAT1 and STAT3 Signaling. Int. J. Mol. Sci. 2020, 21, 7034. [Google Scholar] [CrossRef]
- Charras, A.; Arvaniti, P.; Le Dantec, C.; Dalekos, G.N.; Zachou, K.; Bordron, A.; Renaudineau, Y. JAK Inhibitors and Oxidative Stress Control. Front. Immunol. 2019, 10, 2814. [Google Scholar] [CrossRef]
- New-Aaron, M.; Ganesan, M.; Dagur, R.S.; Kharbanda, K.K.; Poluektova, L.Y.; Osna, N.A. Obeticholic acid attenuates human immunodeficiency virus/alcohol metabolism-induced pro-fibrotic activation in liver cells. World J. Hepatol. 2020, 12, 965–975. [Google Scholar] [CrossRef]
- Phan, T.K.; Poon, I.K.; Atkin-Smith, G.K. Detection and Isolation of Apoptotic Bodies to High Purity. J. Vis. Exp. 2018, 138, e58317. [Google Scholar] [CrossRef]
- Powers, J.; Zhang, H.; Battrell, L.; Meadows, G.G.; Trobridge, G.D. Establishment of an immunodeficient alcohol mouse model to study the effects of alcohol on human cells in vivo. J. Stud. Alcohol Drugs 2012, 73, 933–937. [Google Scholar] [CrossRef][Green Version]
- Melhem, A.; Muhanna, N.; Bishara, A.; Alvarez, C.E.; Ilan, Y.; Bishara, T.; Horani, A.; Nassar, M.; Friedman, S.L.; Safadi, R. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J. Hepatol. 2006, 45, 60–71. [Google Scholar] [CrossRef]
- Muthiah, M.D.; Huang, D.Q.; Zhou, L.; Jumat, N.H.; Choolani, M.; Chan, J.K.Y.; Wee, A.; Lim, S.G.; Dan, Y.Y. A murine model demonstrating reversal of structural and functional correlates of cirrhosis with progenitor cell transplantation. Sci. Rep. 2019, 9, 15446. [Google Scholar] [CrossRef] [PubMed]
- Maacha, S.; Bhat, A.A.; Jimenez, L.; Raza, A.; Haris, M.; Uddin, S.; Grivel, J.-C. Extracellular vesicles-mediated intercellular communication: Roles in the tumor microenvironment and anti-cancer drug resistance. Mol. Cancer 2019, 18, 55. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.L.N.; Breakefield, X.O.; Weaver, A.M. Extracellular Vesicles: Unique Intercellular Delivery Vehicles. Trends Cell Biol. 2017, 27, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.J.; Kim, O.Y.; Gho, Y.S. Extracellular vesicles as emerging intercellular communicasomes. BMB Rep. 2014, 47, 531–539. [Google Scholar] [CrossRef]
- Battistelli, M.; Falcieri, E. Apoptotic bodies: Particular extracellular vesicles involved in intercellular communication. Biology 2020, 9, 21. [Google Scholar] [CrossRef]
- Naeini, M.B.; Bianconi, V.; Pirro, M.; Sahebkar, A. The role of phosphatidylserine recognition receptors in multiple biological functions. Cell. Mol. Biol. Lett. 2020, 25, 23. [Google Scholar] [CrossRef]
- Miyanishi, M.; Tada, K.; Koike, M.; Uchiyama, Y.; Kitamura, T.; Nagata, S. Identification of Tim4 as a phosphatidylserine receptor. Nature 2007, 450, 435–439. [Google Scholar] [CrossRef]
- Lemke, G. Phosphatidylserine Is the Signal for TAM Receptors and Their Ligands. Trends Biochem. Sci. 2017, 42, 738–748. [Google Scholar] [CrossRef]
- Shao, W.H.; Zhen, Y.; Eisenberg, R.A.; Cohen, P.L. The Mer receptor tyrosine kinase is expressed on discrete macrophage subpopulations and mainly uses Gas6 as its ligand for uptake of apoptotic cells. Clin. Immunol. 2009, 133, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Raggi, F.; Pelassa, S.; Pierobon, D.; Penco, F.; Gattorno, M.; Novelli, F.; Eva, A.; Varesio, L.; Giovarelli, M.; Bosco, M.C. Regulation of Human Macrophage M1–M2 Polarization Balance by Hypoxia and the Triggering Receptor Expressed on Myeloid Cells-1. Front. Immunol. 2017, 8, 1097. [Google Scholar] [CrossRef] [PubMed]
