
The Inflammatory Response after Moderate Contusion Spinal Cord Injury: A Time Study

 , ,
, ,  , ,
, , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. CSF Collection
2.2. Animals
2.3. Induction of Spinal Cord Injury (SCI)
2.4. Basso Mouse Scale (BMS)
2.5. Human SCI Tissue
2.6. Tissue Processing
2.7. Gene Analysis
2.8. In Situ Hybridization for Tnf mRNA
2.9. Protein Purification
2.10. Electrochemiluminescence Analysis
2.11. CSF ELISA
2.12. Immunohistochemistry for TNF
2.13. Immunofluorescence Staining
2.14. Flow Cytometry
2.15. Statistical Analysis
3. Results
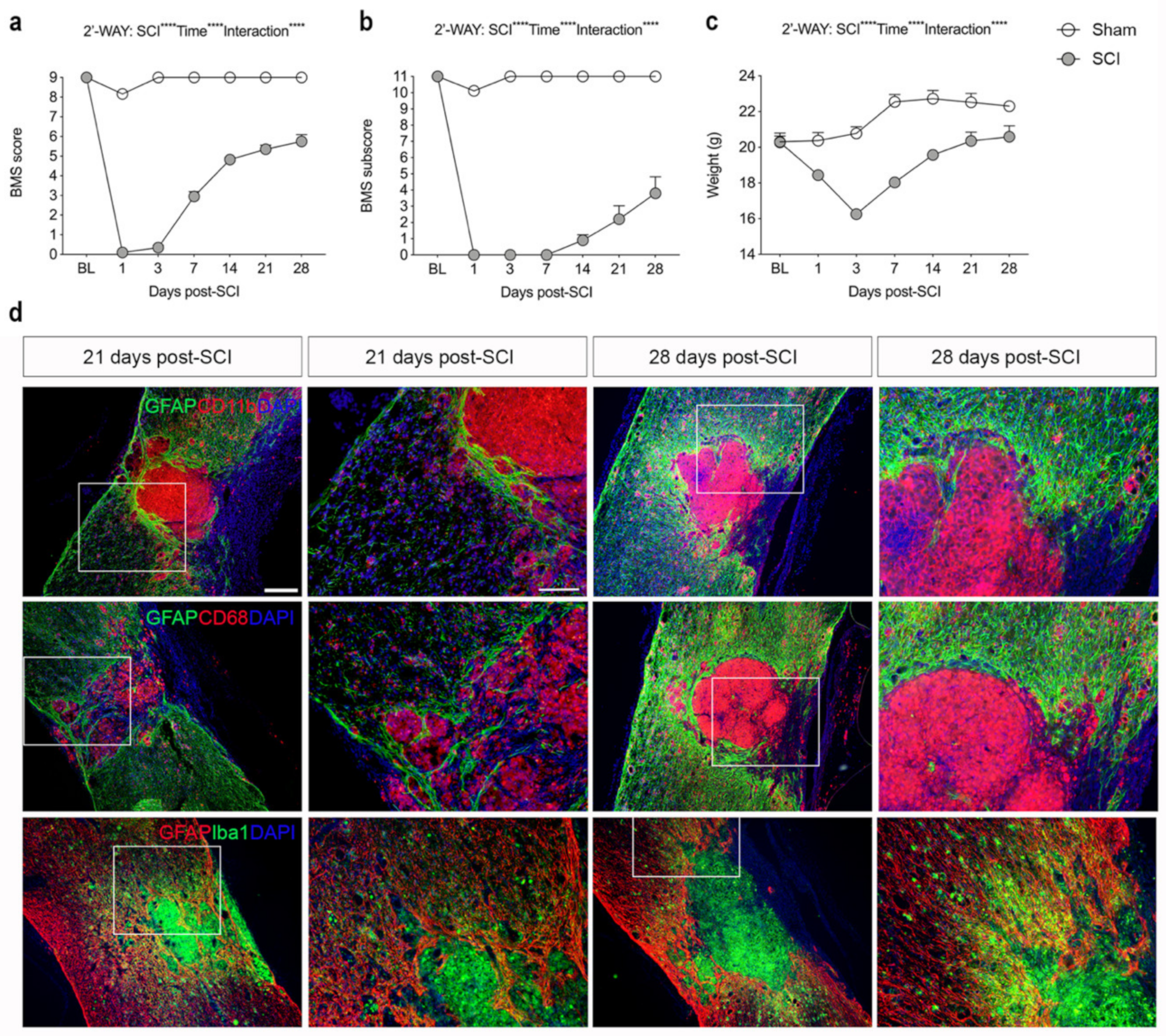
3.1. SCI Leads to Significant Changes in Locomotor Function
3.2. SCI Results in Glial Scar Formation
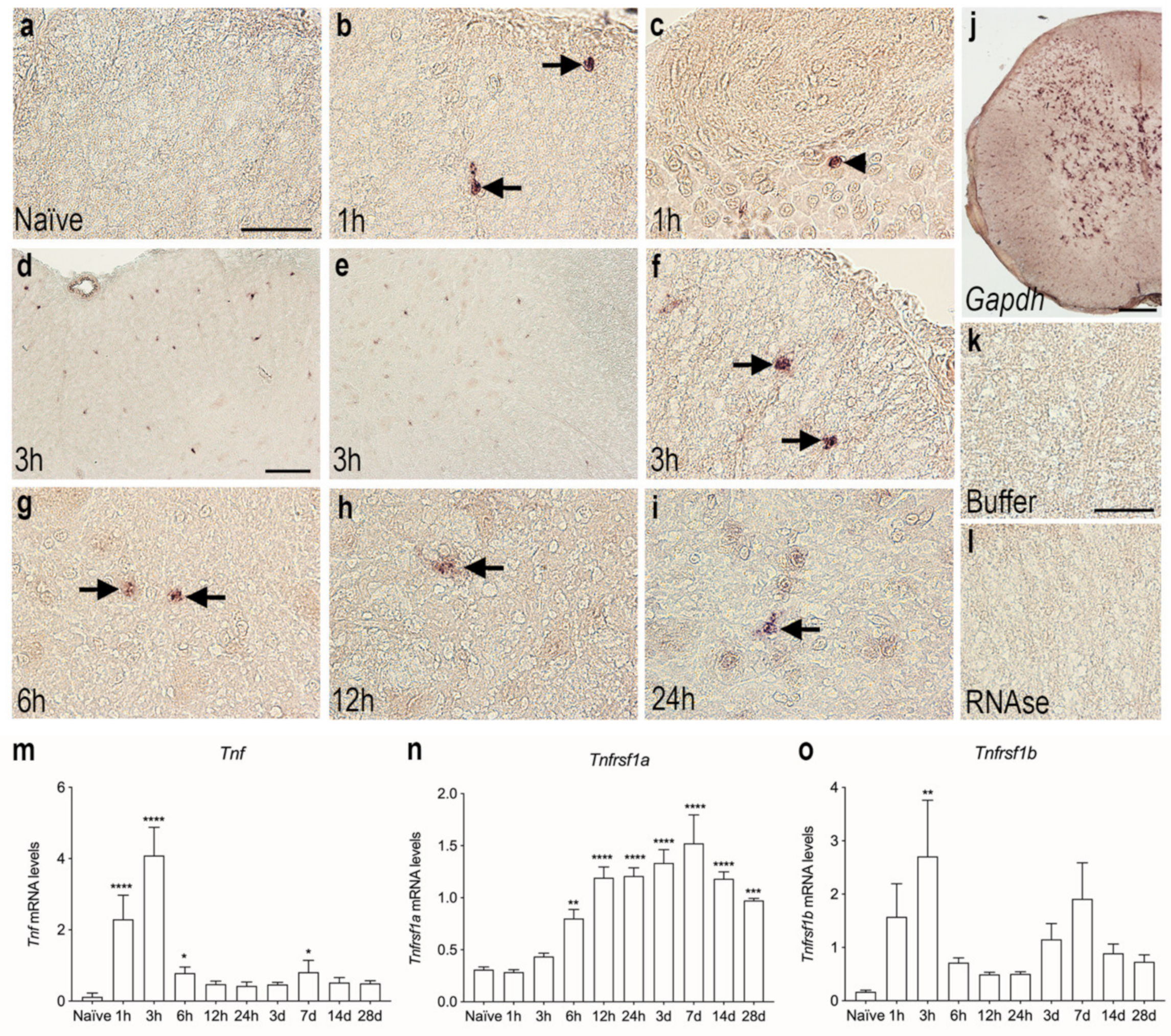
3.3. Tnf mRNA Synthesis Increases in the Acute Phase after SCI
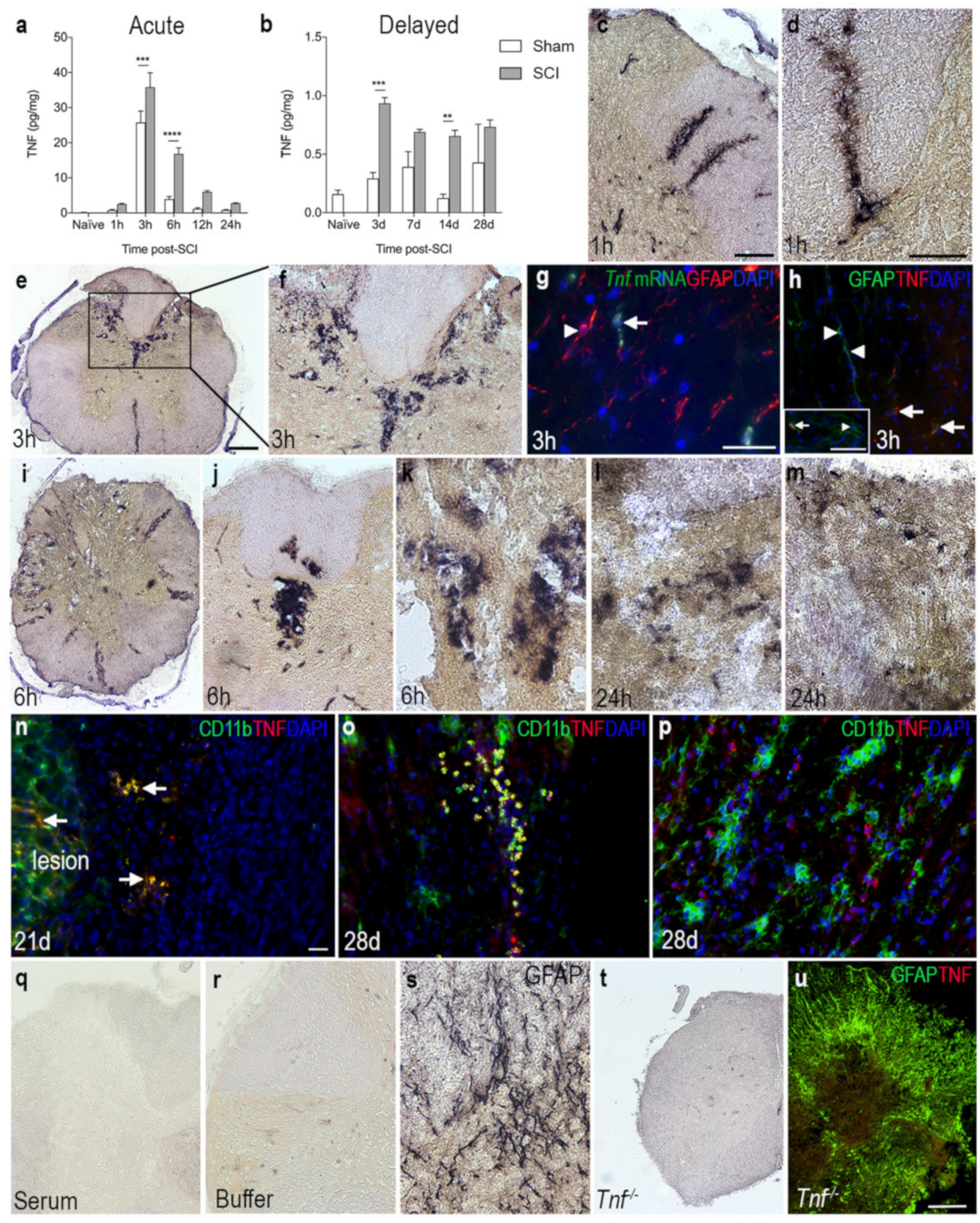
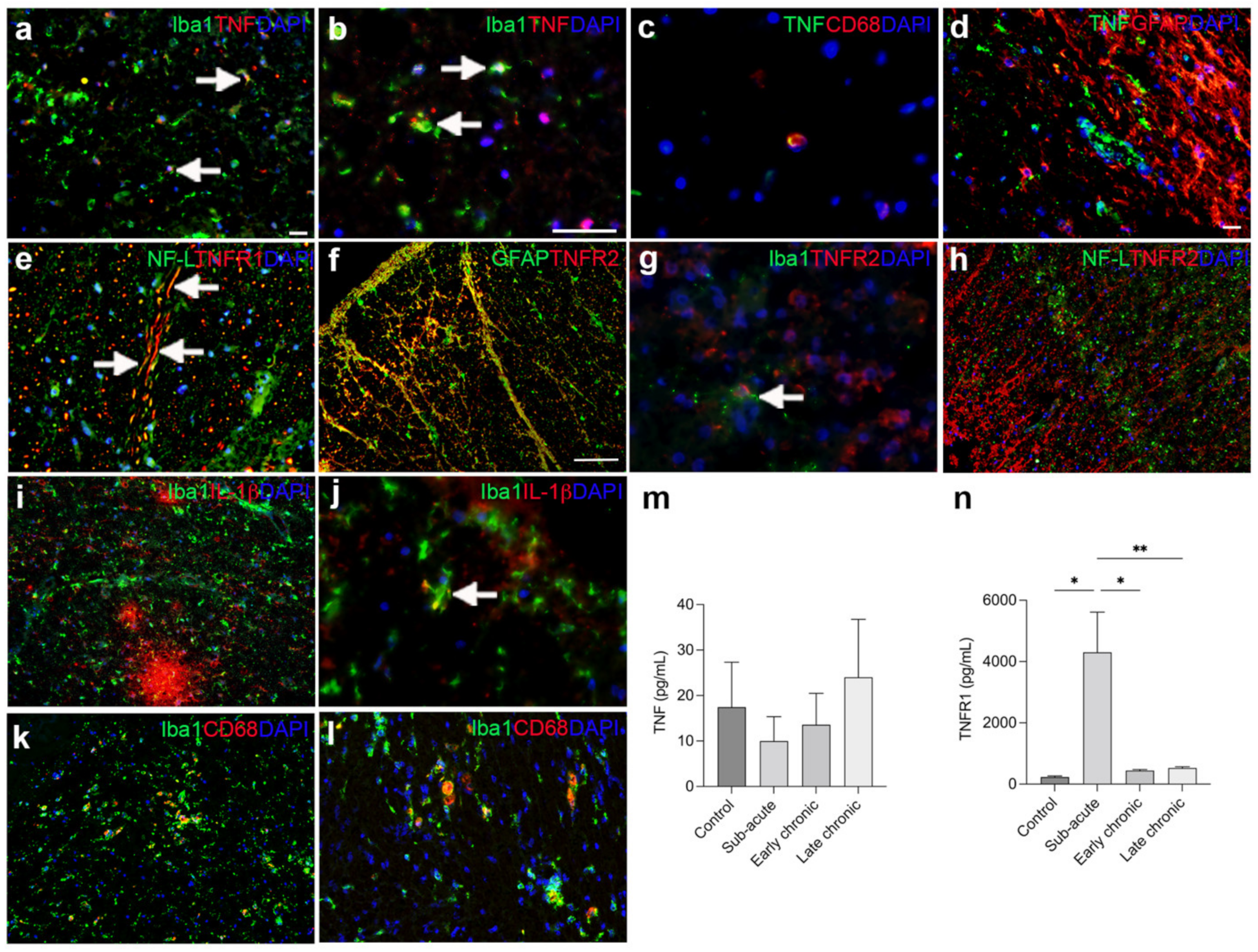
3.4. TNF Is Increased on Glial Cells after SCI
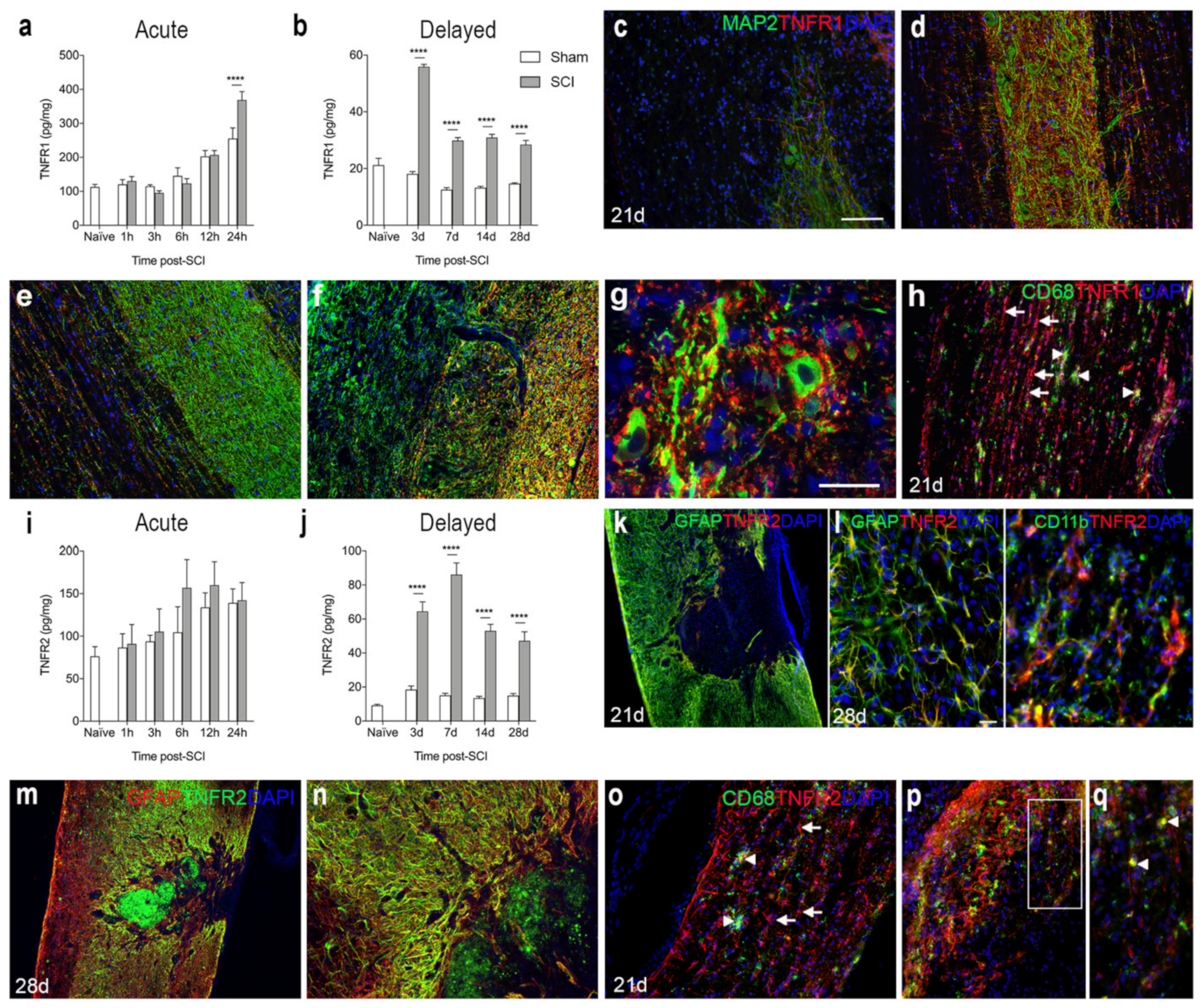
3.5. TNFR1 and TNFR2 Expression Increases in the Lesioned Spinal Cord
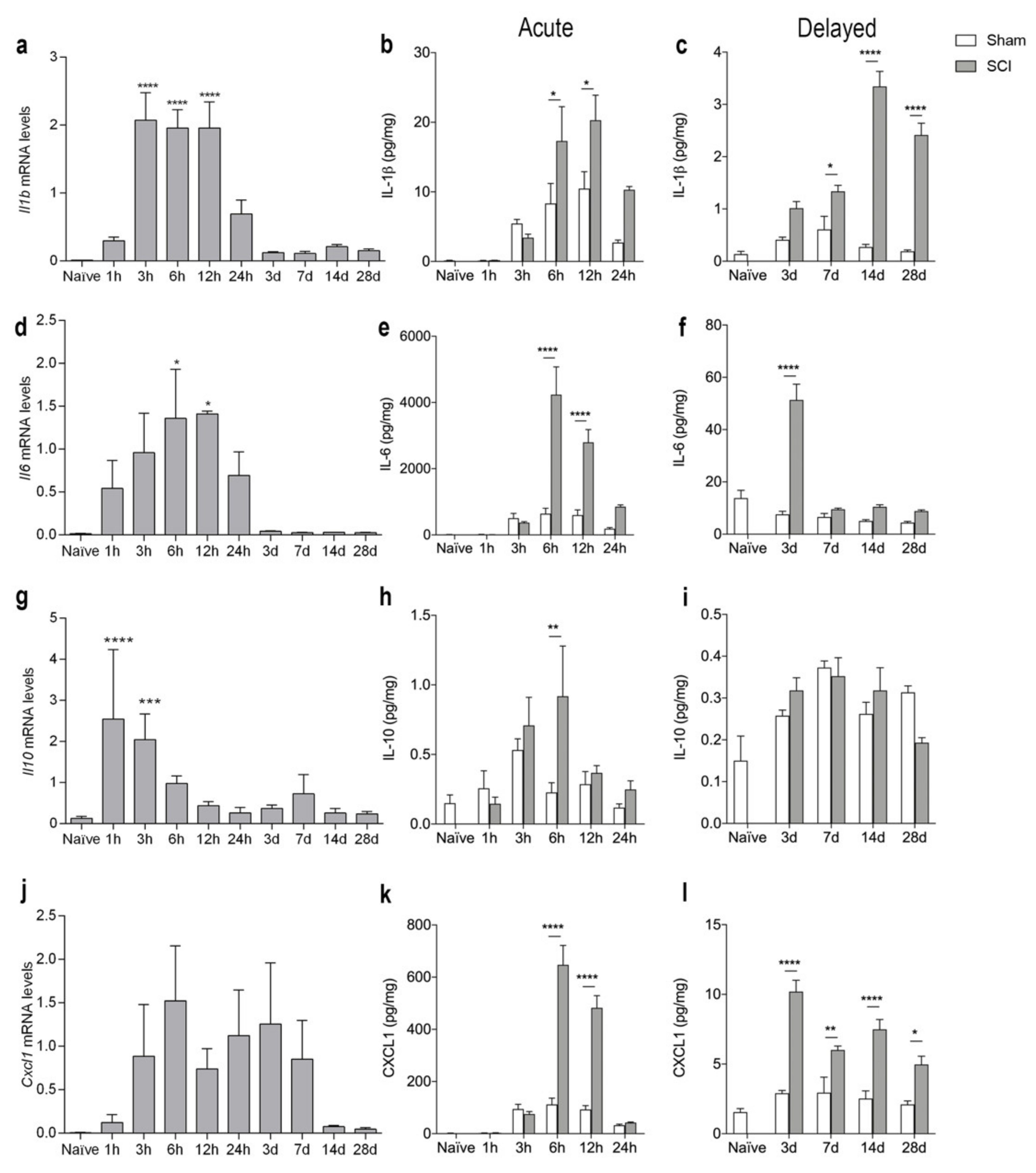
3.6. SCI Results in Increased Levels of Inflammatory Cytokines
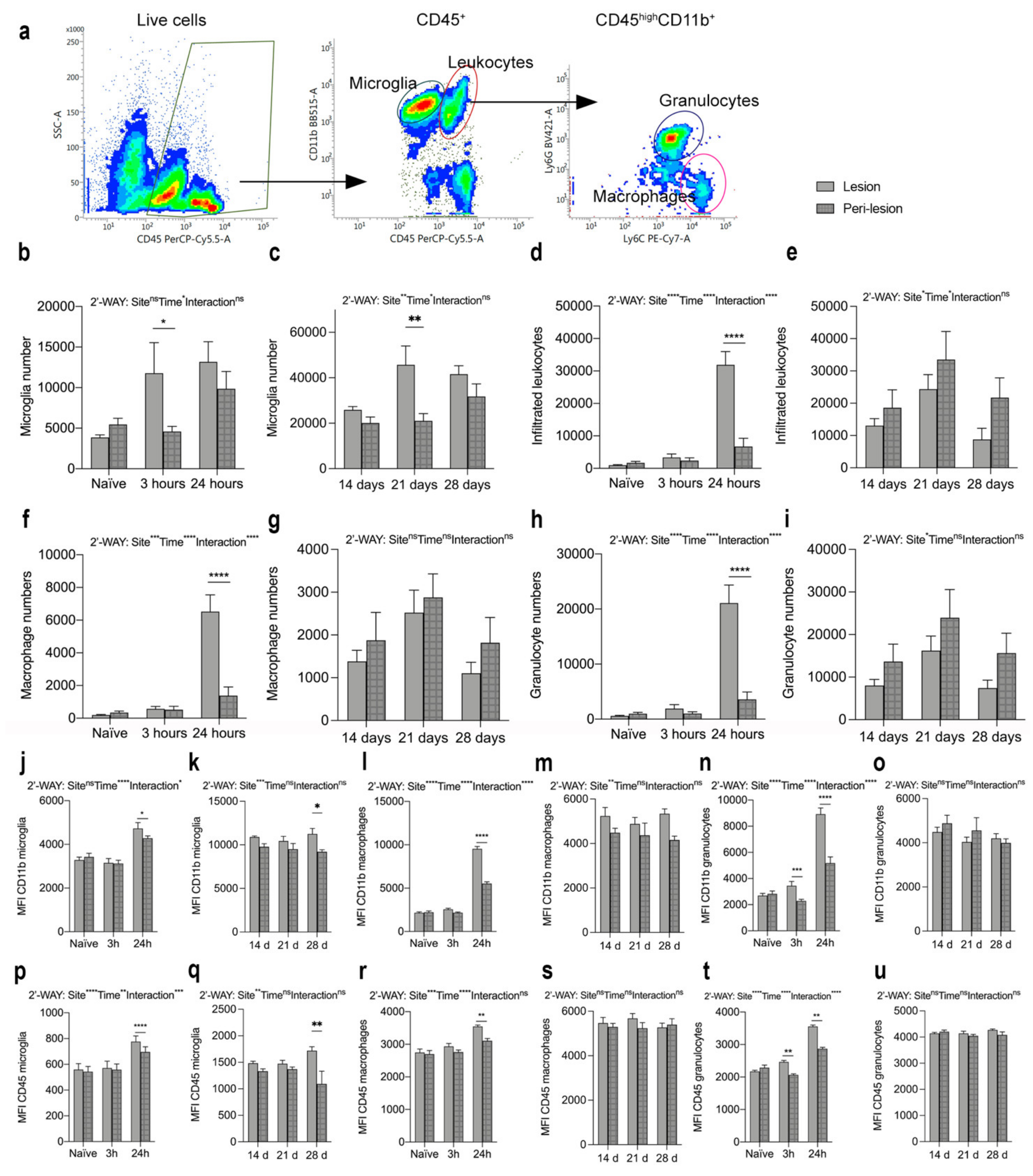
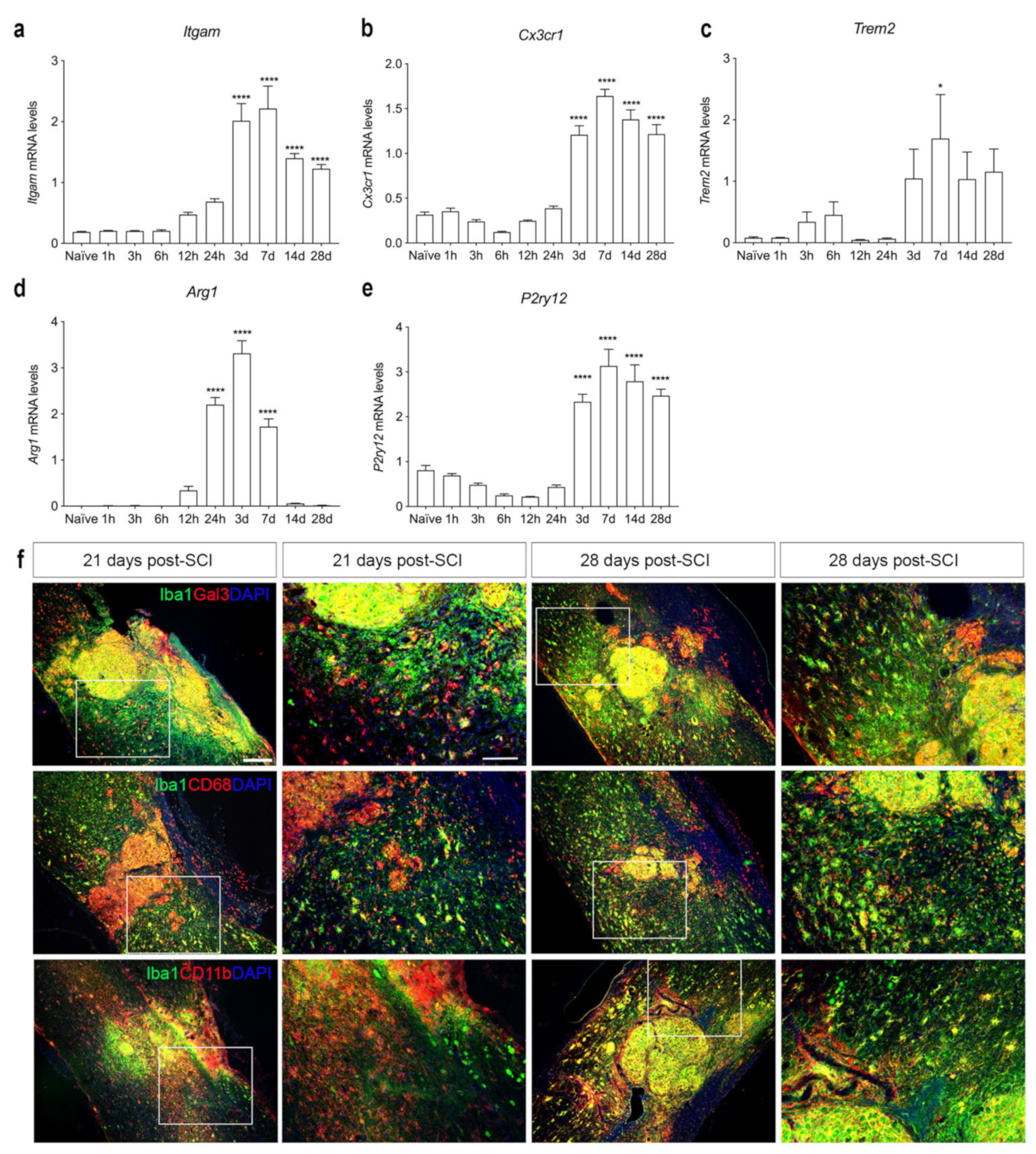
3.7. SCI Results in Microglial Activation and Immune Cell Infiltration into Spinal Cord
3.8. The Cellular Source of TNF and Its Receptors in Human Traumatic Spinal Cord Injury
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, N.K.; Xu, X.M. Neuroprotection and its molecular mechanism following spinal cord injury. Neural Regen. Res. 2012, 7, 2051–2062. [Google Scholar] [CrossRef]
- Trivedi, A.; Olivas, A.D.; Noble-Haeusslein, L.J. Inflammation and Spinal Cord Injury: Infiltrating Leukocytes as Determinants of Injury and Repair Processes. Clin. Neurosci. Res. 2006, 6, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.D.; Nguyen, H.X.; Galvan, M.D.; Salazar, D.L.; Woodruff, T.M.; Anderson, A.J. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: Evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain 2010, 133, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Bastien, D.; Lacroix, S. Cytokine pathways regulating glial and leukocyte function after spinal cord and peripheral nerve injury. Exp. Neurol. 2014, 258, 62–77. [Google Scholar] [CrossRef]
- Fischer, R.; Kontermann, R.E.; Pfizenmaier, K. Selective Targeting of TNF Receptors as a Novel Therapeutic Approach. Front. Cell Dev. Biol. 2020, 8, 401. [Google Scholar] [CrossRef] [PubMed]
- Atretkhany, K.N.; Gogoleva, V.S.; Drutskaya, M.S.; Nedospasov, S.A. Distinct modes of TNF signaling through its two receptors in health and disease. J. Leukoc. Biol. 2020, 107, 893–905. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Vucic, D. Intracellular regulation of TNF activity in health and disease. Cytokine 2018, 101, 26–32. [Google Scholar] [CrossRef]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef]
- Brambilla, R.; Ashbaugh, J.J.; Magliozzi, R.; Dellarole, A.; Karmally, S.; Szymkowski, D.E.; Bethea, J.R. Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain 2011, 134, 2736–2754. [Google Scholar] [CrossRef]
- Wang, C.X.; Nuttin, B.; Heremans, H.; Dom, R.; Gybels, J. Production of tumor necrosis factor in spinal cord following traumatic injury in rats. J. Neuroimmunol. 1996, 69, 151–156. [Google Scholar] [CrossRef]
- Bartholdi, D.; Schwab, M.E. Expression of pro-inflammatory cytokine and chemokine mRNA upon experimental spinal cord injury in mouse: An in situ hybridization study. Eur. J. Neurosci. 1997, 9, 1422–1438. [Google Scholar] [CrossRef]
