
Integrated Expression Analysis of Small RNA, Degradome and Microarray Reveals Complex Regulatory Action of miRNA during Prolonged Shade in Swarnaprabha Rice

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Growth Condition and Sample Preparation
2.2. Small RNA Library Preparation and Sequencing Analysis
2.3. Phylogenetic Analysis of MIR Families
2.4. Degradome Sequencing, Target Identification and Analysis
2.5. Transcript Expression Analysis of miRNA and Target
3. Results
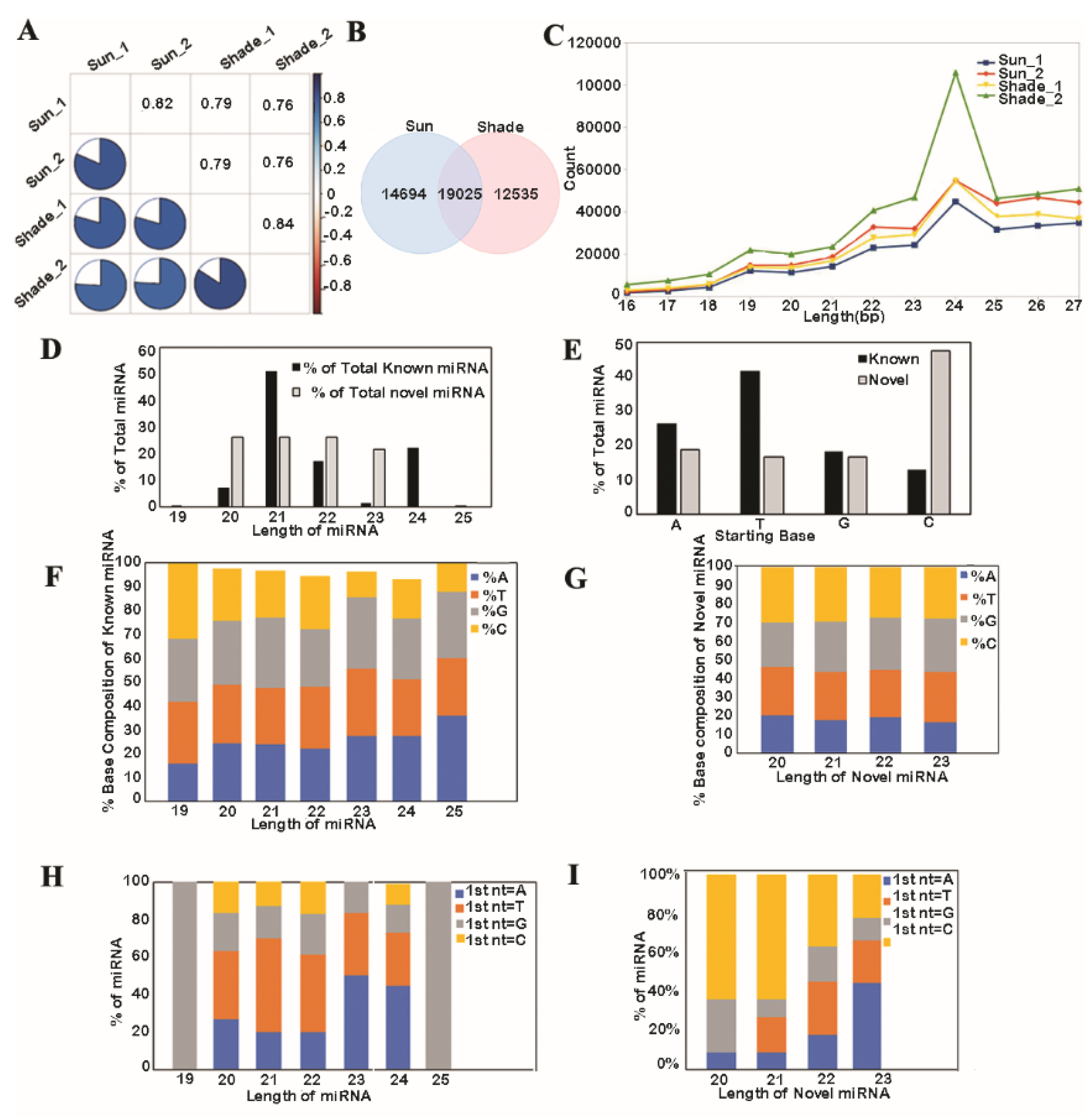
3.1. Analysis of Known and Novel miRNAs
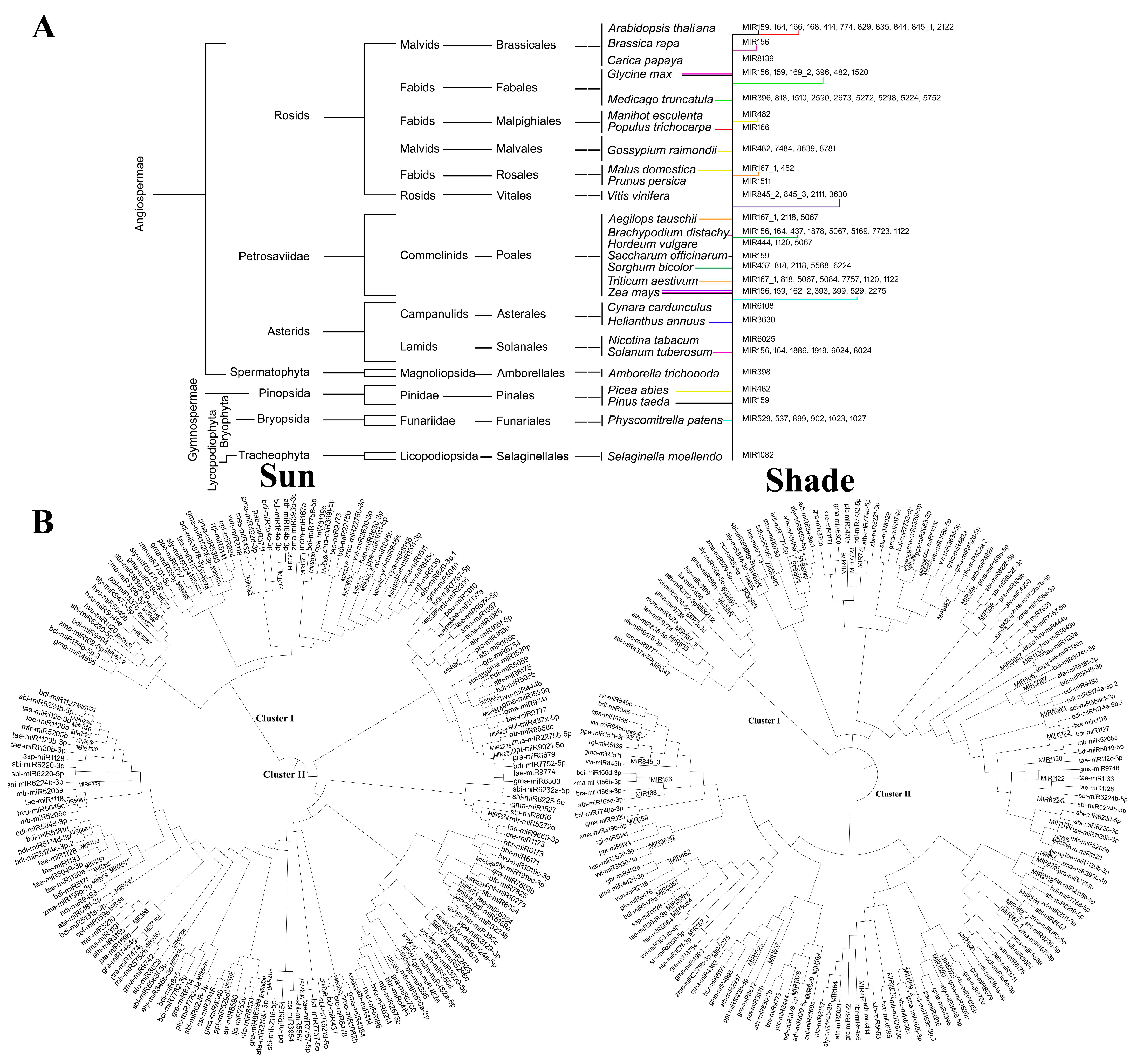
3.2. Similarity Analysis of Rice MIR Families with Other Species
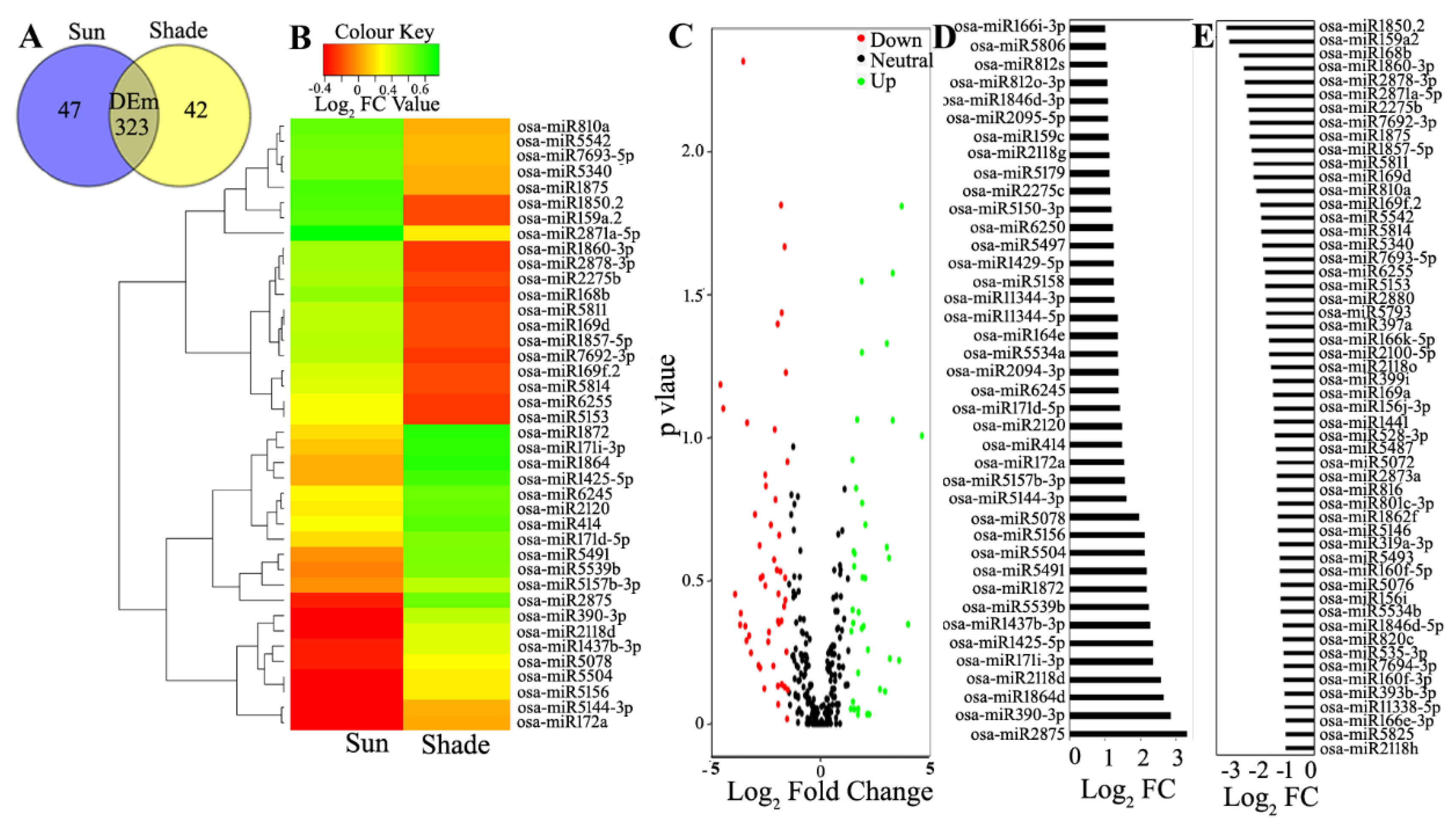
3.3. Differential Expression Analysis of the Known and Novel miRNA
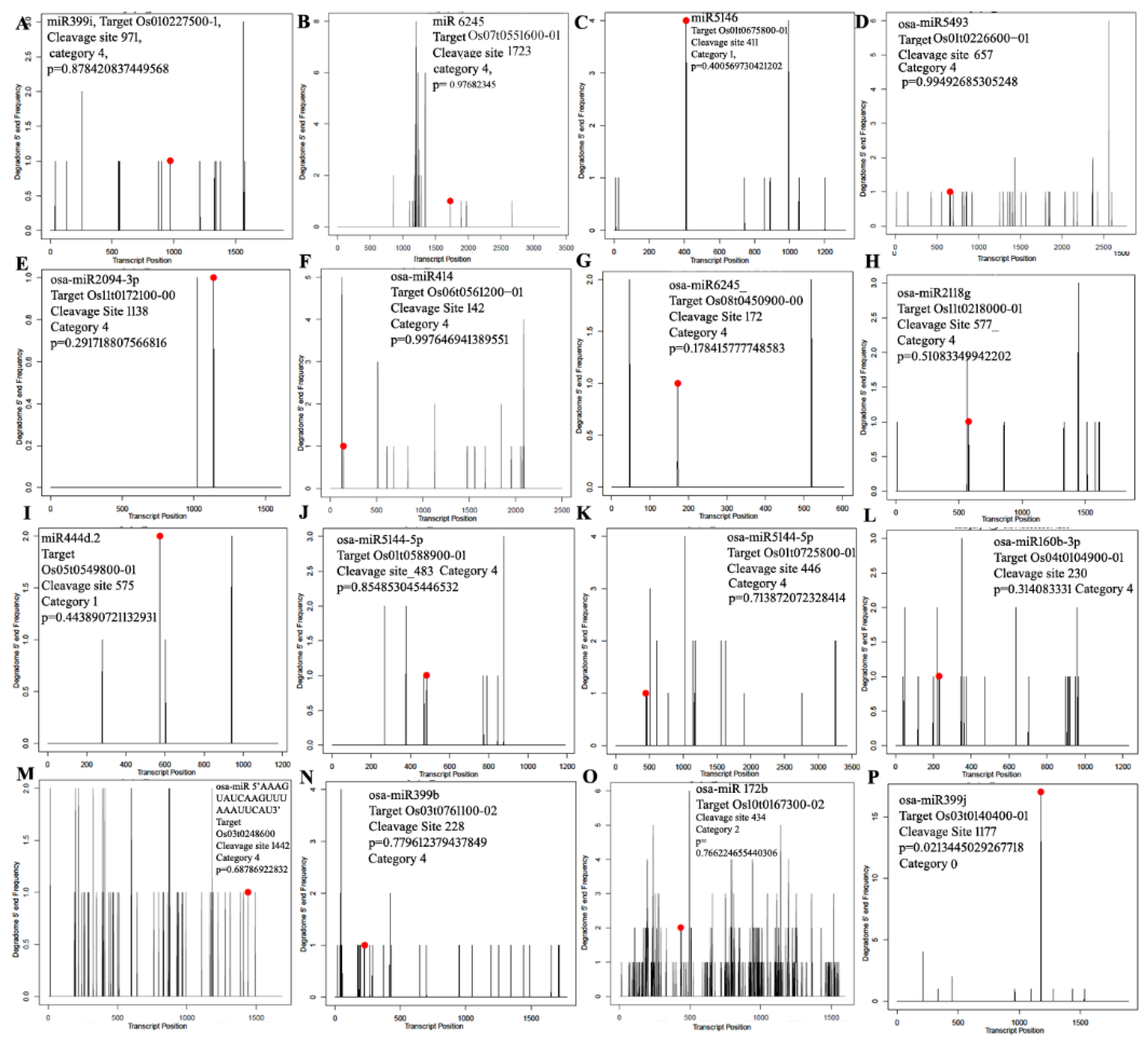
3.4. miRNA Target Identification Using Degradome Sequencing
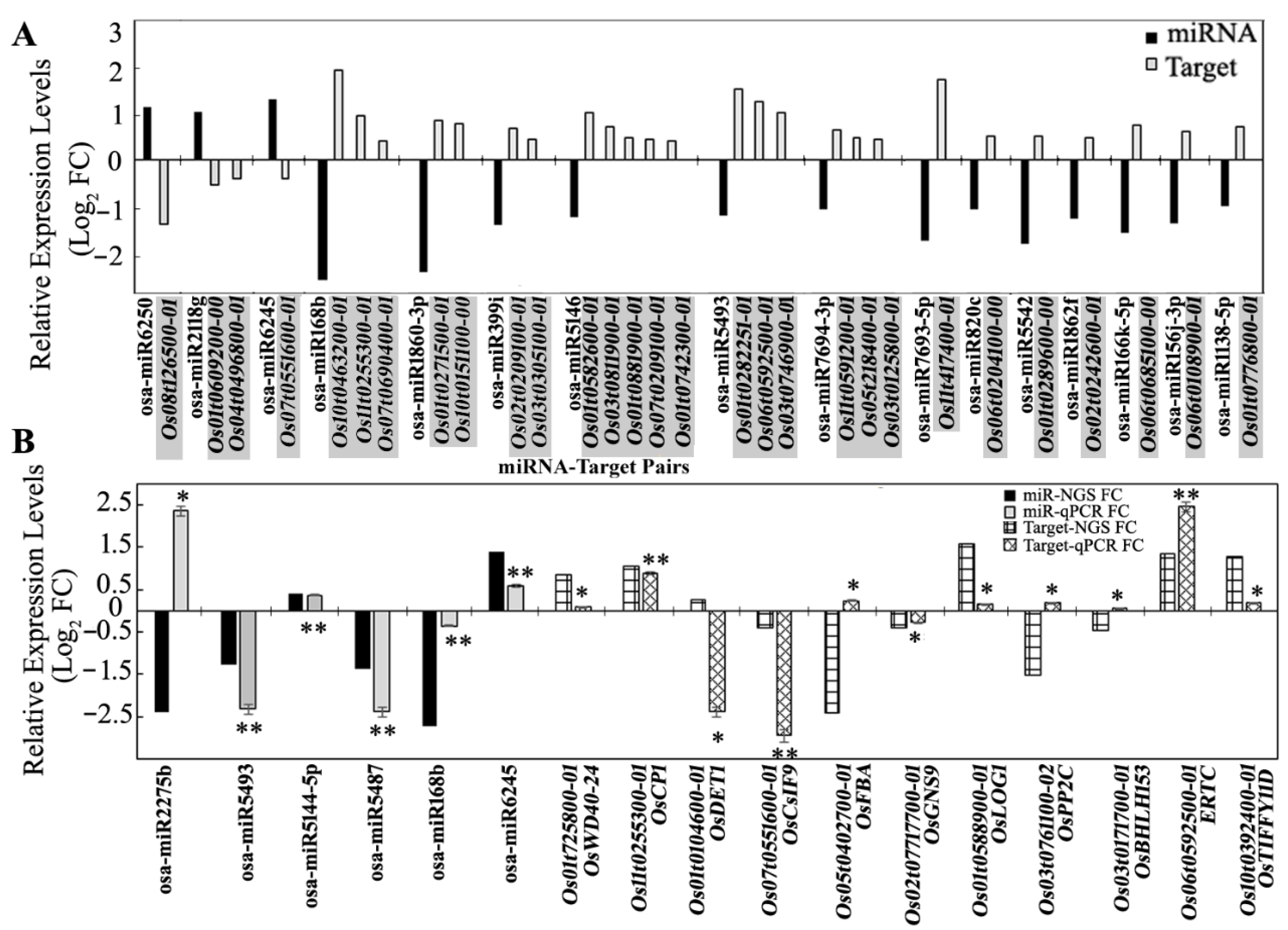
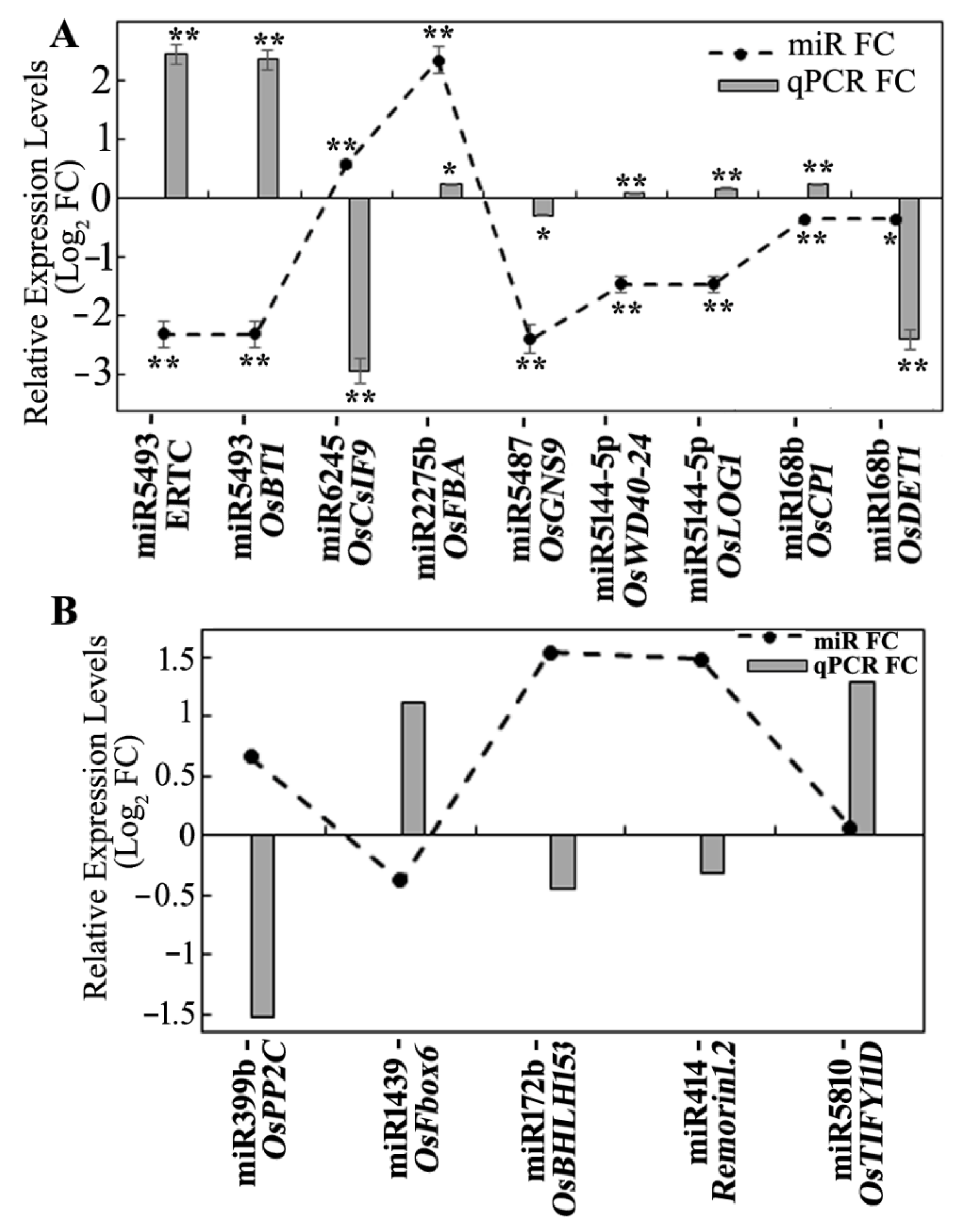
3.5. Expression Analysis of miRNA Targets Using Microarray and qRT-PCR
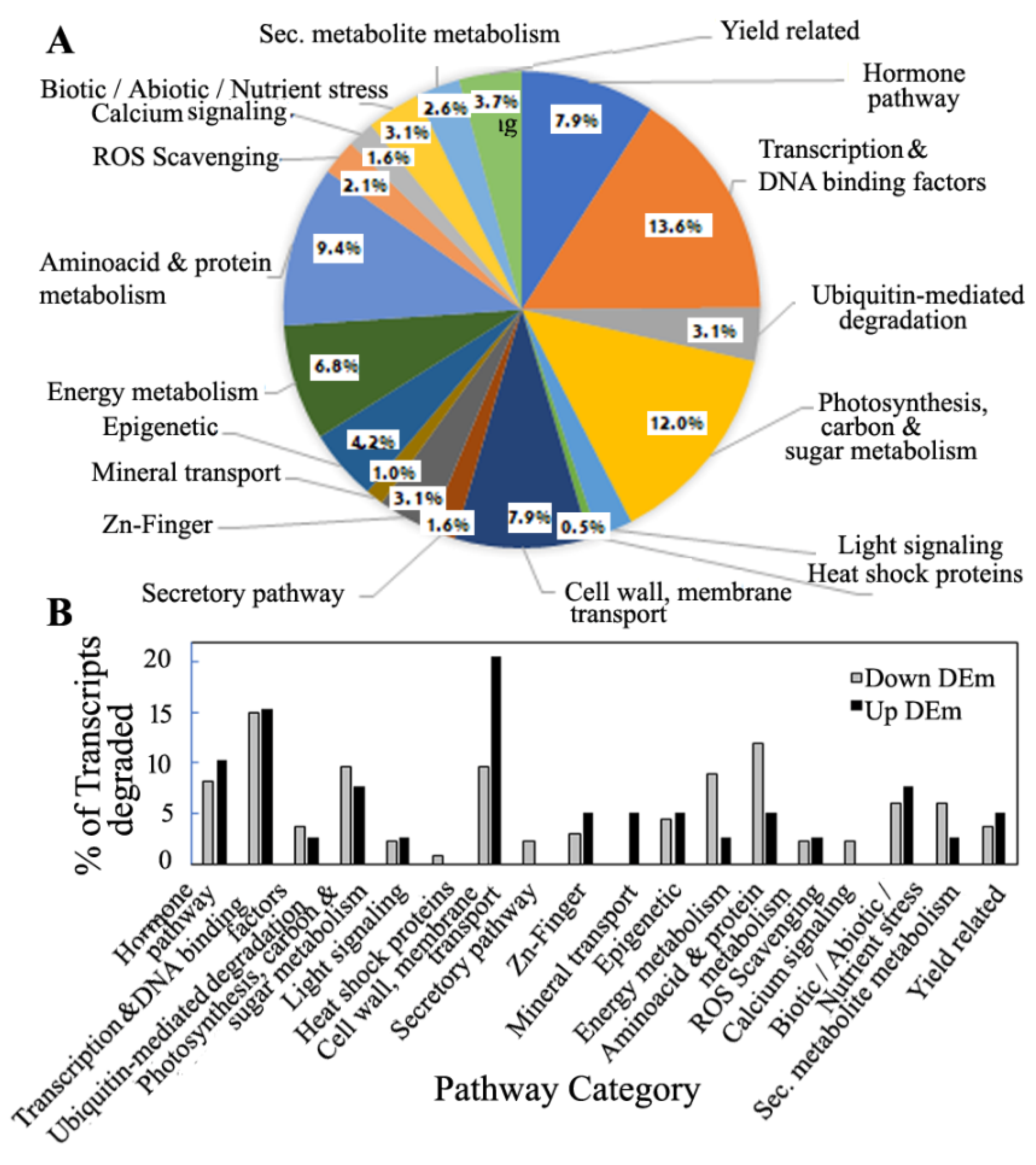
3.6. Pathway Distribution Analysis of Predicted Targets of miRNA and the Degradome Targets
3.7. miRNA Regulation According to Functional Properties of the Targets
3.7.1. miRNA Regulation of Transcription Factors
3.7.2. Regulation by Uniquely Expressed and Neutrally Regulated miRNAs
3.7.3. miRNA Regulation of Transcripts of Cell Wall, Membrane Dynamics or Hormone Signaling
3.7.4. miR Regulation of Photosynthesis and Carbon Metabolism Transcripts
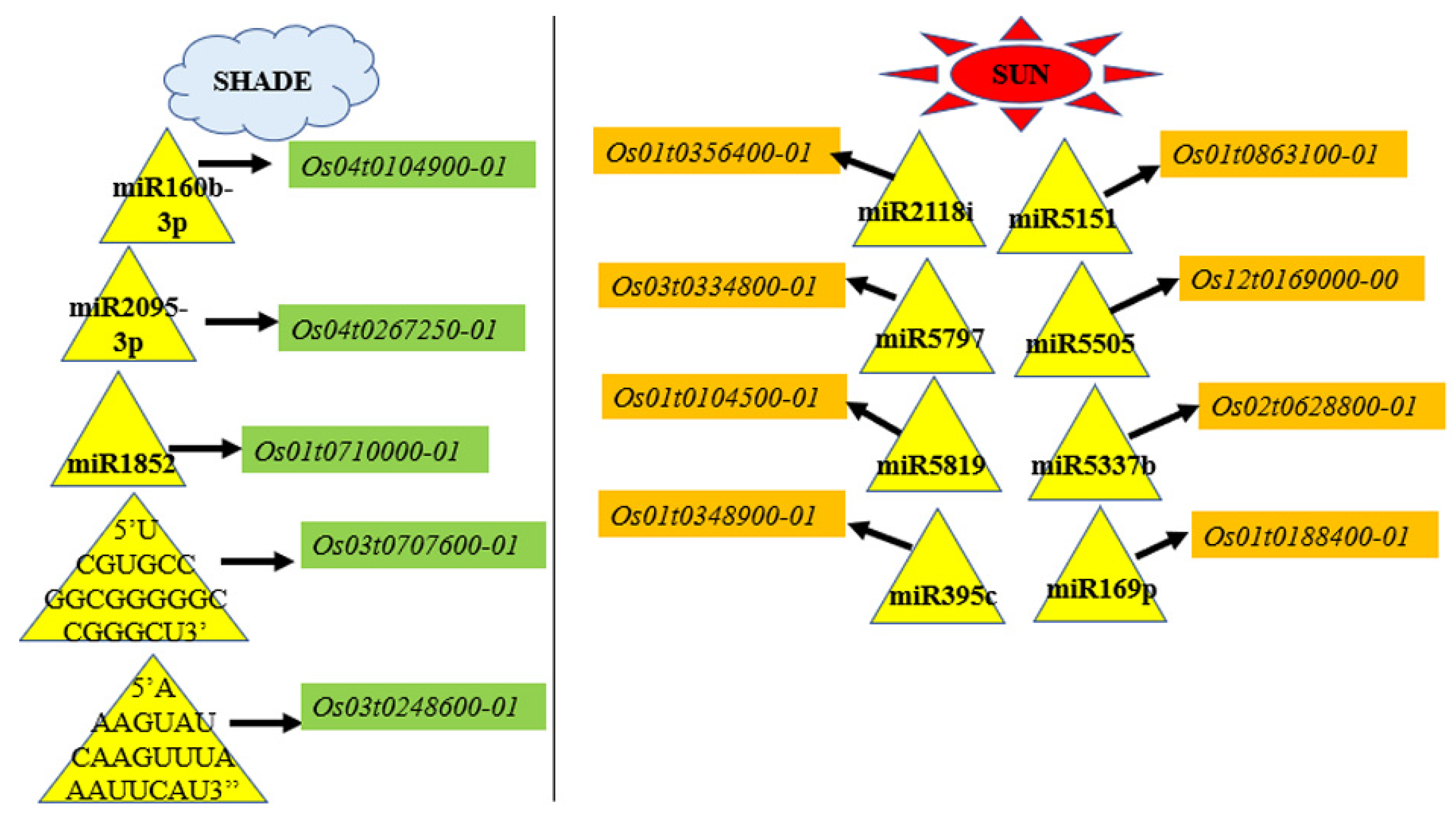
3.7.5. miRNA Regulation of Transcripts of Abiotic Stress, Light Signaling, and Shade Tolerance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Jiao, D.; Li, X. Cultivar differences in photosynthetic tolerance to photooxidation and shading in Rice (Oryza sativa L.). Photosynthetica 2001, 39, 167–175. [Google Scholar]
- Ganguly, S.; Saha, S.; Vangaru, S.; Purkayastha, S.; Das, D.; Saha, A.K.; Roy, A.; Das, S.; Bhattacharyya, P.K.; Mukherjee, S.; et al. Identification and analysis of low light tolerant rice genotypes in field condition and their SSP-based diversity in various abiotic stress tolerant line. J. Genet. 2020, 99, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Narasingarao, C.; Murty, K.S. Swarnaprabha, a physiologically efficient variety [Kerala]. Int. Rice Res. Newsl. Philipp. 1987, 12, 7–8. [Google Scholar]
- Singh, S. Effect of low-light stress at various growth phases on yield and yield components of two rice cultivars. Int. Rice Res. Notes 2005, 30, 36–37. [Google Scholar]
- Adhya, T.K.; Singh, O.N.; Ghosh, A. Rice in Eastern India: Causes for low productivity and available options. J. Rice Res. 2008, 2, 1–5. [Google Scholar]
- Liu, Q.; Wu, X.; Chen, B.; Ma, J.; Gao, J. Effect of low light on agronomic and physiological characteristics of rice including grain yield and quality. Rice Sci. 2014, 21, 243–251. [Google Scholar] [CrossRef]