- Chua, B.A.; Ngo, J.A.; Situ, K.; Ramirez, C.M.; Nakano, H.; Morizono, K. Protein S and Gas6 induce efferocytosis of HIV-1-infected cells. Virology 2018, 515, 176–190. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Lahair, M.M.; Franklin, R.A. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid. Redox Signal. 2006, 8, 1775–1789. [Google Scholar] [CrossRef]
- Xiang, D.M.; Sun, W.; Ning, B.F.; Zhou, T.F.; Li, X.F.; Zhong, W.; Cheng, Z.; Xia, M.Y.; Wang, X.; Deng, X.; et al. The HLF/IL-6/STAT3 feedforward circuit drives hepatic stellate cell activation to promote liver fibrosis. Gut 2018, 67, 1704–1715. [Google Scholar] [CrossRef]
- Bica, I.; McGovern, B.; Dhar, R.; Stone, D.; McGowan, K.; Scheib, R.; Snydman, D.R. Increasing Mortality Due to End-Stage Liver Disease in Patients with Human Immunodeficiency Virus Infection. Clin. Infect. Dis. 2001, 32, 492–497. [Google Scholar] [CrossRef]
- Ganesan, M.; Poluektova, L.Y.; Kharbanda, K.K.; Osna, N.A. Liver as a target of human immunodeficiency virus infection. World J. Gastroenterol. 2018, 24, 4728–4737. [Google Scholar] [CrossRef]
- Sastry, J.; Mohammed, H.; Campos, M.M.; Uetrecht, J.; Abu-Asab, M. Nevirapine-induced liver lipid-SER inclusions and other ultrastructural aberrations. Ultrastruct. Pathol. 2018, 42, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Gwag, T.; Meng, Z.; Sui, Y.; Helsley, R.N.; Park, S.H.; Wang, S.; Greenberg, R.N.; Zhou, C. Non-nucleoside reverse transcriptase inhibitor efavirenz activates PXR to induce hypercholesterolemia and hepatic steatosis. J. Hepatol. 2019, 70, 930–940. [Google Scholar] [CrossRef]
- Ganesan, M.; Poluektova, L.Y.; Enweluzo, C.; Kharbanda, K.K.; Osna, N.A. Hepatitis C Virus-Infected Apoptotic Hepatocytes Program Macrophages and Hepatic Stellate Cells for Liver Inflammation and Fibrosis Development: Role of Ethanol as a Second Hit. Biomolecules 2018, 8, 113. [Google Scholar] [CrossRef]
- Nevola, R.; Rinaldi, L.; Giordano, M.; Marrone, A.; Adinolfi, L.E. Mechanisms and clinical behavior of hepatocellular carcinoma in HBV and HCV infection and alcoholic and non-alcoholic fatty liver disease. Hepatoma Res. 2018, 4, 55. [Google Scholar] [CrossRef]
- Kaspar, M.B.; Sterling, R.K. Mechanisms of liver disease in patients infected with HIV. BMJ Open Gastroenterol. 2017, 4, e000166. [Google Scholar] [CrossRef]
- Chakraborty, J.B.; Oakley, F.; Walsh, M.J. Mechanisms and biomarkers of apoptosis in liver disease and fibrosis. Int. J. Hepatol. 2012, 2012, 648915. [Google Scholar] [CrossRef] [PubMed]
- Witek, R.P.; Stone, W.C.; Karaca, F.G.; Syn, W.K.; Pereira, T.A.; Agboola, K.M.; Omenetti, A.; Jung, Y.; Teaberry, V.; Choi, S.S.; et al. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology 2009, 50, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Barreyro, F.J.; Holod, S.; Finocchietto, P.V.; Camino, A.M.; Aquino, J.B.; Avagnina, A.; Carreras, M.C.; Poderoso, J.J.; Gores, G.J. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2015, 35, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Shiffman, M.L.; Pockros, P.; McHutchison, J.G.; Schiff, E.R.; Morris, M.; Burgess, G. Clinical trial: The efficacy and safety of oral PF-03491390, a pancaspase inhibitor—A randomized placebo-controlled study in patients with chronic hepatitis C. Aliment. Pharmacol. Ther. 2010, 31, 969–978. [Google Scholar] [CrossRef]
- Canbay, A.; Feldstein, A.; Baskin-Bey, E.; Bronk, S.F.; Gores, G.J. The caspase inhibitor IDN-6556 attenuates hepatic injury and fibrosis in the bile duct ligated mouse. J. Pharmacol. Exp. Ther. 2004, 308, 1191–1196. [Google Scholar] [CrossRef]
- Guo, R.; Ren, J. Alcohol and acetaldehyde in public health: From marvel to menace. Int. J. Environ. Res. Public Health 2010, 7, 1285–1301. [Google Scholar] [CrossRef]