- Yan, P.; Li, Q.; Kim, G.M.; Xu, J.; Hsu, C.Y.; Xu, X.M. Cellular localization of tumor necrosis factor-alpha following acute spinal cord injury in adult rats. J. Neurotrauma 2001, 18, 563–568. [Google Scholar] [CrossRef]
- Pineau, I.; Lacroix, S. Proinflammatory cytokine synthesis in the injured mouse spinal cord: Multiphasic expression pattern and identification of the cell types involved. J. Comp. Neurol. 2007, 500, 267–285. [Google Scholar] [CrossRef]
- Yune, T.Y.; Chang, M.J.; Kim, S.J.; Lee, Y.B.; Shin, S.W.; Rhim, H.; Kim, Y.C.; Shin, M.L.; Oh, Y.J.; Han, C.T.; et al. Increased production of tumor necrosis factor-alpha induces apoptosis after traumatic spinal cord injury in rats. J. Neurotrauma 2003, 20, 207–219. [Google Scholar] [CrossRef]
- Yang, L.; Blumbergs, P.C.; Jones, N.R.; Manavis, J.; Sarvestani, G.T.; Ghabriel, M.N. Early expression and cellular localization of proinflammatory cytokines interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in human traumatic spinal cord injury. Spine 2004, 29, 966–971. [Google Scholar] [CrossRef]
- Yan, P.; Liu, N.; Kim, G.M.; Xu, J.; Li, Q.; Hsu, C.Y.; Xu, X.M. Expression of the type 1 and type 2 receptors for tumor necrosis factor after traumatic spinal cord injury in adult rats. Exp. Neurol. 2003, 183, 286–297. [Google Scholar] [CrossRef]
- Lund, M.C.; Clausen, B.H.; Brambilla, R.; Lambertsen, K.L. The Role of Tumor Necrosis Factor Following Spinal Cord Injury: A Systematic Review. Cell. Mol. Neurobiol. 2022. [Google Scholar] [CrossRef]
- Vidal, P.M.; Lemmens, E.; Geboes, L.; Vangansewinkel, T.; Nelissen, S.; Hendrix, S. Late blocking of peripheral TNF-alpha is ineffective after spinal cord injury in mice. Immunobiology 2013, 218, 281–284. [Google Scholar] [CrossRef]
- Esposito, E.; Cuzzocrea, S. Anti-TNF therapy in the injured spinal cord. Trends Pharmacol. Sci. 2011, 32, 107–115. [Google Scholar] [CrossRef]
- Kroner, A.; Greenhalgh, A.D.; Zarruk, J.G.; Passos Dos Santos, R.; Gaestel, M.; David, S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron 2014, 83, 1098–1116. [Google Scholar] [CrossRef]
- Genovese, T.; Mazzon, E.; Crisafulli, C.; Di Paola, R.; Muia, C.; Esposito, E.; Bramanti, P.; Cuzzocrea, S. TNF-alpha blockage in a mouse model of SCI: Evidence for improved outcome. Shock 2008, 29, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Huie, J.R.; Ferguson, A.R.; Kyritsis, N.; Pan, J.Z.; Irvine, K.A.; Nielson, J.L.; Schupp, P.G.; Oldham, M.C.; Gensel, J.C.; Lin, A.; et al. Machine intelligence identifies soluble TNFa as a therapeutic target for spinal cord injury. Sci. Rep. 2021, 11, 3442. [Google Scholar] [CrossRef] [PubMed]
- Chi, L.Y.; Yu, J.; Zhu, H.; Li, X.G.; Zhu, S.G.; Kindy, M.S. The dual role of tumor necrosis factor-alpha in the pathophysiology of spinal cord injury. Neurosci. Lett. 2008, 438, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.M.; Xu, J.; Song, S.K.; Yan, P.; Ku, G.; Xu, X.M.; Hsu, C.Y. Tumor necrosis factor receptor deletion reduces nuclear factor-kappaB activation, cellular inhibitor of apoptosis protein 2 expression, and functional recovery after traumatic spinal cord injury. J. Neurosci. 2001, 21, 6617–6625. [Google Scholar] [CrossRef]
- Ellman, D.G.; Degn, M.; Lund, M.C.; Clausen, B.H.; Novrup, H.G.; Flaeng, S.B.; Jorgensen, L.H.; Suntharalingam, L.; Svenningsen, A.F.; Brambilla, R.; et al. Genetic Ablation of Soluble TNF Does Not Affect Lesion Size and Functional Recovery after Moderate Spinal Cord Injury in Mice. Mediat. Inflamm. 2016, 2016, 2684098. [Google Scholar] [CrossRef]
