
Comprehensive In Silico Analysis of Retrotransposon Insertions within the Survival Motor Neuron Genes Involved in Spinal Muscular Atrophy

 , , , and
, , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. SMA Genomic Sequence Cohort
2.2. In Silico Analysis of Sequencing Data
3. Results
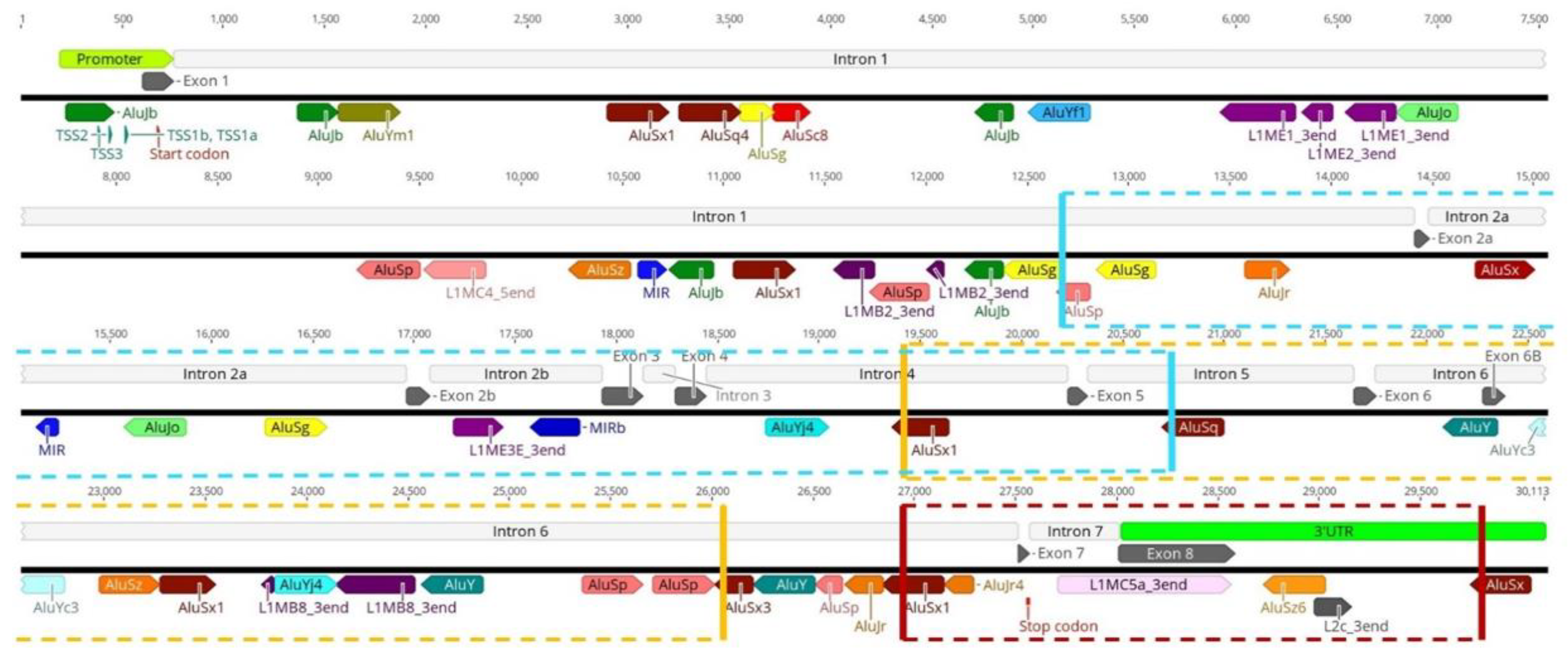
3.1. Transposable Elements and SMN1 Transcription
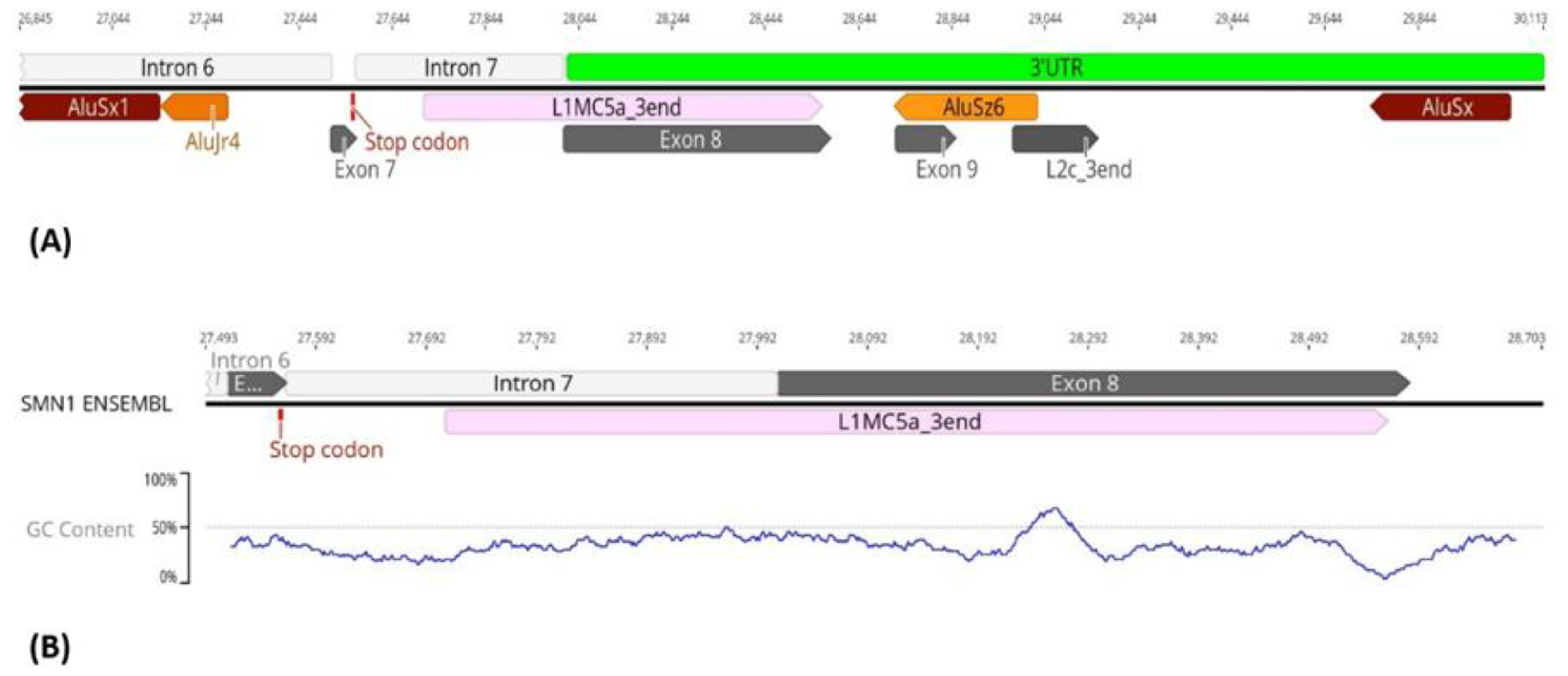
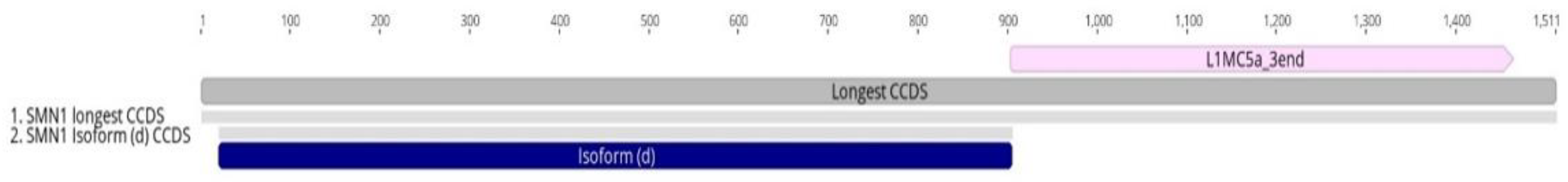
3.2. Transposable Elements and Alternative Splicing by Exonization
3.3. Transposable Elements and Partial Deletions of SMN1
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hancks, D.C.; Kazazian, H.H. Roles for retrotransposon insertions in human disease. Mob. DNA 2016, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Jangam, D.; Feschotte, C.; Betrán, E. Transposable element domestication as an adaptation to evolutionary conflicts. Trends Genet. 2017, 33, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef]
- Platt, R.N.; Vandewege, M.W.; Ray, D.A. Mammalian transposable elements and their impacts on genome evolution. Chromosome Res. 2018, 26, 25–43. [Google Scholar] [CrossRef]
- Cosby, R.L.; Chang, N.-C.; Feschotte, C. Host-transposon interactions: Conflict, cooperation, and cooption. Genes Dev. 2019, 33, 1098–1116. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Perez, J.L.; Widmann, T.J.; Adams, I.R. The impact of transposable elements on mammalian development. Development 2016, 143, 4101–4114. [Google Scholar] [CrossRef] [PubMed]
- Paço, A.; Adega, F.; Chaves, R. LINE-1 retrotransposons: From ‘parasite’ sequences to functional elements. J. Appl. Genet. 2015, 56, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Payer, L.M.; Burns, K.H. Transposable elements in human genetic disease. Nat. Rev. Genet. 2019, 20, 760–772. [Google Scholar] [CrossRef]
- Burns, K.H. Our Conflict with Transposable Elements and Its Implications for Human Disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 51–70. [Google Scholar] [CrossRef]
- Batzer, M.A.; Deininger, P.L.; Hellmann-Blumberg, U.; Jurka, J.; Labuda, D.; Rubin, C.M.; Schmid, C.W.; Zietkiewicz, E.; Zuckerkandl, E. Standardized nomenclature for Alu repeats. J. Mol. Evol. 1996, 42, 3–6. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2017, 18, 71. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, R.; Farivar, S.; Mager, D.L. C-GATE-catalogue of genes affected by transposable elements. Mob. DNA 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Friedli, M.; Trono, D. The developmental control of transposable elements and the evolution of higher species. Annu. Rev. Cell Dev. Biol. 2015, 31, 429–451. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Pederson, S.M.; Kortschak, R.D.; Adelson, D.L. Transposable elements and gene expression during the evolution of amniotes. Mob. DNA 2018, 9, 17. [Google Scholar] [CrossRef]