- Teschke, R.; Neuman, M.G.; Liangpunsakul, S.; Seitz, H.-K. Alcoholic Liver Disease and the co-triggering Role of MEOS with Its CYP 2E1 Catalytic Cycle and ROS. Arch. Gastroenterol. Res. 2021, 2, 9–25. [Google Scholar]
- Doody, E.E.; Groebner, J.L.; Walker, J.R.; Frizol, B.M.; Tuma, D.J.; Fernandez, D.J.; Tuma, P.L. Ethanol metabolism by alcohol dehydrogenase or cytochrome P(450) 2E1 differentially impairs hepatic protein trafficking and growth hormone signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G558–G569. [Google Scholar] [CrossRef]
- Koop, D.R. Alcohol metabolism’s damaging effects on the cell: A focus on reactive oxygen generation by the enzyme cytochrome P450 2E1. Alcohol Res. Health 2006, 29, 274–280. [Google Scholar] [PubMed]
- Urtasun, R.; de la Rosa, L.C.; Nieto, N. Oxidative and nitrosative stress and fibrogenic response. Clin. Liver Dis. 2008, 12, 769–790. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, M.; Natarajan, S.K.; Zhang, J.; Mott, J.L.; Poluektova, L.I.; McVicker, B.L.; Kharbanda, K.K.; Tuma, D.J.; Osna, N.A. Role of apoptotic hepatocytes in HCV dissemination: Regulation by acetaldehyde. Am. J. Physiol.-Gastrointest. Liver Physiol. 2016, 310, G930–G940. [Google Scholar] [CrossRef] [PubMed]
- Poon, I.K.; Parkes, M.A.; Jiang, L.; Atkin-Smith, G.K.; Tixeira, R.; Gregory, C.D.; Ozkocak, D.C.; Rutter, S.F.; Caruso, S.; Santavanond, J.P. Moving beyond size and phosphatidylserine exposure: Evidence for a diversity of apoptotic cell-derived extracellular vesicles in vitro. J. Extracell. Vesicles 2019, 8, 1608786. [Google Scholar] [CrossRef]
- Johnston, D.E.; Kroening, C. Mechanism of early carbon tetrachloride toxicity in cultured rat hepatocytes. Pharm. Toxicol. 1998, 83, 231–239. [Google Scholar] [CrossRef]
- Higuchi, H.; Kurose, I.; Kato, S.; Miura, S.; Ishii, H. Ethanol-induced apoptosis and oxidative stress in hepatocytes. Alcohol Clin. Exp. Res. 1996, 20, 340a–346a. [Google Scholar] [CrossRef]
- Jiang, J.X.; Mikami, K.; Venugopal, S.; Li, Y.; Török, N.J. Apoptotic body engulfment by hepatic stellate cells promotes their survival by the JAK/STAT and Akt/NF-kappaB-dependent pathways. J. Hepatol. 2009, 51, 139–148. [Google Scholar] [CrossRef]
- Xu, X.; Lai, Y.; Hua, Z.-C. Apoptosis and apoptotic body: Disease message and therapeutic target potentials. Biosci. Rep. 2019, 39, BSR20180992. [Google Scholar] [CrossRef]
- Depraetere, V. “Eat me” signals of apoptotic bodies. Nat. Cell Biol. 2000, 2, E104. [Google Scholar] [CrossRef]
- Fadok, V.A.; De Cathelineau, A.; Daleke, D.L.; Henson, P.M.; Bratton, D.L. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J. Biol. Chem. 2001, 276, 1071–1077. [Google Scholar] [CrossRef]
- Canbay, A.; Taimr, P.; Torok, N.; Higuchi, H.; Friedman, S.; Gores, G.J. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab. Investig. 2003, 83, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.K.; Wilhelm, A.; Antoniades, C.G. TAM receptor tyrosine kinase function and the immunopathology of liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G899–G905. [Google Scholar] [CrossRef]
- Myers, K.V.; Amend, S.R.; Pienta, K.J. Targeting Tyro3, Axl and MerTK (TAM receptors): Implications for macrophages in the tumor microenvironment. Mol. Cancer 2019, 18, 94. [Google Scholar] [CrossRef]
- Ortmayr, G.; Brunnthaler, L.; Pereyra, D.; Huber, H.; Santol, J.; Rumpf, B.; Najarnia, S.; Smoot, R.; Ammon, D.; Sorz, T.; et al. Immunological Aspects of AXL/GAS-6 in the Context of Human Liver Regeneration. Hepatol. Commun. 2022, 6, 576–592. [Google Scholar] [CrossRef] [PubMed]