- Izumi, M.; Hidema, J.; Wada, S.; Kondo, E.; Kurusu, T.; Kuchitsu, K.; Makino, A.; Ishida, H. Establishment of monitoring methods for autophagy in rice reveals autophagic recycling of chloroplasts and root plastids during energy limitation. Plant Physiol. 2015, 167, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, M.; Ranga, A.; Das, J.; Panigrahi, K.C.S. Shade tolerance in Swarnaprabha rice is associated with higher rate of panicle emergence and positively regulated by genes of ethylene and cytokinin pathway. Sci. Rep. 2019, 9, 6817–6834. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Gautam, V.; Singh, S.; Das, S.S.; Verma, S.; Mishra, V.; Mukherjee, S.; Sarkar, A.K. Plant small RNAs: Advancement in the understanding of biogenesis and role in plant development. Planta 2018, 248, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Waheed, S.; Zeng, L. The critical role of miRNAs in regulation of flowering time and flower development. Genes 2020, 11, 319. [Google Scholar] [CrossRef] [PubMed]
- Spanudakis, E.; Jackson, S. The role of microRNAs in the control of flowering time. J. Exp. Bot. 2014, 65, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Teotia, S.; Tang, G. To Bloom or Not to Bloom: Role of MicroRNAs in Plant Flowering. Mol. Plant 2015, 8, 359–377. [Google Scholar] [CrossRef]
- Nag, A.; Jack, T. Sculpting the Flower; the Role of microRNAs in Flower Development. Curr. Top. Dev. Biol. 2010, 91, 349–378. [Google Scholar] [PubMed]
- Chen, D.; Yan, W.; Fu, L.-Y.; Kaufmann, K. Architecture of gene regulatory networks controlling flower development in Arabidopsis thaliana. Nat. Commun. 2018, 9, 4534. [Google Scholar] [CrossRef]
- Sun, W.; Xu, X.H.; Wu, X.; Wang, Y.; Lu, X.; Sun, H.; Xie, X. Genome-wide identification of microRNAs and their targets in wild type and phyB mutant provides a key link between microRNAs and the PhyB-mediated light signaling pathway in rice. Front. Plant Sci. 2015, 6, 372. [Google Scholar] [CrossRef]
- Choi, S.W.; Ryu, M.Y.; Viczián, A.; Jung, H.J.; Kim, G.M.; Arce, A.L.; Achkar, N.P.; Manavella, P.; Dolde, U.; Wenkel, S.; et al. Light triggers the miRNA-biogenetic inconsistency for de-etiolated seedling survivability in Arabidopsis thaliana. Mol. Plant 2020, 13, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Retuerta, C.; Suárez-López, P.; Henriques, R. Under a New Light: Regulation of Light-Dependent Pathways by Non-coding RNAs. Front. Plant Sci. 2018, 9, 962. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Li, Y.-F.; Jagadeeswaran, G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012, 17, 196–203. [Google Scholar] [CrossRef]
- Varsha, S.; Vinay, K.; Rachayya, D.M.; Khare, T.S.; Wani, S.H. MicroRNAs as potential targets for abiotic stress tolerance in plants. Front. Plant Sci. 2016, 7, 817. [Google Scholar]
- Shukla, L.; Chinnusamy, V.; Sunkar, R. The role of microRNAs and other endogenous small RNAs in plant stress responses. Biochim. Biophys. Acta 2008, 1779, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Kantar, M.; Lucas, S.J.; Budak, H. miRNA expression patterns of Triticum dicoccoides in response to shock drought stress. Planta 2011, 233, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; Sharma, N.L.; Sharma, V.D.; Sopory, S.K.; Sananmishra, N. Role of microRNAs in rice plant under salt stress. Ann. Appl. Biol. 2016, 168, 2–18. [Google Scholar] [CrossRef]