- Sundaram, V.; Wysocka, J. Transposable elements as a potent source of diverse cis-regulatory sequences in mammalian genomes. Philos. Trans. R. Soc. B 2020, 375, 20190347. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- O’Donnell, K.A.; Burns, K.H. Mobilizing diversity: Transposable element insertions in genetic variation and disease. Mob. DNA 2010, 1, 21. [Google Scholar] [CrossRef]
- Vorechovsky, I. Transposable elements in disease-associated cryptic exons. Hum. Genet. 2010, 127, 135–154. [Google Scholar] [CrossRef]
- Saleh, A.; Macia, A.; Muotri, A.R. Transposable elements, inflammation, and neurological disease. Front. Neurol. 2019, 10, 894. [Google Scholar] [CrossRef]
- Hedges, D.J.; Deininger, P.L. Inviting instability: Transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2007, 616, 46–59. [Google Scholar] [CrossRef]
- Lapp, H.E.; Hunter, R.G. Early life exposures, neurodevelopmental disorders, and transposable elements. Neurobiol. Stress 2019, 11, 100174. [Google Scholar] [CrossRef] [PubMed]
- Percharde, M.; Sultana, T.; Ramalho-Santos, M. What doesn’t kill you makes you stronger: Transposons as dual players in chromatin regulation and genomic variation. BioEssays 2020, 42, 1900232. [Google Scholar] [CrossRef] [PubMed]
- Jönsson, M.E.; Garza, R.; Johansson, P.A.; Jakobsson, J. Transposable elements: A common feature of neurodevelopmental and neurodegenerative disorders. Trends Genet. 2020, 36, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Crawford, T.O.; Pardo, C.A. The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis. 1996, 3, 97–110. [Google Scholar] [CrossRef]
- Kolb, S.J.; Kissel, J.T. Spinal muscular atrophy. Neurol. Clin. 2015, 33, 831–846. [Google Scholar] [CrossRef]
- Butchbach, M.E. Genomic variability in the survival motor neuron genes (Smn1 and smn2): Implications for spinal muscular atrophy phenotype and therapeutics development. Int. J. Mol. Sci. 2021, 22, 7896. [Google Scholar] [CrossRef]
- Wirth, B. Spinal muscular atrophy: In the challenge lies a solution. Trends Neurosci. 2021, 44, 306–322. [Google Scholar] [CrossRef]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- Blasco-Pérez, L.; Paramonov, I.; Leno, J.; Bernal, S.; Alias, L.; Fuentes-Prior, P.; Cuscó, I.; Tizzano, E.F. Beyond copy number: A new, rapid, and versatile method for sequencing the entire SMN2 gene in SMA patients. Hum Mutat 2021, 42, 787–795. [Google Scholar] [CrossRef]
- Campbell, L.; Potter, A.; Ignatius, J.; Dubowitz, V.; Davies, K. Genomic variation and gene conversion in spinal muscular atrophy: Implications for disease process and clinical phenotype. Am. J. Hum. Genet. 1997, 61, 40. [Google Scholar] [CrossRef]