- Kluwe, J.; Pradere, J.P.; Gwak, G.Y.; Mencin, A.; De Minicis, S.; Osterreicher, C.H.; Colmenero, J.; Bataller, R.; Schwabe, R.F. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology 2010, 138, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Hong, I.H.; Park, S.J.; Goo, M.J.; Lee, H.R.; Park, J.K.; Ki, M.R.; Kim, S.H.; Lee, E.M.; Kim, A.Y.; Jeong, K.S. JNK1 and JNK2 regulate α-SMA in hepatic stellate cells during CCl4-induced fibrosis in the rat liver. Pathol. Int. 2013, 63, 483–491. [Google Scholar] [CrossRef]
- Parola, M.; Robino, G.; Marra, F.; Pinzani, M.; Bellomo, G.; Leonarduzzi, G.; Chiarugi, P.; Camandola, S.; Poli, G.; Waeg, G. HNE interacts directly with JNK isoforms in human hepatic stellate cells. J. Clin. Investig. 1998, 102, 1942–1950. [Google Scholar] [CrossRef]
- Novo, E.; Marra, F.; Zamara, E.; Valfrè di Bonzo, L.; Monitillo, L.; Cannito, S.; Petrai, I.; Mazzocca, A.; Bonacchi, A.; De Franco, R.S.; et al. Overexpression of Bcl-2 by activated human hepatic stellate cells: Resistance to apoptosis as a mechanism of progressive hepatic fibrogenesis in humans. Gut 2006, 55, 1174–1182. [Google Scholar] [CrossRef]
- Novo, E.; Marra, F.; Zamara, E.; Valfrè di Bonzo, L.; Caligiuri, A.; Cannito, S.; Antonaci, C.; Colombatto, S.; Pinzani, M.; Parola, M. Dose dependent and divergent effects of superoxide anion on cell death, proliferation, and migration of activated human hepatic stellate cells. Gut 2006, 55, 90–97. [Google Scholar] [CrossRef]
- Pessayre, D.; Fromenty, B.; Mansouri, A. Mitochondrial injury in steatohepatitis. Eur. J. Gastroenterol. Hepatol. 2004, 16, 1095–1105. [Google Scholar] [CrossRef]
- Gandhi, C.R. Oxidative Stress and Hepatic Stellate Cells: A Paradoxical Relationship. Trends Cell Mol. Biol. 2012, 7, 1–10. [Google Scholar] [PubMed]
- Jiang, J.X.; Venugopal, S.; Serizawa, N.; Chen, X.; Scott, F.; Li, Y.; Adamson, R.; Devaraj, S.; Shah, V.; Gershwin, M.E.; et al. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology 2010, 139, 1375–1384. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; van Boxel-Dezaire, A.H.; Cheon, H.; Yang, J.; Stark, G.R. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 16975–16980. [Google Scholar] [CrossRef] [PubMed]
- Flowers, L.O.; Subramaniam, P.S.; Johnson, H.M. A SOCS-1 peptide mimetic inhibits both constitutive and IL-6 induced activation of STAT3 in prostate cancer cells. Oncogene 2005, 24, 2114–2120. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
New-Aaron, M.; Dagur, R.S.; Koganti, S.S.; Ganesan, M.; Wang, W.; Makarov, E.; Ogunnaike, M.; Kharbanda, K.K.; Poluektova, L.Y.; Osna, N.A. Alcohol and HIV-Derived Hepatocyte Apoptotic Bodies Induce Hepatic Stellate Cell Activation. Biology 2022, 11, 1059. https://doi.org/10.3390/biology11071059
New-Aaron M, Dagur RS, Koganti SS, Ganesan M, Wang W, Makarov E, Ogunnaike M, Kharbanda KK, Poluektova LY, Osna NA. Alcohol and HIV-Derived Hepatocyte Apoptotic Bodies Induce Hepatic Stellate Cell Activation. Biology. 2022; 11(7):1059. https://doi.org/10.3390/biology11071059
Chicago/Turabian StyleNew-Aaron, Moses, Raghubendra Singh Dagur, Siva Sankar Koganti, Murali Ganesan, Weimin Wang, Edward Makarov, Mojisola Ogunnaike, Kusum K. Kharbanda, Larisa Y. Poluektova, and Natalia A. Osna. 2022. "Alcohol and HIV-Derived Hepatocyte Apoptotic Bodies Induce Hepatic Stellate Cell Activation" Biology 11, no. 7: 1059. https://doi.org/10.3390/biology11071059
APA StyleNew-Aaron, M., Dagur, R. S., Koganti, S. S., Ganesan, M., Wang, W., Makarov, E., Ogunnaike, M., Kharbanda, K. K., Poluektova, L. Y., & Osna, N. A. (2022). Alcohol and HIV-Derived Hepatocyte Apoptotic Bodies Induce Hepatic Stellate Cell Activation. Biology, 11(7), 1059. https://doi.org/10.3390/biology11071059