- Lv, D.-K.; Bai, X.; Li, Y.; Ding, X.-D.; Ge, Y.; Cai, H.; Ji, W.; Wu, N.; Zhu, Y.-M. Profiling of cold-stress-responsive miRNAs in rice by microarrays. Gene 2010, 459, 39–47. [Google Scholar] [CrossRef]
- Zeng, X.; Xu, Y.; Jiang, J.; Zhang, F.; Ma, L.; Wu, D.; Wang, Y.; Sun, W. Identification of cold stress responsive microRNAs in two winter turnip rape (Brassica rapa L.) by high throughput sequencing. BMC Plant Biol. 2018, 18, 52. [Google Scholar] [CrossRef]
- Li, Z.; Tian, P.; Huang, T.; Huang, J. Noncoding-RNA-Mediated Regulation in Response to Macronutrient Stress in Plants. Int. J. Mol. Sci. 2021, 22, 11205. [Google Scholar] [CrossRef]
- Kuo, H.-F.; Chiou, T.-J. The Role of MicroRNAs in Phosphorus Deficiency Signaling. Plant Physiol. 2011, 156, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, M.; Rao, D.N.; Sarla, N. Molecular mechanisms in response to phosphate starvation in rice. Biotechnol. Adv. 2009, 27, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ye, J.; Tang, W.; Liu, Z.; Zhu, C.; Wang, M.; Wan, J. Loop nucleotide polymorphism in a putative miRNA precursor associated with seed length in rice (Oryza sativa L.). Int. J. Biol. Sci. 2013, 9, 578. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Yang, J.; Mathioni, S.M.; Yu, J.; Shen, J.; Yang, X.; Wang, L.; Zhang, Q.; Cai, Z.; Xu, C.; et al. PMS1T, producing phased small-interfering RNAs, regulates photoperiod-sensitive male sterility in rice. Proc. Natl. Acad. Sci. USA 2016, 113, 15144–15149. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, S.; Panigrahy, M.; Kariali, E.; Dash, S.K.; Sahu, B.B.; Sahu, S.K.; Mohapatra, P.K.; Panigrahi, K.C.S. MicroRNAs modulate ethylene induced retrograde signal for rice endosperm starch biosynthesis by default expression of transcriptome. Sci. Rep. 2021, 11, 5573–5590. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.W.; Weigel, D.; Poethig, R.S. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and Their Regulatory Roles in Plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Liu, Y.; Wang, H.; Ma, X.; Wang, B.; Wu, G.; Wang, H. Phytochrome-interacting factors directly suppress miR156 expression to enhance shade-avoidance syndrome in Arabidopsis. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Adam, H.; Díaz-Mendoza, M.; Zurczak, M.; González-Schain, N.D.; Suárez-López, P. Graft-transmissible induction of potato tuberization by the microRNA miR172. Development 2009, 136, 2873–2881. [Google Scholar] [CrossRef]
- Achkar, N.P.; Cho, S.K.; Poulsen, C.; Arce, A.L.; Ré, D.A.; Giudicatti, A.J.; Karayekov, E.; Ryu, M.Y.; Choi, S.; Harholt, J.; et al. A Quick HYL1-Dependent Reactivation of MicroRNA Production Is Required for a Proper Developmental Response after Extended Periods of Light Deprivation. Dev. Cell 2018, 46, 236–247. [Google Scholar] [PubMed]
- Panigrahy, M.; Majeed, N.; Panigrahi, K.C.S. Low-light and its effects on crop yield: Genetic and genomic implications. J. Biosci. 2020, 45, 102–117. [Google Scholar] [CrossRef]
- Liu, Y.; Pan, T.; Tang, Y.; Zhuang, Y.; Liu, Z.; Li, P.; Li, H.; Huang, W.; Tu, S.; Ren, G.; et al. Proteomic Analysis of Rice Subjected to Low Light Stress and Overexpression of OsGAPB Increases the Stress Tolerance. Rice 2020, 13, 30. [Google Scholar] [CrossRef]