- Courseaux, A.; Richard, F.; Grosgeorge, J.; Ortola, C.; Viale, A.; Turc-Carel, C.; Dutrillaux, B.; Gaudray, P.; Nahon, J.-L. Segmental duplications in euchromatic regions of human chromosome 5: A source of evolutionary instability and transcriptional innovation. Genome Res. 2003, 13, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; Martin, J.; Terry, A.; Couronne, O.; Grimwood, J.; Lowry, S.; Gordon, L.A.; Scott, D.; Xie, G.; Huang, W.; et al. The DNA sequence and comparative analysis of human chromosome 5. Nature 2004, 431, 268–274. [Google Scholar] [CrossRef]
- Ottesen, E.W.; Singh, R.N. Characteristics of circular RNAs generated by human Survival Motor Neuron genes. Cell. Signal. 2020, 73, 109696. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.W.; Seo, J.; Singh, N.N.; Singh, R.N. A multilayered control of the human survival motor neuron gene expression by Alu elements. Front. Microbiol. 2017, 8, 2252. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, V.; Jolly, A.; Bielli, P.; Di Rosa, V.; De la Grange, P.; Sette, C. Sam68 binds Alu-rich introns in SMN and promotes pre-mRNA circularization. Nucleic Acids Res. 2020, 48, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Ruhno, C.; McGovern, V.L.; Avenarius, M.R.; Snyder, P.J.; Prior, T.W.; Nery, F.C.; Muhtaseb, A.; Roggenbuck, J.S.; Kissel, J.T.; Sansone, V.A.; et al. Complete sequencing of the SMN2 gene in SMA patients detects SMN gene deletion junctions and variants in SMN2 that modify the SMA phenotype. Hum. Genet. 2019, 138, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Wirth, B.; Herz, M.; Wetter, A.; Moskau, S.; Hahnen, E.; Rudnik-Schöneborn, S.; Wienker, T.; Zerres, K. Quantitative analysis of survival motor neuron copies: Identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am. J. Hum. Genet. 1999, 64, 1340–1356. [Google Scholar] [CrossRef]
- Jedličková, I.; Přistoupilová, A.; Nosková, L.; Majer, F.; Stránecký, V.; Hartmannová, H.; Hodaňová, K.; Trešlová, H.; Hýblová, M.; Solár, P.; et al. Spinal muscular atrophy caused by a novel Alu-mediated deletion of exons 2a-5 in SMN1 undetectable with routine genetic testing. Mol. Genet. Genom. Med. 2020, 8, e1238. [Google Scholar] [CrossRef]
- Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef]
- Larson, J.L.; Silver, A.J.; Chan, D.; Borroto, C.; Spurrier, B.; Silver, L.M. Validation of a high resolution NGS method for detecting spinal muscular atrophy carriers among phase 3 participants in the 1000 Genomes Project. BMC Med. Genet. 2015, 16, 100. [Google Scholar] [CrossRef]
- Chen, X.; Sanchis-Juan, A.; French, C.E.; Connell, A.J.; Delon, I.; Kingsbury, Z.; Chawla, A.; Halpern, A.L.; Taft, R.J.; Bentley, D.R. Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genet. Med. 2020, 22, 945–953. [Google Scholar] [CrossRef]
