
Compositional Shifts in Microbial Diversity under Traditional Banana Cropping Systems of Sub-Saharan Africa

 , and
, and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Study Area and Sample Collection
2.2. Soil Physico-Chemical Analysis and Enzymatic Activities
2.3. Genomic DNA Extraction and Amplicon Generation
2.4. Sequencing Data Processing
2.5. OTU Clustering and Taxonomic Annotation
2.6. Data Analysis
3. Results
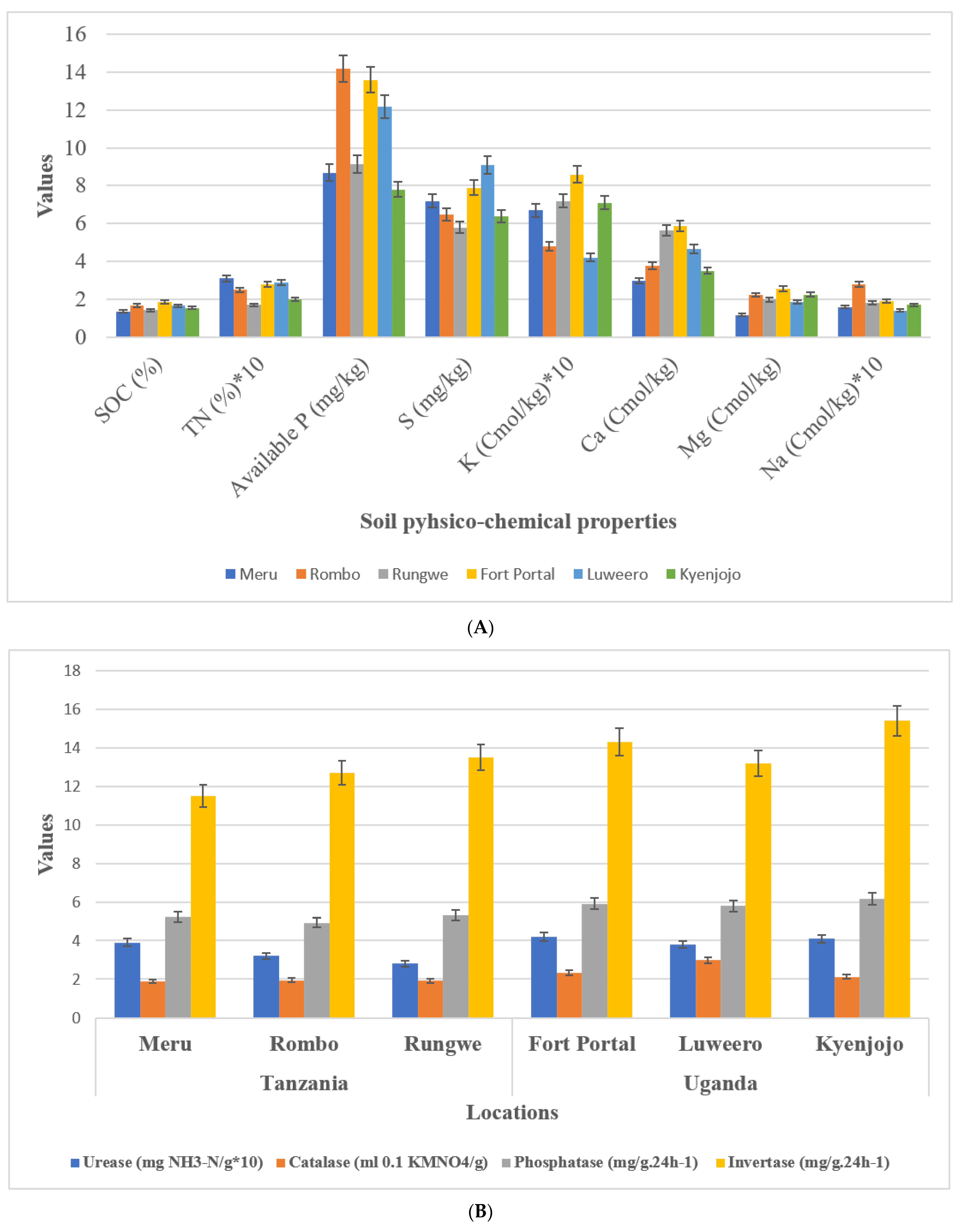
3.1. Physico-Chemical Parameters of Study Sites
3.2. Enzymatic Activities in Study Sites
3.3. Species Richness of Microbial Communities in Cropping Systems
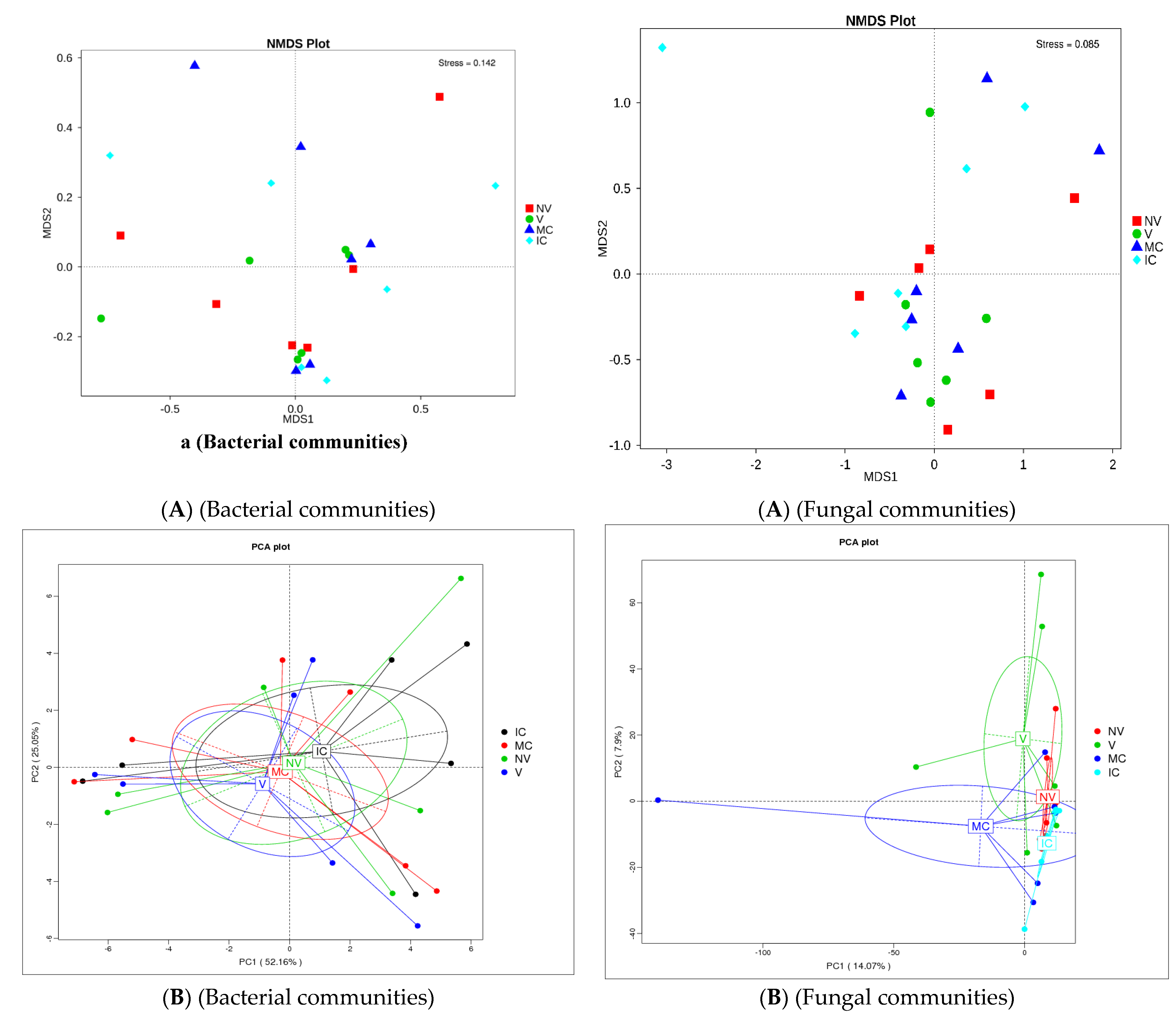