- Gad, A.G.; Habiba; Zheng, X.; Miao, Y. Low Light/Darkness as Stressors of Multifactor-Induced Senescence in Rice Plants. Int. J. Mol. Sci. 2021, 22, 3936. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Hu, J.; Zeng, T.; Xia, Q.; Qian, Q.; Yang, C.; Ding, Y.; Chen, L.; Wang, W. Unravelling miRNA regulation in yield of rice (Oryza sativa) based on differential network model. Sci. Rep. 2018, 8, 8498. [Google Scholar]
- Yu, D.; Xu, M.; Ito, H.; Shao, W.; Ma, X.; Wang, H.; Meng, Y. Tracking microRNA Processing Signals by Degradome Sequencing Data Analysis. Front. Genet. 2018, 9, 1664. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.; Singh, A.; Gandhi, N.; Sarkar Das, S.; Yadav, S.; Kumar, A.; Sarkar, A. MicroRNA775 and target Galactosyltransferase (GALT9) module regulates recovery from submergence induced senescence by modulating SAGs in Arabidopsis thaliana. BioRxiv 2021, 428849. [Google Scholar] [CrossRef]
- Jin, J.; Xu, Y.; Lu, P.; Chen, Q.; Liu, P.; Wang, J.; Jhang, J.; Li, Z.; Yang, A.; Li, F.; et al. Degradome, small RNAs and transcriptome sequencing of a high-nicotine cultivated tobacco uncovers miRNA’s function in nicotine biosynthesis. Sci. Rep. 2020, 10, 11751. [Google Scholar] [CrossRef]
- Xu, X.; Yin, L.; Ying, Q.; Song, H.; Xue, D.; Lai, T.; Xu, M.; Shen, B.; Wang, H.; Shi, X. High-Throughput Sequencing and Degradome Analysis Identify miRNAs and Their Targets Involved in Fruit Senescence of Fragaria ananassa. PLoS ONE 2013, 8, e70959. [Google Scholar]
- Stocks, M.B.; Moxon, S.; Mapleson, D.; Woolfenden, H.C.; Mohorianu, I.; Folkes, L.; Schwach, F.; Dalmay, T.; Moulton, V. The UEA sRNA workbench: A suite of tools for analysing and visualizing next generation sequencing microRNA and small RNA datasets. Bioinformatics 2012, 28, 2059–2061. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Bateman, A.; Marshall, M.; Khanna, A.; Eddy, S.R. Rfam: An RNA family database. Nucleic Acids Res. 2003, 31, 439–441. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Qibin, L.; Jiang, W. MIREAP: MicroRna Discovery by Deep Sequencing. 2008. Available online: http://sourceforge.net/project/mireap/ (accessed on 1 January 2022).
- Zuker, M.; Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981, 9, 133–148. [Google Scholar] [CrossRef]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4. 0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef] [PubMed]
- Addo Quaye, C.; Miller, W.; Ax Tell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Yamasaki, K.; Kigawa, T.; Seki, M.; Shinozaki, K.; Yokoyama, S. DNA-binding domains of plant-specific transcription factors: Structure, function, and evolution. Trends Plant Sci. 2013, 18, 267–276. [Google Scholar] [CrossRef]
- Mann, M.; Wright, P.R.; Backofen, R. IntaRNA 2.0: Enhanced and customizable prediction of RNA–RNA interactions. Nucleic Acids Res. 2017, 45, W435–W439. [Google Scholar] [CrossRef]
- Zhang, X.; Li, J.; Liu, A.; Zou, J.; Zhou, X.; Xiang, J.; Rerksiri, W.; Peng, Y.; Xiong, X.; Chen, X. Expression profile in rice panicle: Insights into heat response mechanism at reproductive stage. PLoS ONE 2012, 7, e49652. [Google Scholar] [CrossRef]