- Ellman, D.G.; Lund, M.C.; Nissen, M.; Nielsen, P.S.; Sorensen, C.; Lester, E.B.; Thougaard, E.; Jorgensen, L.H.; Nedospasov, S.A.; Andersen, D.C.; et al. Conditional Ablation of Myeloid TNF Improves Functional Outcome and Decreases Lesion Size after Spinal Cord Injury in Mice. Cells 2020, 9, 2407. [Google Scholar] [CrossRef]
- Farooque, M.; Isaksson, J.; Olsson, Y. Improved recovery after spinal cord injury in neuronal nitric oxide synthase-deficient mice but not in TNF-alpha-deficient mice. J. Neurotrauma 2001, 18, 105–114. [Google Scholar] [CrossRef]
- Novrup, H.G.; Bracchi-Ricard, V.; Ellman, D.G.; Ricard, J.; Jain, A.; Runko, E.; Lyck, L.; Yli-Karjanmaa, M.; Szymkowski, D.E.; Pearse, D.D.; et al. Central but not systemic administration of XPro1595 is therapeutic following moderate spinal cord injury in mice. J. Neuroinflamm. 2014, 11, 159. [Google Scholar] [CrossRef]
- Chen, K.B.; Uchida, K.; Nakajima, H.; Yayama, T.; Hirai, T.; Watanabe, S.; Guerrero, A.R.; Kobayashi, S.; Ma, W.Y.; Liu, S.Y.; et al. Tumor necrosis factor-alpha antagonist reduces apoptosis of neurons and oligodendroglia in rat spinal cord injury. Spine 2011, 36, 1350–1358. [Google Scholar] [CrossRef]
- Pasparakis, M.; Alexopoulou, L.; Episkopou, V.; Kollias, G. Immune and inflammatory responses in TNF alpha-deficient mice: A critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J. Exp. Med. 1996, 184, 1397–1411. [Google Scholar] [CrossRef]
- Lambertsen, K.L.; Clausen, B.H.; Babcock, A.A.; Gregersen, R.; Fenger, C.; Nielsen, H.H.; Haugaard, L.S.; Wirenfeldt, M.; Nielsen, M.; Dagnaes-Hansen, F.; et al. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J. Neurosci. 2009, 29, 1319–1330. [Google Scholar] [CrossRef]
- Harrison, M.; O’Brien, A.; Adams, L.; Cowin, G.; Ruitenberg, M.J.; Sengul, G.; Watson, C. Vertebral landmarks for the identification of spinal cord segments in the mouse. Neuroimage 2013, 68, 22–29. [Google Scholar] [CrossRef]
- Basso, D.M.; Fisher, L.C.; Anderson, A.J.; Jakeman, L.B.; McTigue, D.M.; Popovich, P.G. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J. Neurotrauma 2006, 23, 635–659. [Google Scholar] [CrossRef]
- Kibbe, W.A. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res. 2007, 35, W43–W46. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Lambertsen, K.L.; Meldgaard, M.; Ladeby, R.; Finsen, B. A quantitative study of microglial-macrophage synthesis of tumor necrosis factor during acute and late focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 2005, 25, 119–135. [Google Scholar] [CrossRef]
- Lambertsen, K.L.; Gregersen, R.; Drojdahl, N.; Owens, T.; Finsen, B. A specific and sensitive method for visualization of tumor necrosis factor in the murine central nervous system. Brain Res. Protoc. 2001, 7, 175–191. [Google Scholar] [CrossRef]
- Clausen, B.H.; Wirenfeldt, M.; Hogedal, S.S.; Frich, L.H.; Nielsen, H.H.; Schroder, H.D.; Ostergaard, K.; Finsen, B.; Kristensen, B.W.; Lambertsen, K.L. Characterization of the TNF and IL-1 systems in human brain and blood after ischemic stroke. Acta Neuropathol. Commun. 2020, 8, 81. [Google Scholar] [CrossRef]
- Clausen, B.; Degn, M.; Martin, N.; Couch, Y.; Karimi, L.; Ormhoj, M.; Mortensen, M.L.; Gredal, H.; Gardiner, C.; Sargent, I.I.; et al. Systemically administered anti-TNF therapy ameliorates functional outcomes after focal cerebral ischemia. J. Neuroinflamm. 2014, 11, 203. [Google Scholar] [CrossRef]