- Stabley, D.L.; Harris, A.W.; Holbrook, J.; Chubbs, N.J.; Lozo, K.W.; Crawford, T.O.; Swoboda, K.J.; Funanage, V.L.; Wang, W.; Mackenzie, W. SMN1 and SMN2 copy numbers in cell lines derived from patients with spinal muscular atrophy as measured by array digital PCR. Mol. Genet. Genom. Med. 2015, 3, 248–257. [Google Scholar] [CrossRef]
- Jiang, L.; Lin, R.; Gallagher, S.; Zayac, A.; Butchbach, M.E.R.; Hung, P. Development and validation of a 4-color multiplexing spinal muscular atrophy (SMA) genotyping assay on a novel integrated digital PCR instrument. Sci. Rep. 2020, 10, 19892. [Google Scholar] [CrossRef]
- Stabley, D.L.; Holbrook, J.; Scavina, M.; Crawford, T.O.; Swoboda, K.J.; Robbins, K.M.; Butchbach, M.E.R. Detection of SMN1 to SMN2 gene conversion events and partial SMN1 gene deletions using array digital PCR. Neurogenetics 2021, 22, 53–64. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Pujar, S.; O’Leary, N.A.; Farrell, C.M.; Loveland, J.E.; Mudge, J.M.; Wallin, C.; Girón, C.G.; Diekhans, M.; Barnes, I.; Bennett, R.; et al. Consensus coding sequence (CCDS) database: A standardized set of human and mouse protein-coding regions supported by expert curation. Nucleic Acids Res 2018, 46, D221–D228. [Google Scholar] [CrossRef]
- Hubley, R.; Finn, R.D.; Clements, J.; Eddy, S.R.; Jones, T.A.; Bao, W.; Smit, A.F.; Wheeler, T.J. The Dfam database of repetitive DNA families. Nucleic Acids Res. 2016, 44, D81–D89. [Google Scholar] [CrossRef]
- Storer, J.; Hubley, R.; Rosen, J.; Wheeler, T.J.; Smit, A.F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA 2021, 12, 2. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Germain-Desprez, D.; Brun, T.; Rochette, C.; Semionov, A.; Rouget, R.; Simard, L.R. The SMN genes are subject to transcriptional regulation during cellular differentiation. Gene 2001, 279, 109–117. [Google Scholar] [CrossRef]
- Monani, U.R.; McPherson, J.D.; Burghes, A.H. Promoter analysis of the human centromeric and telomeric survival motor neuron genes (SMNC and SMNT). Biochim. Biophys. Acta 1999, 1445, 330–336. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. TIG 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Singh, N.N.; Ottesen, E.W.; Singh, R.N. A survey of transcripts generated by spinal muscular atrophy genes. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194562. [Google Scholar] [CrossRef]
- Sorek, R.; Ast, G.; Graur, D. Alu-containing exons are alternatively spliced. Genome Res. 2002, 12, 1060–1067. [Google Scholar] [CrossRef]
- Piriyapongsa, J.; Rutledge, M.T.; Patel, S.; Borodovsky, M.; Jordan, I.K. Evaluating the protein coding potential of exonized transposable element sequences. Biol. Direct 2007, 2, 31. [Google Scholar] [CrossRef]