- Fukayama, H.; Masumoto, C.; Taniguchi, Y.; Baba-Kasai, A.; Katoh, Y.; Ohkawa, H.; Miyao, M. Characterization and expression analyses of two plastidic enolase genes in rice. Biosci. Biotech. Bioch. 2015, 79, 402–409. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xue, T.; Wang, D.; Zhang, S.; Ehlting, J.; Ni, F.; Jakab, S.; Zheng, C.; Zhong, Y. Genome-wide and expression analysis of protein phosphatase 2C in rice and Arabidopsis. BMC Genom. 2008, 9, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Du, H.; Tang, N.; Li, X.; Xiong, L. Identification and expression profiling analysis of TIFY family genes involved in stress and phytohormone responses in rice. Pl. Mol. Biol. 2009, 1, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Yamburenko, M.; Kieber, J.J.; Schaller, G.E. Dynamic patterns of expression for genes regulating cytokinin metabolism and signaling during rice inflorescence development. PLoS ONE 2017, 12, e0176060. [Google Scholar]
- Yang, J.; Li, C.; Kong, D.; Guo, F.; Wei, H. Light-Mediated Signaling and Metabolic Changes Coordinate Stomatal Opening and Closure. Front. Plant Sci. 2020, 11, 1915. [Google Scholar] [CrossRef]
- Lee, H.-J.; Zheng, J.J. PDZ domains and their binding partners: Structure, specificity, and modification. Cell Commun. Signal. 2010, 8, 8. [Google Scholar] [CrossRef]
- Hu, X.Q.; Zhang, Y.Y.; Li, X.; Yan, H.F. Research Progress of Remorin Protein in Plants. Biotechnol. Bull. 2020, 36, 136. [Google Scholar]
- Sarkar, N.K.; Thapar, U.; Kundnani, P.; Panwar, P.; Grover, A. Functional relevance of J-protein family of rice (Oryza sativa). Cell Stress Chaperones 2013, 18, 321–331. [Google Scholar] [CrossRef]
- Dong, H.; Zhao, H.; Li, S.; Han, Z.; Hu, G.; Liu, C.; Yang, G.; Wang, G.; Xie, W.; Xing, Y. Genome-wide association studies reveal that members of bHLH subfamily 16 share a conserved function in regulating flag leaf angle in rice (Oryza sativa). PLoS Genetics 2018, 14, e1007323. [Google Scholar] [CrossRef]
- Yang, C.; Li, D.; Mao, D.; Liu, X.; Ji, C.; Li, X.; Zhao, X.; Cheng, Z.; Chen, C.; Zhu, L. Overexpression of microRNA319 impacts leaf morphogenesis and leads to enhanced cold tolerance in rice (Oryza sativa L.). Plant Cell Environ. 2013, 36, 2207–2218. [Google Scholar] [CrossRef]
- Liu, H.-H.; Tian, X.; Li, Y.-J.; Wu, C.-A.; Zheng, C.-C. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 2008, 14, 836–843. [Google Scholar] [CrossRef]
- Zang, G.; Zou, H.; Zhang, Y.; Xiang, Z.; Huang, J.; Luo, L.; Wang, C.; Lei, K.; Li, X.; Song, D.; et al. The De-Etiolated 1 Homolog of Arabidopsis Modulates the ABA Signaling Pathway and ABA Biosynthesis in Rice. Plant Physiol. 2016, 171, 1259–1276. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, W.; Ding, Y.; Zhang, J.; Cambula, E.D.; Weng, F.; Liu, Z.; Ding, C.; Tang, S.; Chen, L.; et al. Shading contributes to the reduction of stem mechanical strength by decreasing cell wall synthesis in Japonica rice (Oryza sativa L.). Front. Plant Sci. 2017, 8, 881. [Google Scholar] [CrossRef]