- Donnelly, D.J.; Popovich, P.G. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp. Neurol. 2008, 209, 378–388. [Google Scholar] [CrossRef]
- Tan, Y.; Zheng, Y.; Xu, D.; Sun, Z.; Yang, H.; Yin, Q. Galectin-3: A key player in microglia-mediated neuroinflammation and Alzheimer’s disease. Cell Biosci. 2021, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- White, R.E.; McTigue, D.M.; Jakeman, L.B. Regional heterogeneity in astrocyte responses following contusive spinal cord injury in mice. J. Comp. Neurol. 2010, 518, 1370–1390. [Google Scholar] [CrossRef]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 2009, 29, 13435–13444. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.L.; Shih, K.; Bao, P.; Ghirnikar, R.S.; Eng, L.F. Cytokine chemokine expression in contused rat spinal cord. Neurochem. Int. 2000, 36, 417–425. [Google Scholar] [CrossRef]
- Wang, C.X.; Reece, C.; Wrathall, J.R.; Shuaib, A.; Olschowka, J.A.; Hao, C. Expression of tumor necrosis factor alpha and its mRNA in the spinal cord following a weight-drop injury. Neuroreport 2002, 13, 1391–1393. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Bracchi-Ricard, V.; Hu, W.H.; Frydel, B.; Bramwell, A.; Karmally, S.; Green, E.J.; Bethea, J.R. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J. Exp. Med. 2005, 202, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Simmons, R.D.; Willenborg, D.O. Direct injection of cytokines into the spinal cord causes autoimmune encephalomyelitis-like inflammation. J. Neurol. Sci. 1990, 100, 37–42. [Google Scholar] [CrossRef]
- D’Souza, S.; Alinauskas, K.; McCrea, E.; Goodyer, C.; Antel, J.P. Differential susceptibility of human CNS-derived cell populations to TNF-dependent and independent immune-mediated injury. J. Neurosci. 1995, 15, 7293–7300. [Google Scholar] [CrossRef]
- Akassoglou, K.; Bauer, J.; Kassiotis, G.; Pasparakis, M.; Lassmann, H.; Kollias, G.; Probert, L. Oligodendrocyte apoptosis and primary demyelination induced by local TNF/p55TNF receptor signaling in the central nervous system of transgenic mice: Models for multiple sclerosis with primary oligodendrogliopathy. Am. J. Pathol. 1998, 153, 801–813. [Google Scholar] [CrossRef]
- McCoy, M.K.; Tansey, M.G. TNF signaling inhibition in the CNS: Implications for normal brain function and neurodegenerative disease. J. Neuroinflamm. 2008, 5, 45. [Google Scholar] [CrossRef]
- Fischer, R.; Maier, O.; Siegemund, M.; Wajant, H.; Scheurich, P.; Pfizenmaier, K. A TNF receptor 2 selective agonist rescues human neurons from oxidative stress-induced cell death. PLoS ONE 2011, 6, e27621. [Google Scholar] [CrossRef]
- Papazian, I.; Tsoukala, E.; Boutou, A.; Karamita, M.; Kambas, K.; Iliopoulou, L.; Fischer, R.; Kontermann, R.E.; Denis, M.C.; Kollias, G.; et al. Fundamentally different roles of neuronal TNF receptors in CNS pathology: TNFR1 and IKKbeta promote microglial responses and tissue injury in demyelination while TNFR2 protects against excitotoxicity in mice. J. Neuroinflamm. 2021, 18, 222. [Google Scholar] [CrossRef]
- Veroni, C.; Gabriele, L.; Canini, I.; Castiello, L.; Coccia, E.; Remoli, M.E.; Columba-Cabezas, S.; Arico, E.; Aloisi, F.; Agresti, C. Activation of TNF receptor 2 in microglia promotes induction of anti-inflammatory pathways. Mol. Cell. Neurosci. 2010, 45, 234–244. [Google Scholar] [CrossRef]
- Gao, H.; Danzi, M.C.; Choi, C.S.; Taherian, M.; Dalby-Hansen, C.; Ellman, D.G.; Madsen, P.M.; Bixby, J.L.; Lemmon, V.P.; Lambertsen, K.L.; et al. Opposing Functions of Microglial and Macrophagic TNFR2 in the Pathogenesis of Experimental Autoimmune Encephalomyelitis. Cell Rep. 2017, 18, 198–212. [Google Scholar] [CrossRef]