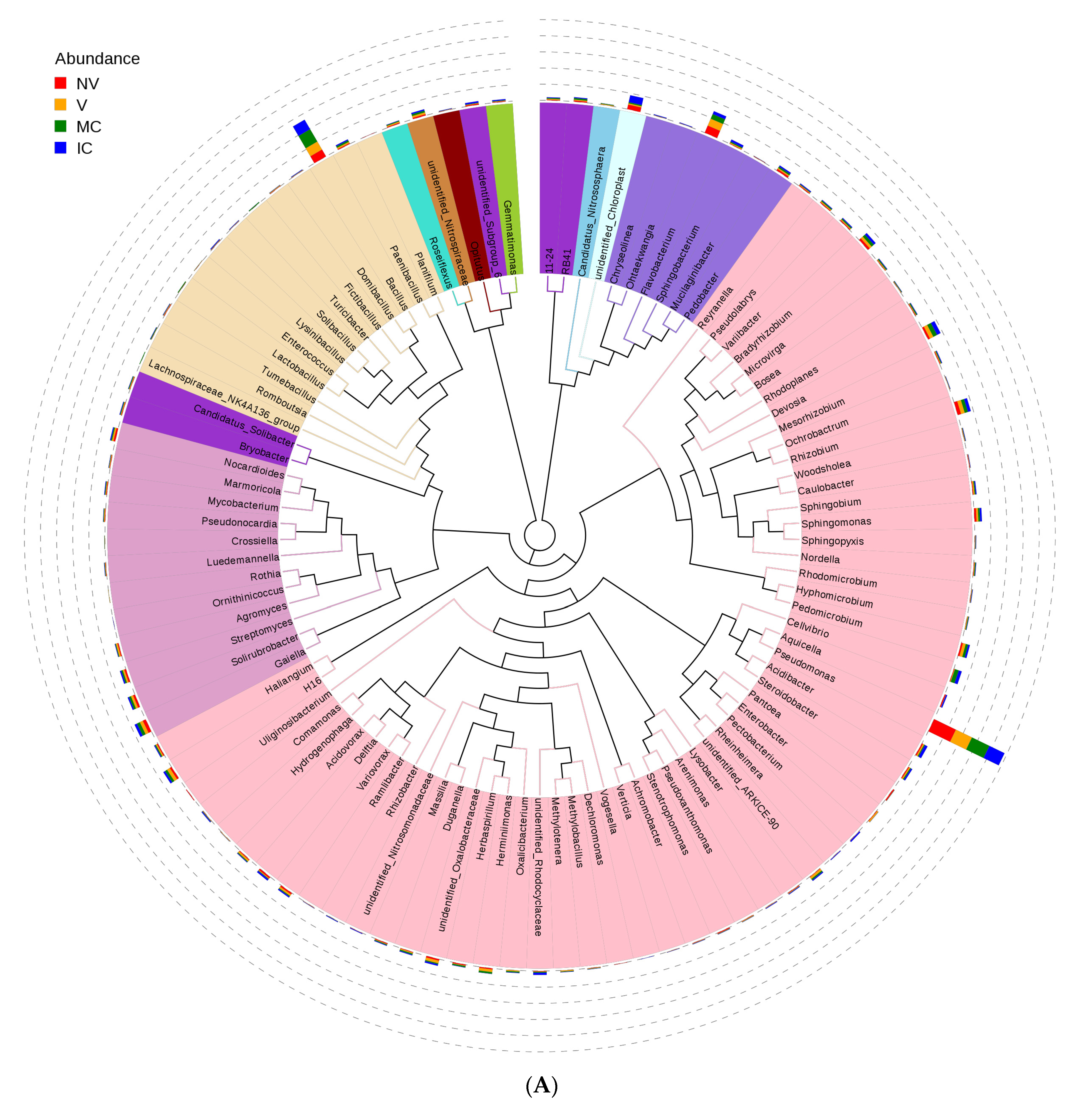
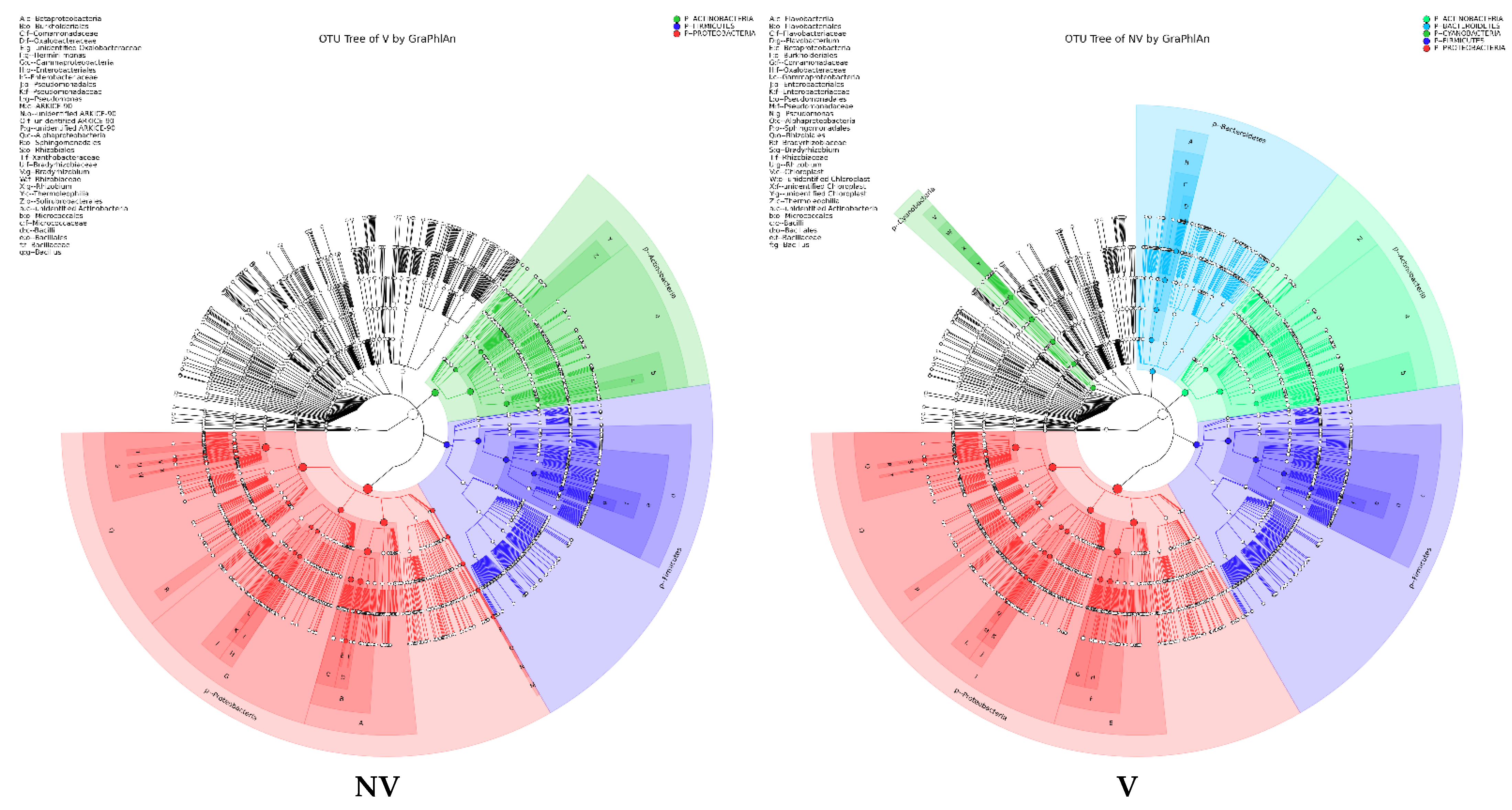
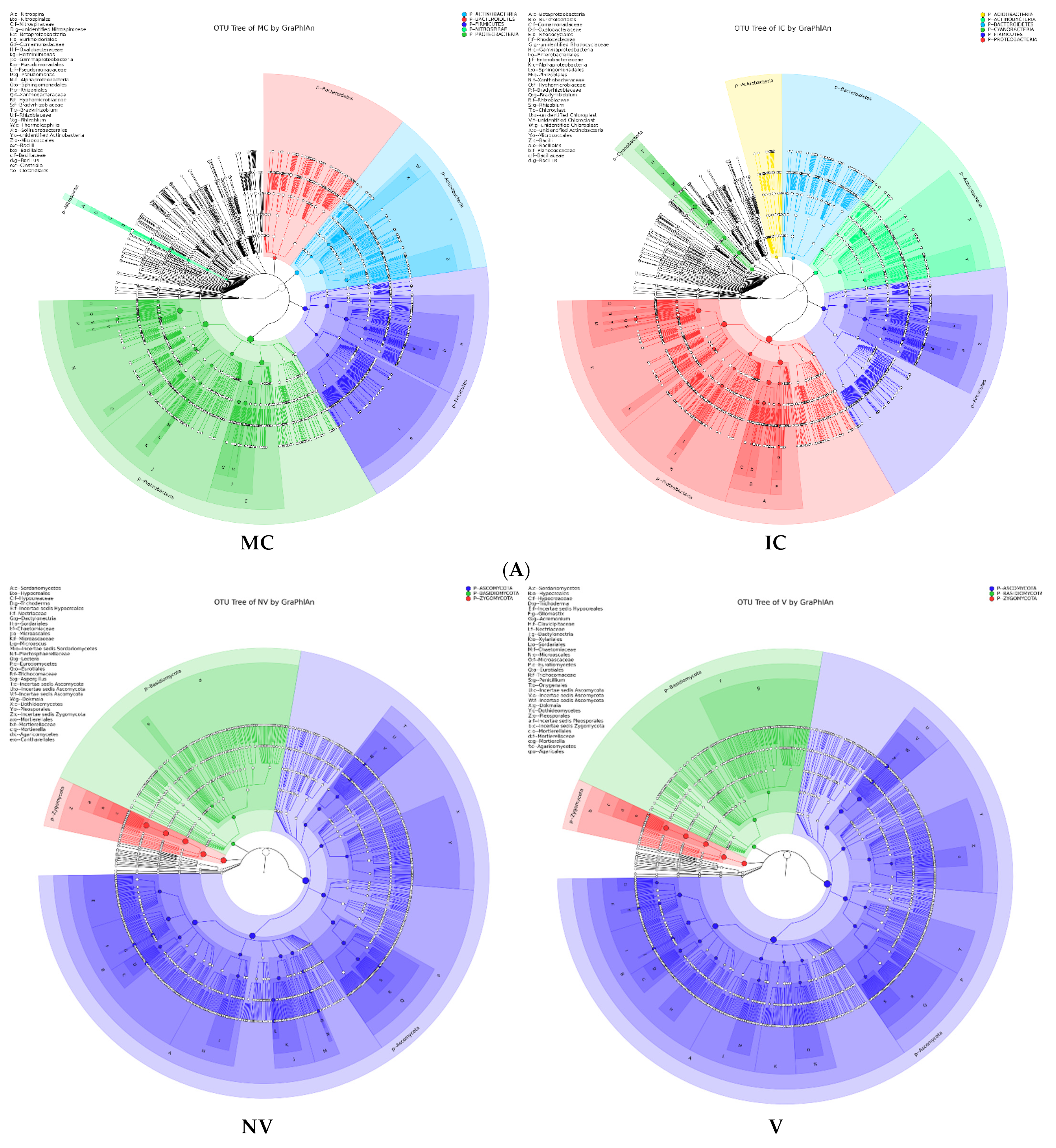
3.4. Cropping Systems (NV-V and MC-IC) Impact on Bacterial Microbiome Structure and Composition
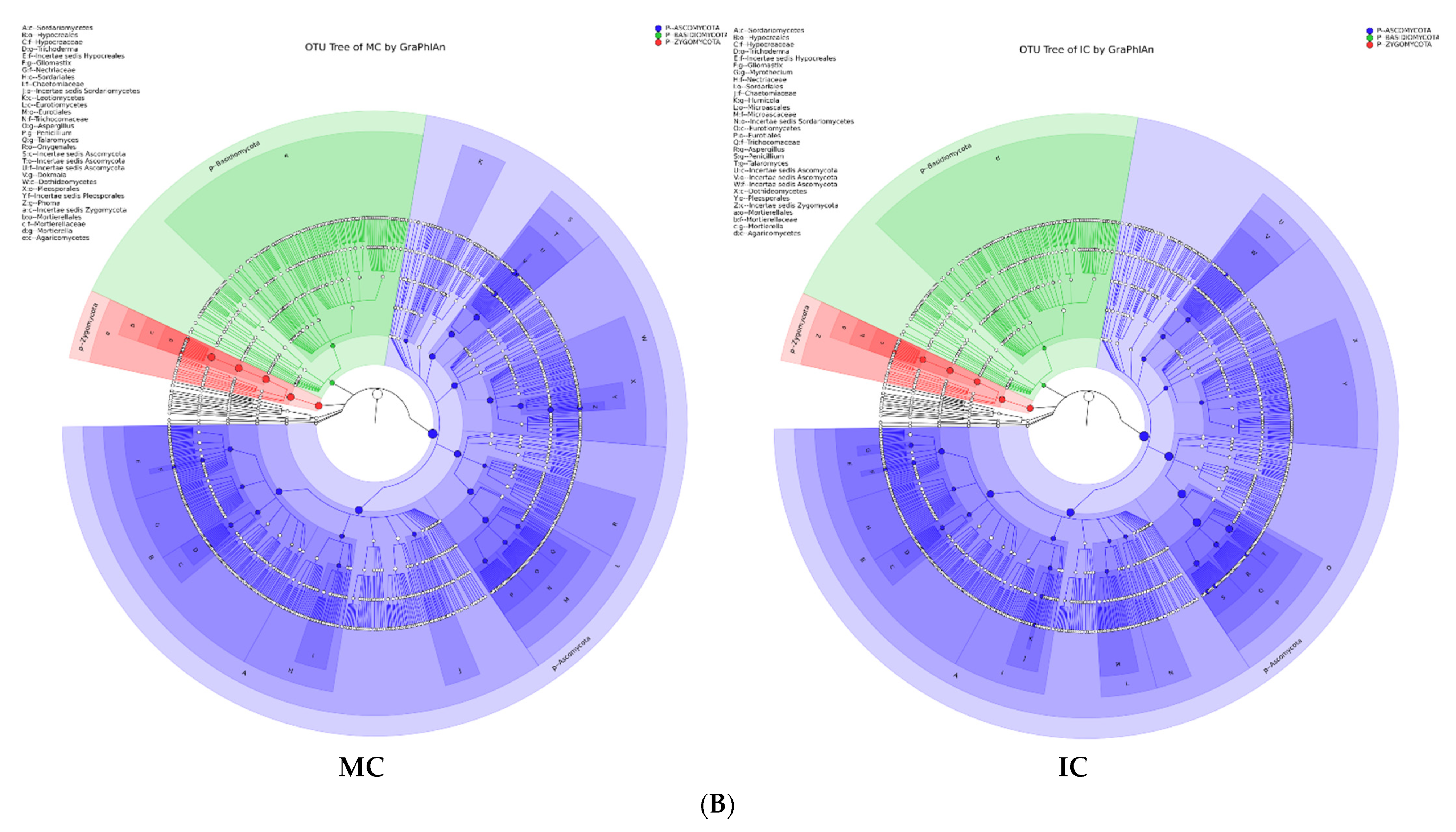
3.5. Cropping Systems (NV-V and MC-IC) Impact on Fungal Microbiome Structure and Composition
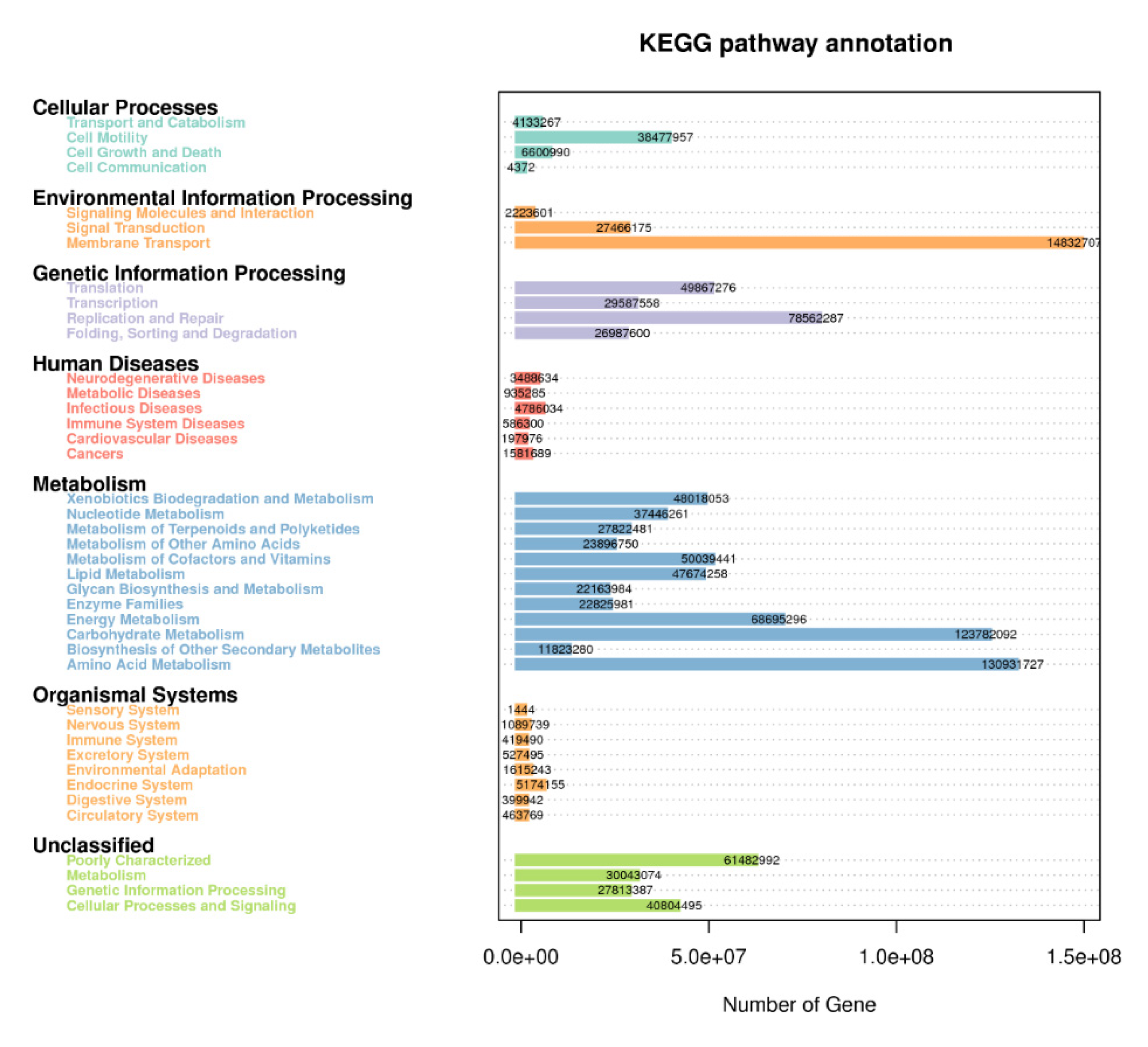
3.6. KEGG Metabolic Pathways Enrichment
4. Discussion
4.1. Physico-Chemical and Enzaymatic Caharcteristics
4.2. Cropping Systems Richeness and Diversity
4.3. Metabolic Pathways Revealed in Cropping Systems