- Heng, Y.; Wu, C.; Long, Y.; Luo, S.; Ma, J.; Chen, J.; Liu, J.; Zhang, H.; Ren, Y.; Wang, M.; et al. OsALMT7 Maintains Panicle Size and Grain Yield in Rice by Mediating Malate Transport. Plant Cell 2018, 30, 889–906. [Google Scholar] [CrossRef]
- Li, S.; Wei, X.; Ren, Y.; Qiu, J.; Jiao, G.; Guo, X.; Tang, S.; Wan, J.; Hu, P. OsBT1 encodes an ADP-glucose transporter involved in starch synthesis and compound granule formation in rice endosperm. Sci. Rep. 2017, 7, 40124. [Google Scholar] [CrossRef]
- Sharma, R.; Cao, P.; Jung, K.H.; Sharma, M.K.; Ronald, P.C. Construction of a rice glycoside hydrolase phylogenomic database and identification of targets for biofuel research. Front. Plant Sci. 2013, 4, 330. [Google Scholar] [CrossRef]
- Xu, X.; Feng, Y.; Fang, S.; Xu, J.; Wang, X.; Guo, W. Genome-wide characterization of the β-1,3-glucanase gene family in Gossypium by comparative analysis. Sci. Rep. 2016, 6, 29044. [Google Scholar] [CrossRef]
- Trindade, I.; Capitão, C.; Dalmay, T.; Fevereiro, M.P.; dos Santos, D.M. miR398 and miR408 are up-regulated in response to water deficit in Medicago truncatula. Planta 2010, 231, 705–716. [Google Scholar] [CrossRef]
- Mangrauthia, S.K.; Bhogireddy, S.; Agarwal, S.; Prasanth, V.V.; Voleti, S.R.; Neelamraju, S.; Subrahmanyam, D. Genome-wide changes in microRNA expression during short and prolonged heat stress and recovery in contrasting rice cultivars. J. Exp. Bot. 2017, 68, 2399–2412. [Google Scholar] [CrossRef]
- Sekhar, S.; Panda, D.; Kumar, J.; Mohanty, N.; Biswal, M.; Baig, M.J.; Kumar, A.; Umakanta, N.; Samantaray, S.; Pradhan, S.K.; et al. Comparative transcriptome profiling of low light tolerant and sensitive rice varieties induced by low light stress at active tillering stage. Sci. Rep. 2019, 9, 5753. [Google Scholar] [CrossRef]
- Li, Y.T.; Li, Y.; Li, Y.N.; Liang, Y.; Sun, Q.; Li, G.; Liu, P.; Zhang, Z.S.; Gao, H.Y. Dynamic light caused less photosynthetic suppression, rather than more, under nitrogen deficit conditions than under sufficient nitrogen supply conditions in soybean. BMC Plant Biol. 2020, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, K.; Suzuki, N.; Iwamae, T.; Inui, M.; Yukawa, H. Increased fructose 1,6-bisphosphate aldolase in plastids enhances growth and photosynthesis of tobacco plants. J. Exp. Bot. 2012, 63, 3001–3009. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Yamane, H.; Maeshima, M.; Komatsu, S. Characterization of fructose-bisphosphate aldolase regulated by gibberellin in roots of rice seedling. Plant. Mol. Biol. 2004, 56, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Eremina, M.; Rozhon, W.; Yang, S.; Poppenberger, B. ENO2 activity is required for the development and reproductive suc-cess of plants, and is feedback-repressed by AtMBP-1. Plant J. 2015, 81, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zheng, L.; Pu, L.; Ma, X.; Wang, X.; Wu, Y.; Ming, H.; Wang, Q.; Zhang, G. ENO2 Affects the Seed Size and Weight by Adjusting Cytokinin Content and Forming ENO2-bZIP75 Complex in Arabidopsis thaliana. Front. Plant Sci. 2020, 11, 1331. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Target ID/Name | Gene Name/Gene ID | Function | T-Range Start-Stop (nt) | Cleavage Site on the Target (nt) | Validated Degradome Category | Allen Score | p-Value | MFE Perfect | MFE Site | Sequence | Structure |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| miR5493 | Os06t0592500-01 | Heat stress response | 239–259 | 249 | 4 | 9 | 0.999999999998832 | −48.2 | −32 | CGGGGGCGGCGGCGCCCGCGCG&AGC-CGGGCUCUGUCGCGCGUG | ((.(.(((((((.(((((.((.&.))-))))).))))))).).)) | |