- Seo, J.; Singh, N.N.; Ottesen, E.W.; Lee, B.M.; Singh, R.N. A novel human-specific splice isoform alters the critical C-terminus of Survival Motor Neuron protein. Sci. Rep. 2016, 6, 30778. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Harahap, N.I.F.; Hamamura, Y.; Ar Rochmah, M.; Shima, A.; Morisada, N.; Shinohara, M.; Saito, T.; Saito, K.; Lai, P.S.; et al. Alternative splicing of a cryptic exon embedded in intron 6 of SMN1 and SMN2. Hum. Genome Var 2016, 3, 16040. [Google Scholar] [CrossRef]
- Ottesen, E.W.; Luo, D.; Seo, J.; Singh, N.N.; Singh, R.N. Human Survival Motor Neuron genes generate a vast repertoire of circular RNAs. Nucleic Acids Res. 2019, 47, 2884–2905. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef]
- Martinez-Gomez, L.; Abascal, F.; Jungreis, I.; Pozo, F.; Kellis, M.; Mudge, J.M.; Tress, M.L. Few SINEs of life: Alu elements have little evidence for biological relevance despite elevated translation. NAR Genom. Bioinform. 2020, 2, lqz023. [Google Scholar] [CrossRef]
- Jahic, A.; Erichsen, A.K.; Deufel, T.; Tallaksen, C.M.; Beetz, C. A polymorphic Alu insertion that mediates distinct disease-associated deletions. Eur. J. Hum. Genet. EJHG 2016, 24, 1371–1374. [Google Scholar] [CrossRef]
- Van de Lagemaat, L.N.; Landry, J.R.; Mager, D.L.; Medstrand, P. Transposable elements in mammals promote regulatory variation and diversification of genes with specialized functions. Trends Genet. TIG 2003, 19, 530–536. [Google Scholar] [CrossRef]
- Jordan, I.K.; Rogozin, I.B.; Glazko, G.V.; Koonin, E.V. Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet. TIG 2003, 19, 68–72. [Google Scholar] [CrossRef]
- Huh, J.W.; Kim, D.S.; Ha, H.S.; Lee, J.R.; Kim, Y.J.; Ahn, K.; Lee, S.R.; Chang, K.T.; Kim, H.S. Cooperative exonization of MaLR and AluJo elements contributed an alternative promoter and novel splice variants of RNF19. Gene 2008, 424, 63–70. [Google Scholar] [CrossRef]
- Thornburg, B.G.; Gotea, V.; Makałowski, W. Transposable elements as a significant source of transcription regulating signals. Gene 2006, 365, 104–110. [Google Scholar] [CrossRef]
- Wells, J.N.; Feschotte, C. A Field Guide to Eukaryotic Transposable Elements. Annu. Rev. Genet. 2020, 54, 539–561. [Google Scholar] [CrossRef]
- Faulkner, G.J.; Kimura, Y.; Daub, C.O.; Wani, S.; Plessy, C.; Irvine, K.M.; Schroder, K.; Cloonan, N.; Steptoe, A.L.; Lassmann, T.; et al. The regulated retrotransposon transcriptome of mammalian cells. Nat. Genet. 2009, 41, 563–571. [Google Scholar] [CrossRef]
- Branco, M.R.; Chuong, E.B. Crossroads between transposons and gene regulation. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2020, 375, 20190330. [Google Scholar] [CrossRef]
- Marasca, F.; Gasparotto, E.; Polimeni, B.; Vadalà, R.; Ranzani, V.; Bodega, B. The Sophisticated Transcriptional Response Governed by Transposable Elements in Human Health and Disease. Int. J. Mol. Sci. 2020, 21, 3201. [Google Scholar] [CrossRef]
- Hu, S.; Wang, X.; Shan, G. Insertion of an Alu element in a lncRNA leads to primate-specific modulation of alternative splicing. Nat. Struct. Mol. Biol. 2016, 23, 1011–1019. [Google Scholar] [CrossRef]