- Patel, J.R.; Williams, J.L.; Muccigrosso, M.M.; Liu, L.; Sun, T.; Rubin, J.B.; Klein, R.S. Astrocyte TNFR2 is required for CXCL12-mediated regulation of oligodendrocyte progenitor proliferation and differentiation within the adult CNS. Acta Neuropathol. 2012, 124, 847–860. [Google Scholar] [CrossRef]
- Fischer, R.; Wajant, H.; Kontermann, R.; Pfizenmaier, K.; Maier, O. Astrocyte-specific activation of TNFR2 promotes oligodendrocyte maturation by secretion of leukemia inhibitory factor. Glia 2014, 62, 272–283. [Google Scholar] [CrossRef]
- Hellenbrand, D.J.; Reichl, K.A.; Travis, B.J.; Filipp, M.E.; Khalil, A.S.; Pulito, D.J.; Gavigan, A.V.; Maginot, E.R.; Arnold, M.T.; Adler, A.G.; et al. Sustained interleukin-10 delivery reduces inflammation and improves motor function after spinal cord injury. J. Neuroinflamm. 2019, 16, 93. [Google Scholar] [CrossRef]
- Bethea, J.R.; Nagashima, H.; Acosta, M.C.; Briceno, C.; Gomez, F.; Marcillo, A.E.; Loor, K.; Green, J.; Dietrich, W.D. Systemically administered interleukin-10 reduces tumor necrosis factor-alpha production and significantly improves functional recovery following traumatic spinal cord injury in rats. J. Neurotrauma 1999, 16, 851–863. [Google Scholar] [CrossRef]
- Kuno, R.; Wang, J.; Kawanokuchi, J.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Autocrine activation of microglia by tumor necrosis factor-alpha. J. Neuroimmunol. 2005, 162, 89–96. [Google Scholar] [CrossRef]
- Wang, C.X.; Olschowka, J.A.; Wrathall, J.R. Increase of interleukin-1beta mRNA and protein in the spinal cord following experimental traumatic injury in the rat. Brain Res. 1997, 759, 190–196. [Google Scholar] [CrossRef]
- Plunkett, J.A.; Yu, C.G.; Easton, J.M.; Bethea, J.R.; Yezierski, R.P. Effects of interleukin-10 (IL-10) on pain behavior and gene expression following excitotoxic spinal cord injury in the rat. Exp. Neurol. 2001, 168, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Giulian, D.; Woodward, J.; Young, D.G.; Krebs, J.F.; Lachman, L.B. Interleukin-1 injected into mammalian brain stimulates astrogliosis and neovascularization. J. Neurosci. 1988, 8, 2485–2490. [Google Scholar] [CrossRef]
- Bayrakli, F.; Kurtuncu, M.; Karaarslan, E.; Ozgen, S. Perineural cyst presenting like cubital tunnel syndrome. Eur. Spine J. 2012, 21 (Suppl. S4), S387–S389. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sato, A.; Ohtaki, H.; Tsumuraya, T.; Song, D.; Ohara, K.; Asano, M.; Iwakura, Y.; Atsumi, T.; Shioda, S. Interleukin-1 participates in the classical and alternative activation of microglia/macrophages after spinal cord injury. J. Neuroinflamm. 2012, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Bastien, D.; Bellver Landete, V.; Lessard, M.; Vallieres, N.; Champagne, M.; Takashima, A.; Tremblay, M.E.; Doyon, Y.; Lacroix, S. IL-1alpha Gene Deletion Protects Oligodendrocytes after Spinal Cord Injury through Upregulation of the Survival Factor Tox3. J. Neurosci. 2015, 35, 10715–10730. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.G.; Jogia, T.; Gillespie, E.R.; Couch, Y.; Ruitenberg, M.J.; Anthony, D.C. Acute IL-1RA treatment suppresses the peripheral and central inflammatory response to spinal cord injury. J. Neuroinflamm. 2021, 18, 15. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.R.; Uchida, K.; Nakajima, H.; Watanabe, S.; Nakamura, M.; Johnson, W.E.; Baba, H. Blockade of interleukin-6 signaling inhibits the classic pathway and promotes an alternative pathway of macrophage activation after spinal cord injury in mice. J. Neuroinflamm. 2012, 9, 40. [Google Scholar] [CrossRef]