| miR5493 | Os01t0226600-01 | OsSLAC | panicle size and grain yield | 646–668 | 657 | 4 | 10 | 0.99492685305248 | −48.2 | −34.6 | CGCGCGCGGCGGCGGCGGCGGCG&AGCCG-GGCU-CUGUCGCGCGUG | ((((((((((((.(((..((((.&.))))-.)))-)))))))))))) |
| miR5493 | Os02t0202400-01 | OsBT1-1 | grain formation by controlling starch synthesis | 1026–1048 | 1038 | 2 | 9.5 | 0.957334082969076 | −48.2 | −33.4 | GGCGCGCCGACGUCGGCCCGGCC&AGCCGGGCU-CUGUCG-CGCGUG | .(((((.(((((..((((((((.&.))))))))-.)))))-))))). |
| miR160b-3p (shade) | Os04t0104900-01 | OsCOMTL2 | methyltransferase | 218–240 | 230 | 4 | 7.5 | 0.69392523364486 | −42.8 | −29.7 | CAUGCUGAGGCUCCUCGCGUCGU&GCG-UGCAAGGAGCC-AAGCAUG | ((((((..(((((((.(((.(((&)))-))).)))))))-.)))))) |
| miR169-p (sun) | Os01t0188400-01 | OschlME | NADP-dependent malic enzyme | 1621–1645 | 1635 | 4 | 13.5 | 0.906795694004373 | −39.1 | −27.7 | GAGCCAGGGUCGUGCAGUAUUUGCC&GGCAAGU-CUGU----CCUUGGCUA | .((((((((....((((.(((((((&)))))))-))))----)))))))). |
| miR399b | Os03t0761100-02 | OsPP2C | stress tolerance. ABA-signalling | 217–236 | 228 | 4 | 9 | 0.779612379437849 | −39.6 | −26 | CGGGGGAGUUCUCGA-UGGCG&UGCCAAAGGAGAAUUGCCCUG | (((((.(((((((..-((((.&.))))...))))))).))))) |
| miR1439 | Os01t0281000-01 | OsFbox6 OsFBX5 OsSTA12 | cyclin F-box containing protein | 2027–2049 | 2038 | 4 | 9 | 0.969782287321394 | −31.6 | −20.7 | AUUGCUCAUUCUGUAUUCUGAAA&UUUUGGA—ACAGAGUGAGUAUU | ..((((((((((((..(((((((&)))))))--)))))))))))).. |
| miR414 | Os06t0561200-01 | Potassium/proton antiporter | 132–151 | 142 | 4 | 4.5 | 0.997646941389551 | −36.4 | −26.8 | UCCUCCUCGUCCUCGUCGUU&GACGAUGAUGACGAGGAUGA | ((.((((((((.((((((((&)))))))).)))))))).)) | |
| miR414 | Os10t0503800-01 | OsREM1.2 | Membrane protein, plant growth, development, stress responses | 734–753 | 744 | 4 | 3.5 | 0.809937142647283 | −36.4 | −30.7 | UCGUCGUCGUCGUCGUCGUU&GACGAUGAUGACGAGGAUGA | (((((.((((((((((((((&)))))))))))))).))))) |
| miR6245 | Os07t0551600-01 | OsCslF9 | mechanical Strength of stem | 1716–1732 | 1723 | 4 | 11 | 0.97682345 | −19.6 | −10.2 | AGGCCGGCGCC&GGUGUCGGCACU | (((((((((((&))))))))).)) |
| miR5075 | Os02t0285300-01 | OsDREPP2 | plasma membrane protein | 361–371 | 369 | 4 | 5 | 1 | −40.2 | −27.5 | CUCCGCCGCCGUCA-CCA&CGGAUGGCGGCGACGGAG | (((((.((((((((-((.&.)).)))))))).))))) |
| miR529a | Os03t0787300-01 | OsDjA5 | co-chaperones | 1235–1255 | 1246 | 4 | 9 | 0.999999999991589 | −36.7 | −24.6 | GAGGAGGAGAUGAGGAGGCGG&CUGUACCCUC-UCUCUUCUUC | ((((((((((.((((..((((&))))..))))-)))))))))) |
| miR444.2 | Os05t0549800-01 | OsEREBP96 | transcription factor in ethylene signalling | 564–584 | 575 | 1 | 8 | 0.443890721132931 | −40.1 | −26.7 | CCGCCGGCGGCGGCGAUUGCA&UGCAGUUGCUGCCUCAAGCUU | ..((..(.(((((((((((((&))))))))))))).)..)).. |