- Lee, J.Y.; Ji, Z.; Tian, B. Phylogenetic analysis of mRNA polyadenylation sites reveals a role of transposable elements in evolution of the 3′-end of genes. Nucleic Acids Res. 2008, 36, 5581–5590. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.; Oakey, R.J. Transposable elements re-wire and fine-tune the transcriptome. PLoS Genet. 2013, 9, e1003234. [Google Scholar] [CrossRef] [PubMed]
- Roy-Engel, A.M.; El-Sawy, M.; Farooq, L.; Odom, G.L.; Perepelitsa-Belancio, V.; Bruch, H.; Oyeniran, O.O.; Deininger, P.L. Human retroelements may introduce intragenic polyadenylation signals. Cytogenet. Genome Res. 2005, 110, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Sela, N.; Kim, E.; Ast, G. The role of transposable elements in the evolution of non-mammalian vertebrates and invertebrates. Genome Biol. 2010, 11, R59. [Google Scholar] [CrossRef]
- Chen, L.-L.; Yang, L. ALUternative regulation for gene expression. Trends Cell Biol. 2017, 27, 480–490. [Google Scholar] [CrossRef]
- Deininger, P.L.; Moran, J.V.; Batzer, M.A.; Kazazian, H.H., Jr. Mobile elements and mammalian genome evolution. Curr. Opin. Genet. Dev. 2003, 13, 651–658. [Google Scholar] [CrossRef]
- Medstrand, P.; van de Lagemaat, L.N.; Mager, D.L. Retroelement distributions in the human genome: Variations associated with age and proximity to genes. Genome Res. 2002, 12, 1483–1495. [Google Scholar] [CrossRef]
- Kabelitz, T.; Bäurle, I. Get the jump—Do 3′UTRs protect transposable elements from silencing? Mob. Genet. Elem. 2015, 5, 51–54. [Google Scholar] [CrossRef][Green Version]
- Lavi, E.; Carmel, L. Alu exaptation enriches the human transcriptome by introducing new gene ends. RNA Biol. 2018, 15, 715–725. [Google Scholar] [CrossRef]
- Jjingo, D.; Huda, A.; Gundapuneni, M.; Mariño-Ramírez, L.; Jordan, I.K. Effect of the transposable element environment of human genes on gene length and expression. Genome Biol. Evol. 2011, 3, 259–271. [Google Scholar] [CrossRef]
- Hughes, T.A. Regulation of gene expression by alternative untranslated regions. Trends Genet. TIG 2006, 22, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Sela, N.; Mersch, B.; Gal-Mark, N.; Lev-Maor, G.; Hotz-Wagenblatt, A.; Ast, G. Comparative analysis of transposed element insertion within human and mouse genomes reveals Alu’s unique role in shaping the human transcriptome. Genome Biol. 2007, 8, R127. [Google Scholar] [CrossRef] [PubMed]
- Petri, R.; Brattås, P.L.; Sharma, Y.; Jönsson, M.E.; Pircs, K.; Bengzon, J.; Jakobsson, J. LINE-2 transposable elements are a source of functional human microRNAs and target sites. PLoS Genet. 2019, 15, e1008036. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Greene, J.; Baird, A.M.; Brady, L.; Lim, M.; Gray, S.G.; McDermott, R.; Finn, S.P. Circular RNAs: Biogenesis, Function and Role in Human Diseases. Front. Mol. Biosci. 2017, 4, 38. [Google Scholar] [CrossRef]