- Mukaino, M.; Nakamura, M.; Yamada, O.; Okada, S.; Morikawa, S.; Renault-Mihara, F.; Iwanami, A.; Ikegami, T.; Ohsugi, Y.; Tsuji, O.; et al. Anti-IL-6-receptor antibody promotes repair of spinal cord injury by inducing microglia-dominant inflammation. Exp. Neurol. 2010, 224, 403–414. [Google Scholar] [CrossRef]
- Cafferty, W.B.; Gardiner, N.J.; Das, P.; Qiu, J.; McMahon, S.B.; Thompson, S.W. Conditioning injury-induced spinal axon regeneration fails in interleukin-6 knock-out mice. J. Neurosci. 2004, 24, 4432–4443. [Google Scholar] [CrossRef]
- Okada, S.; Nakamura, M.; Katoh, H.; Miyao, T.; Shimazaki, T.; Ishii, K.; Yamane, J.; Yoshimura, A.; Iwamoto, Y.; Toyama, Y.; et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat. Med. 2006, 12, 829–834. [Google Scholar] [CrossRef]
- Neirinckx, V.; Coste, C.; Franzen, R.; Gothot, A.; Rogister, B.; Wislet, S. Neutrophil contribution to spinal cord injury and repair. J. Neuroinflamm. 2014, 11, 150. [Google Scholar] [CrossRef]
- Masuda, T.; Amann, L.; Sankowski, R.; Staszewski, O.; Lenz, M.; Errico, P.D.; Snaidero, N.; Costa Jordao, M.J.; Bottcher, C.; Kierdorf, K.; et al. Novel Hexb-based tools for studying microglia in the CNS. Nat. Immunol. 2020, 21, 802–815. [Google Scholar] [CrossRef]
- Bellver-Landete, V.; Bretheau, F.; Mailhot, B.; Vallieres, N.; Lessard, M.; Janelle, M.E.; Vernoux, N.; Tremblay, M.E.; Fuehrmann, T.; Shoichet, M.S.; et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat. Commun. 2019, 10, 518. [Google Scholar] [CrossRef]
- Milich, L.M.; Choi, J.; Ryan, C.; Yahn, S.L.; Tsoulfas, P.; Lee, J.K. Single cell analysis of the cellular heterogeneity and interactions in the injured mouse spinal cord. J. Exp. Med. 2021, 218, e20210040. [Google Scholar] [CrossRef]
- Thomas, L.; Pasquini, L.A. Galectin-3-Mediated Glial Crosstalk Drives Oligodendrocyte Differentiation and (Re)myelination. Front. Cell. Neurosci. 2018, 12, 297. [Google Scholar] [CrossRef]
- Hoyos, H.C.; Rinaldi, M.; Mendez-Huergo, S.P.; Marder, M.; Rabinovich, G.A.; Pasquini, J.M.; Pasquini, L.A. Galectin-3 controls the response of microglial cells to limit cuprizone-induced demyelination. Neurobiol. Dis. 2014, 62, 441–455. [Google Scholar] [CrossRef]
- Nugent, A.A.; Lin, K.; van Lengerich, B.; Lianoglou, S.; Przybyla, L.; Davis, S.S.; Llapashtica, C.; Wang, J.; Kim, D.J.; Xia, D.; et al. TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron 2020, 105, 837–854. [Google Scholar] [CrossRef]
- Greenhalgh, A.D.; David, S. Differences in the phagocytic response of microglia and peripheral macrophages after spinal cord injury and its effects on cell death. J. Neurosci. 2014, 34, 6316–6322. [Google Scholar] [CrossRef]
- Nakajima, H.; Honjoh, K.; Watanabe, S.; Kubota, A.; Matsumine, A. Distribution and polarization of microglia and macrophages at injured sites and the lumbar enlargement after spinal cord injury. Neurosci. Lett. 2020, 737, 135152. [Google Scholar] [CrossRef]
- Pang, Q.M.; Chen, S.Y.; Xu, Q.J.; Fu, S.P.; Yang, Y.C.; Zou, W.H.; Zhang, M.; Liu, J.; Wan, W.H.; Peng, J.C.; et al. Neuroinflammation and Scarring After Spinal Cord Injury: Therapeutic Roles of MSCs on Inflammation and Glial Scar. Front. Immunol. 2021, 12, 751021. [Google Scholar] [CrossRef]
- Khazaei, M.; Siddiqui, A.M.; Fehlings, M.G. The Potential for iPS-Derived Stem Cells as a Therapeutic Strategy for Spinal Cord Injury: Opportunities and Challenges. J. Clin. Med. 2014, 4, 37–65. [Google Scholar] [CrossRef]