| miR5810 | Os10t0392400-01 | OsTIFY11D | jasmonate signalling | 692–704 | 699 | 4 | 13 | 0.999999999324921 | −28.98 | −6.48 | AUUGUUGUUUUCC&GGAACCCUAACAGCGAU | (((((((((((((&))))….))))))))) |
| miR2275b | Os05t0402700-01 | OsFBA | formation of SBP and FBP, increase photosynthetic carbon flux, RUBP regeneration, promote gibberellin mediated root growth | 337–350 | 341 | 2 | 5 | 0.440673027304968 | −34 | −22.18 | CCUCCAGUACCUCA&UGAGAUACUGGAGG | ((...(((((((((.((((&)))).)))))))))...)) |
| miR5487 | Os02t0771700-01 | OsGns9 | glycoside hydrolase pollen development, seed germination, cold response | 862–884 | 878 | 4 | 20 | 0.652419432123166 | −34.6 | −24.6 | GGAACUACAAUGCCGUGCGCGUCGUG&GAUGUGCAUGUAGUUCC | (((((((((……((((((((&))))))))))))))))) |
| miR5144-5p | Os01t0725800-01 | OsWD40-24 | repressors of photomorphogeneis, stomatal closure, mesophyll photosynthesis, sucrose breakdown | 435–455 | 446 | 4 | 9 | 0.713872072328414 | −37.9 | −25.8 | GUCUCGGCAGCAGCGGGUGGA&UUCUUGUGCUGCUGAAGAGAC | (((((..((((((((.(.(((&))).).))))))))..))))) |
| miR5144-5p | Os01t0588900-01 | OsLOG1 | Cytokinin-activating enzyme | 474–492 | 483 | 4 | 6 | 0.854853045446532 | −37.9 | −25.8 | GUCUCUGGCAUGCACGAGA&UUCUUGUGCUGCUGAAGAGAC | ((((((.(((.((((((((&)))))))))))…)))))) |
| miR172b | Os03t0171700-01 | OsbHLH153 | flag leaf angle | 303–321 | 313 | 4 | 6.5 | 0.926716620410872 | −36 | −24.6 | UGCAGCUACAUCAAGAGCC&GGAAUCUUGAUGAUGCUGCA | ((((((..((((((((.((&))..))))))))..)))))) |
| miR168b | Os11t0255300-01 | OsCP1 | cysteine protease pollen development | 1036–1052 | 1044 | 4 | 6 | 0.999999999999982 | −30 | −20.3 | ACUCGAGACGGCACCAAG&CUUGGUGCAGCUCGGG | ((((((...((((((((&))))))))..)))))) |
| miR168b | Os01t0104600-01 | OsDET1 | repressors of photomorphogeneis | 358–375 | 366 | 4 | 6 | 0.999999999973558 | −38.4 | −25.9 | GCCCGCGCCGCAGCAAGC&GCUUGGUGCAGCUCGGGA | .((((.((.(((.(((((&))))).))).)).)))). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panigrahy, M.; Panigrahi, K.C.S.; Poli, Y.; Ranga, A.; Majeed, N. Integrated Expression Analysis of Small RNA, Degradome and Microarray Reveals Complex Regulatory Action of miRNA during Prolonged Shade in Swarnaprabha Rice. Biology 2022, 11, 798. https://doi.org/10.3390/biology11050798
Panigrahy M, Panigrahi KCS, Poli Y, Ranga A, Majeed N. Integrated Expression Analysis of Small RNA, Degradome and Microarray Reveals Complex Regulatory Action of miRNA during Prolonged Shade in Swarnaprabha Rice. Biology. 2022; 11(5):798. https://doi.org/10.3390/biology11050798
Chicago/Turabian StylePanigrahy, Madhusmita, Kishore Chandra Sekhar Panigrahi, Yugandhar Poli, Aman Ranga, and Neelofar Majeed. 2022. "Integrated Expression Analysis of Small RNA, Degradome and Microarray Reveals Complex Regulatory Action of miRNA during Prolonged Shade in Swarnaprabha Rice" Biology 11, no. 5: 798. https://doi.org/10.3390/biology11050798
APA StylePanigrahy, M., Panigrahi, K. C. S., Poli, Y., Ranga, A., & Majeed, N. (2022). Integrated Expression Analysis of Small RNA, Degradome and Microarray Reveals Complex Regulatory Action of miRNA during Prolonged Shade in Swarnaprabha Rice. Biology, 11(5), 798. https://doi.org/10.3390/biology11050798