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sainia, V.K.; Bhandarib, S.C.; Tarafdar, J.C. Comparison of crop yield, soil microbial C, N and P, N-fixation, nodulation and mycorrhizal infection in inoculated and non-inoculated sorghum and chickpea crops. Field Crops Res. 2004, 89, 39–47. [Google Scholar] [CrossRef]
- Wakelin, S.A.; Macdonald, L.M.; Rogers, S.L.; Gregg, A.L.; Bolger, T.P.; Baldock, J.A. Habitat selective factors influencing the structural composition and functional capacity of microbial communities in agricultural soils. Soil Biol. Biochem. 2008, 40, 803–813. [Google Scholar] [CrossRef]
- Pieterse, C.M.J.; Zamioudis, C.; Berendsen, R.L.; Weller, D.M.; Van Wees, S.C.; Bakker, P.A. Induced systemic resistance by beneficial microbes. Annu. Rev. Phytopathol. 2014, 52, 347–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Baquerizo, M.; Maestre, F.T.; Reich, P.B.; Jeffries, T.C.; Gaitan, J.J.; Encinar, D.; Berdugo, M.; Campbell, C.D.; Singh, B.K. Microbial diversity drives multifunctionality in terrestrial ecosystems. Nat. Commun. 2016, 7, 10541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastida, F.; Eldridge, D.J.; García, C.; Png, G.K.; Bardgett, R.D.; Delgado-Baquerizo, M. Soil microbial diversity–biomass relationships are driven by soil carbon content across global biomes. ISME J. 2021, 15, 2081–2091. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tilman, D.; Lambers, H.; Zhang, F.S. Plant diversity and overyielding, insights from belowground facilitation of intercropping in agriculture. New Phytol. 2014, 203, 63–69. [Google Scholar] [CrossRef]
- Singh, K.; Mish, A.K.; Singh, B.; Singh, R.P.; Patra, D.D. Tillage effects on crop yield and physicochemical properties of sodic soils. Land Degrad. Dev. 2016, 27, 223–230. [Google Scholar] [CrossRef]
- Corre-Hellou, G.; Dibet, A.; Hauggaard-Nielsen, H.; Crozat, Y.; Gooding, M.; Ambus, P.; Dahlmann, C.; von Fragstein, P.; Pristeri, A.; Monti, M.; et al. The competitive ability of pea-barley intercrops against weeds and the interactions with crop productivity and soil N availability. Field Crops Res. 2011, 122, 264–272. [Google Scholar] [CrossRef] [Green Version]
- van Asten, P.J.A.; Wairegi, L.W.I.; Mukasa, D.; Uringi, N.O. Agronomic and economic benefits of coffee-banana intercropping in Uganda’s smallholder farming systems. Agric. Syst. 2011, 104, 326–334. [Google Scholar] [CrossRef]
- Zhang, H.; Mallik, A.; Zeng, R.S. Control of Panama disease of banana by rotating and intercropping with Chinese chive (Allium tuberosum Rottler): Role of plant volatiles. J. Chem. Ecol. 2013, 39, 243–252. [Google Scholar] [CrossRef]