- Kawabata, S.; Takano, M.; Numasawa-Kuroiwa, Y.; Itakura, G.; Kobayashi, Y.; Nishiyama, Y.; Sugai, K.; Nishimura, S.; Iwai, H.; Isoda, M.; et al. Grafted Human iPS Cell-Derived Oligodendrocyte Precursor Cells Contribute to Robust Remyelination of Demyelinated Axons after Spinal Cord Injury. Stem Cell Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef]
- Stewart, A.N.; Lowe, J.L.; Glaser, E.P.; Mott, C.A.; Shahidehpour, R.K.; McFarlane, K.E.; Bailey, W.M.; Zhang, B.; Gensel, J.C. Acute inflammatory profiles differ with sex and age after spinal cord injury. J. Neuroinflamm. 2021, 18, 113. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.N.; MacLean, S.M.; Stromberg, A.J.; Whelan, J.P.; Bailey, W.M.; Gensel, J.C.; Wilson, M.E. Considerations for Studying Sex as a Biological Variable in Spinal Cord Injury. Front. Neurol. 2020, 11, 802. [Google Scholar] [CrossRef]
- Del Rivero, T.; Fischer, R.; Yang, F.; Swanson, K.A.; Bethea, J.R. Tumor necrosis factor receptor 1 inhibition is therapeutic for neuropathic pain in males but not in females. Pain 2019, 160, 922–931. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Controls | Sub-Acute | Early Chronic | Late Chronic | |
|---|---|---|---|---|
| Number of cases | 5 | 12 | 10 | 11 |
| Sex, n (%) men | 4 (80) | 11 (92) | 10 (100) | 11 (100) |
| Age, years, median (IQR) | 31 (23.0; 44.5) | 30.5 (28.3; 47.0) | 30.0 (26.8; 36.3) | 45.0 (39.0; 54.0) |
| Case | Age/Sex | Level of Injury | Cause of Injury | Post-SCI Survival Time |
|---|---|---|---|---|
| #1 | 80/F | C6–T1 | Fall | 15 h |
| #2 | 61/M | C1–2 | Dive accident | 2 weeks |
| #3 | 43/M | C7 | Fall | 16 days |
| #4 | 33/M | C6–7 | MVA | 3 weeks |
| #5 | 65/M | C4 | MVA | 5 weeks |
| #6 | 67/M | C5–7 | Fall | 6 weeks |
| Gene | Primer Sequences (5′-3′) | Accession No. |
|---|---|---|
| Tnf | F- AGGCACTCCCCCAAAAGATG | NM_001278601.1 |
| R- TCACCCCGAAGTTCAGTAGACAGA | ||
| Tnfrsf1a | F- GCCCGAAGTCTACTCCATCATTTG | NM_011609.4 |
| R- GGCTGGGGAGGGGGCTGGAGTTAG | ||
| Tnfrsf1b | F- GCCCAGCCAAACTCCAAGCATC | NM_011610.3 |
| R- TCCTAACATCAGCAGACCCAGTG | ||
| Il1b | F- TGCCACCTTTTGACAGTGATG | NM_008361.4 |
| R- CAAAGGTTTGGAAGCAGCCC | ||
| Il6 | F- AGGATACCACTCCCAACAGA | NM_001314054.1 |
| R- ACTCCAGGTAGCTATGGTACTC | ||
| Il10 | F- GCCAGGTGAAGACTTTCTTTCAAAC | NM_010548.2 |
| R- AGTCCAGCAGACTCAATACACAC | ||
| Cxcl1 | F- GCTGGGATTCACCTCAAGAAC | NM_008176.3 |
| R- TGTGGCTATGACTTCGGTTTG | ||
| Itgam | F- GCCTGTCACACTGAGCAGAA | NM_008401.2 |
| R- TGCAACAGAGCAGTTCAGCA | ||
| Cx3cr1 | F- TCCCATCTGCTCAGGACCTC | NM_009987.4 |
| R- GGCCTCAGCAGAATCGTCAT | ||
| Trem2 | F- TGCTGGAGATCTCTGGGTCC | NM_031254.3 |
| R- AGGTCTCTTGATTCCTGGAGGT | ||
| Arg1 | F- ATGAAGAGCTGGCTGGTGTG | NM_007482.3 |
| R- CCAACTGCCAGACTGTGGTC | ||
| P2ry12 | F- GCCAGTGTCATTTGCTGTCAC | NM_027571.4 |
| R- TAGATGCCACCCCTTGCACT | ||
| Hprt1 | F- TCCTCAGACCGCTTTTTGCC | NM_013556.2 |
| R- TCATCATCGCTAATCACGACGC |
| Antibody | Conjugated | Host (Clone) | Dilution | Source (cat. no.) |
|---|---|---|---|---|
| Anti-TNF | Unconjugated | Rabbit | 1:200 | Thermo Fischer Scientific (P-350) |
| Anti-TNFR1 | Unconjugated | Rabbit (H-271) | 1:50 | Santa Cruz (sc-7895) |
| Anti-TNFR2 | Unconjugated | Rabbit | 1:200 | Sigma Aldrich (HPA004796) |
| Anti-TNFR2 | Unconjugated | Goat | 1:50 | R&D Systems (AF-426-PB) |
| Anti-MAP2 | Unconjugated | Chicken | 1:100 | Abcam (ab5392) |
| Anti-CD68 | Unconjugated | Rat | 1:400 | Bio-Rad (MCA1957) |