- Haque, S.; Harries, L.W. Circular RNAs (circRNAs) in Health and Disease. Genes 2017, 8, 353. [Google Scholar] [CrossRef]
- Chen, Y.J.; Chen, C.Y.; Mai, T.L.; Chuang, C.F.; Chen, Y.C.; Gupta, S.K.; Yen, L.; Wang, Y.D.; Chuang, T.J. Genome-wide, integrative analysis of circular RNA dysregulation and the corresponding circular RNA-microRNA-mRNA regulatory axes in autism. Genome Res. 2020, 30, 375–391. [Google Scholar] [CrossRef]
- Voellenkle, C.; Perfetti, A.; Carrara, M.; Fuschi, P.; Renna, L.V.; Longo, M.; Sain, S.B.; Cardani, R.; Valaperta, R.; Silvestri, G.; et al. Dysregulation of Circular RNAs in Myotonic Dystrophy Type 1. Int. J. Mol. Sci 2019, 20, 1938. [Google Scholar] [CrossRef]
- Florea, L.; Payer, L.; Antonescu, C.; Yang, G.; Burns, K. Detection of Alu Exonization Events in Human Frontal Cortex From RNA-Seq Data. Front. Mol. Biosci. 2021, 8, 727537. [Google Scholar] [CrossRef]
- Sen, S.K.; Han, K.; Wang, J.; Lee, J.; Wang, H.; Callinan, P.A.; Dyer, M.; Cordaux, R.; Liang, P.; Batzer, M.A. Human genomic deletions mediated by recombination between Alu elements. Am. J. Hum. Genet. 2006, 79, 41–53. [Google Scholar] [CrossRef]
- Kim, S.; Cho, C.-S.; Han, K.; Lee, J. Structural Variation of Alu Element and Human Disease. Genom. Inform. 2016, 14, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Kryatova, M.S.; Steranka, J.P.; Burns, K.H.; Payer, L.M. Insertion and deletion polymorphisms of the ancient AluS family in the human genome. Mob. DNA 2017, 8, 6. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Carrier Probability | Carrier Status |
|---|---|---|
| HG02134 | 1 | Carrier |
| NA12383 | 1 | Carrier |
| HG01773 | 1 | Carrier |
| HG00346 | 1 | Carrier |
| HG00281 | 1 | Carrier |
| HG02087 | 1 | Carrier |
| HG01085 | 1 | Carrier |
| HG01893 | 1 | Carrier |
| HG00324 | 0.997 | Carrier |
| NA20764 | 0.982 | Carrier |
| HG02265 | 0.982 | Carrier |
| HG02079 | 0.976 | Carrier |
| HG03953 | 0.972 | Carrier |
| HG01248 | 0.935 | Carrier |
| HG01492 | 0.914 | Carrier |
| HG01892 | 0.902 | Carrier |
| HG00525 | 0.763 | Carrier |
| HG01205 | 0.756 | Carrier |
| HG01094 | 0.738 | Carrier |
| NA11932 | 0.716 | Carrier |
| HG00629 | 0.000165 | Non-Carrier |
| HG03585 | 0.000159 | Non-Carrier |
| HG01341 | 0.000155 | Non-Carrier |
| HG00325 | 0.000151 | Non-Carrier |
| HG00369 | 0.000134 | Non-Carrier |
| NA20878 | 0.000131 | Non-Carrier |
| HG04023 | 0.000127 | Non-Carrier |
| HG02136 | 0.000126 | Non-Carrier |
| NA19732 | 0.000126 | Non-Carrier |
| HG00684 | 0.000126 | Non-Carrier |
| NA18629 | 0.000126 | Non-Carrier |
| NA20864 | 0.000122 | Non-Carrier |
| HG03196 | 0.000112 | Non-Carrier |
| HG00372 | 0.000111 | Non-Carrier |
| NA12889 | 0.0000997 | Non-Carrier |