| Anti-CD68 | Unconjugated | Mouse (PG-M1) | 1:100 | Abcam (ab783) |
| Anti-CD11b | Unconjugated | Rat | 1:500 | Bio-Rad (MCA711) |
| Anti-Iba1 | Unconjugated | Rabbit | 1:500 | Wako (019–19741) |
| Anti-IBA1 | Unconjugated | Mouse (GT10312) | 1:1000 | Sigma Aldrich (SAB2702364) |
| Anti-Galectin-3 | Unconjugated | Rat (M38) | 1:300 | Hakon Leffler’s Lab [38] |
| Anti-NF-L | Alexa Fluor-488 | Mouse (N52) | 1:50 | Sigma Aldrich (MAB5266) |
| Anti-IL-1β | Unconjugated | Mouse (2E8) | 1:50 | Bio-Rad (MCA5542Z) |
| Anti-GFAP | Cy3 | Mouse (G-A-5) | 1:500 | Sigma Aldrich (C9205) |
| Anti-GFAP | Alexa Fluor-488 | Mouse (131–17719) | 1:400 | Invitrogen (A21294) |
| Anti-Rabbit | Alexa Fluor-594 | Donkey | 1:200 | Invitrogen (A21207) |
| Anti-Rabbit | Alexa Fluor-488 | Chicken | 1:200 | Invitrogen (A21441) |
| Anti-Rat | Alexa Fluor-594 | Goat | 1:200 | Invitrogen (A11007) |
| Anti-rabbit | Alexa Fluor-594 | Goat | 1:200 | Invitrogen (A21207) |
| Anti-rabbit | Alexa Fluor-488 | Goat | 1:200 | Invitrogen (A11008) |
| Anti-rabbit | Alexa Fluor-568 | Goat | 1:200 | Invitrogen (A11011) |
| Anti-Rat | Alexa Fluor-488 | Goat | 1:200 | Invitrogen (A11006) |
| Anti-Chicken | Alexa Fluor-488 | Goat | 1:200 | Invitrogen (A11039) |
| Anti-mouse | Alexa Fluor-488 | Goat | 1:200 | Invitrogen (A11001) |
| Anti-mouse | Alexa Fluor-568 | Goat | 1:200 | Invitrogen (A11004) |
| Anti-mouse | Alexa Fluor-555 | Goat | 1:200 | Invitrogen (A21422) |
| Anti-goat | Alexa Fluor-594 | Donkey | 1:200 | Invitrogen (A11058) |
| Antibody | Conjugated | Host (Clone) | Dilution | Source (cat. no.) |
|---|---|---|---|---|
| Anti-CD45 | PerCP-Cy5.5 | Rat (30-F11) | 1:100 | BD Biosciences (561869) |
| Anti-CD11b | BB515 | Rat (M1/70) | 1:200 | BD Biosciences (564454) |
| Anti-Ly-6C | PE-Cy7 | Rat (AL-21) | 1:200 | BD Biosciences (560593) |
| Anti-Ly-6G | BV421 | Rat (1A8) | 1:200 | BD Biosciences (562737) |
| IgG2b, κ | PerCP-Cy5.5 | Rat (A95–1) | 1:100 | BD Biosciences (550764) |
| IgG2b, κ | BB515 | Rat (A95–1) | 1:200 | BD biosciences (564421) |
| IgM, κ | PE-Cy7 | Rat (clone R4–22) | 1:200 | BD biosciences (560572) |
| IgG2a, κ | BV421 | Rat (clone R35–95) | 1:200 | BD Biosciences (562602) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lund, M.C.; Ellman, D.G.; Nissen, M.; Nielsen, P.S.; Nielsen, P.V.; Jørgensen, C.; Andersen, D.C.; Gao, H.; Brambilla, R.; Degn, M.; et al. The Inflammatory Response after Moderate Contusion Spinal Cord Injury: A Time Study. Biology 2022, 11, 939. https://doi.org/10.3390/biology11060939
Lund MC, Ellman DG, Nissen M, Nielsen PS, Nielsen PV, Jørgensen C, Andersen DC, Gao H, Brambilla R, Degn M, et al. The Inflammatory Response after Moderate Contusion Spinal Cord Injury: A Time Study. Biology. 2022; 11(6):939. https://doi.org/10.3390/biology11060939
Chicago/Turabian StyleLund, Minna Christiansen, Ditte Gry Ellman, Maiken Nissen, Pernille Sveistrup Nielsen, Pernille Vinther Nielsen, Carina Jørgensen, Ditte Caroline Andersen, Han Gao, Roberta Brambilla, Matilda Degn, and et al. 2022. "The Inflammatory Response after Moderate Contusion Spinal Cord Injury: A Time Study" Biology 11, no. 6: 939. https://doi.org/10.3390/biology11060939
APA StyleLund, M. C., Ellman, D. G., Nissen, M., Nielsen, P. S., Nielsen, P. V., Jørgensen, C., Andersen, D. C., Gao, H., Brambilla, R., Degn, M., Clausen, B. H., & Lambertsen, K. L. (2022). The Inflammatory Response after Moderate Contusion Spinal Cord Injury: A Time Study. Biology, 11(6), 939. https://doi.org/10.3390/biology11060939