| NA19725 | 0.0000982 | Non-Carrier |
| HG03720 | 0.0000969 | Non-Carrier |
| HG00742 | 0.0000923 | Non-Carrier |
| NA20342 | 0.0000907 | Non-Carrier |
| HG03968 | 0.0000851 | Non-Carrier |
| Copy Number (Exon 7) | Copy Number (Exon 8) | ||||
|---|---|---|---|---|---|
| Sample ID | Phenotype | SMN1 | SMN2 | SMN1 | SMN2 |
| MB109 | healthy | 3 | 1 | 2 | 1 |
| MB342 | healthy | 2 | 0 | 2 | 0 |
| MB106 | SMA II | 0 | 3 | 0 | 3 |
| MB110 | SMA I | 0 | 3 | 0 | 3 |
| MB112 | SMA II | 0 | 3 | 0 | 3 |
| MB114 | SMA I | 0 | 2 | 0 | 2 |
| MB125 | SMA III | 0 | 3 | 0 | 3 |
| MB219 | SMA I | 0 | 2 | 0 | 2 |
| MB222 | SMA II | 0 | 3 | 0 | 3 |
| MB230 | SMA III | 0 | 3 | 0 | 3 |
| MB231 | SMA III | 0 | 4 | 0 | 4 |
| MB232 | SMA II | 0 | 3 | 0 | 3 |
| MB233 | SMA I | 0 | 3 | 0 | 3 |
| MB234 | SMA I | 0 | 2 | 0 | 2 |
| MB352 | SMA II | 0 | 3 | 0 | 3 |
| MB354 | SMA II | 0 | 3 | 0 | 3 |
| MB355 | SMA II | 0 | 3 | 0 | 3 |
| MB356 | SMA III | 0 | 4 | 0 | 4 |
| MB357 | SMA II | 0 | 3 | 0 | 3 |
| MB358 | SMA II | 0 | 3 | 0 | 3 |
| MB361 | SMA II | 0 | 3 | 0 | 3 |
| MB362 | SMA I | 0 | 2 | 0 | 2 |
| MB364 | SMA I | 0 | 2 | 0 | 2 |
| MB375 | SMA II | 0 | 3 | 0 | 3 |
| MB377 | SMA III | 0 | 4 | 0 | 4 |
| MB378 | SMA II | 0 | 3 | 0 | 3 |
| MB388 | SMA III | 0 | 3 | 0 | 3 |
| MB488 | SMA I | 0 | 2 | 0 | 2 |
| MB489 | SMA I | 0 | 2 | 0 | 2 |
| MB501 | SMA I | 0 | 2 | 0 | 2 |
| MB503 | SMA III | 0 | 4 | 0 | 4 |
| MB507 | SMA I | 0 | 2 | 0 | 2 |
| MB509 | SMA I | 0 | 2 | 0 | 2 |
| MB510 | SMA I | 0 | 2 | 0 | 2 |
| MB511 | SMA II | 0 | 3 | 0 | 3 |
| MB513 | SMA III | 0 | 3 | 0 | 3 |
| MB691 | SMA I | 0 | 2 | 0 | 2 |
| MB692 | SMA I | 0 | 2 | 0 | 2 |
| MB693 | SMA I | 0 | 2 | 0 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, A.; Cunha, C.; Chaves, R.; Butchbach, M.E.R.; Adega, F. Comprehensive In Silico Analysis of Retrotransposon Insertions within the Survival Motor Neuron Genes Involved in Spinal Muscular Atrophy. Biology 2022, 11, 824. https://doi.org/10.3390/biology11060824
Pinto A, Cunha C, Chaves R, Butchbach MER, Adega F. Comprehensive In Silico Analysis of Retrotransposon Insertions within the Survival Motor Neuron Genes Involved in Spinal Muscular Atrophy. Biology. 2022; 11(6):824. https://doi.org/10.3390/biology11060824
Chicago/Turabian StylePinto, Albano, Catarina Cunha, Raquel Chaves, Matthew E. R. Butchbach, and Filomena Adega. 2022. "Comprehensive In Silico Analysis of Retrotransposon Insertions within the Survival Motor Neuron Genes Involved in Spinal Muscular Atrophy" Biology 11, no. 6: 824. https://doi.org/10.3390/biology11060824
APA StylePinto, A., Cunha, C., Chaves, R., Butchbach, M. E. R., & Adega, F. (2022). Comprehensive In Silico Analysis of Retrotransposon Insertions within the Survival Motor Neuron Genes Involved in Spinal Muscular Atrophy. Biology, 11(6), 824. https://doi.org/10.3390/biology11060824