- Bainard, L.; Koch, A.; Gordon, A.; Klironomos, J. Growth response of crops to soil microbial communities from conventional monocropping and tree-based intercropping systems. Plant Soil 2013, 363, 345–356. [Google Scholar] [CrossRef]
- Eisenhauer, N.; Hines, J.; Isbell, F.; van der Plas, F.; Hobbie, S.E.; Kazanski, C.E.; Lehmann, A.; Liu, M.; Lochner, A.; Rillig, M.C.; et al. Plant diversity maintains multiple soil functions in future environments. eLife 2018, 7, e41228. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Sanders, N.J.; Shi, Y.; Chu, H.; Classen, A.T.; Zhao, K.; Chen, L.; Shi, Y.; Jiang, Y.; He, J.S. The links between ecosystem multifunctionality and above- and belowground biodiversity are mediated by climate. Nat. Commun. 2015, 6, 8159. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Nuccio, E.E.; Shi, Z.J.; He, Z.; Zhou, J.; Firestone, M.K. The interconnected rhizosphere, high network complexity dominates rhizosphere assemblages. Ecol. Lett. 2016, 19, 926–936. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.R.; Blair, P.L.; Boyd, C.; Cody, B.; Hazel, A.; Hedrick, A.; Kathuria, H.; Khurana, P.; Kramer, B.; Muterspaw, K.; et al. Microbial community responses to soil tillage and crop rotation in a corn/soybean agroecosystem. Ecol. Evol. 2016, 6, 8075–8084. [Google Scholar] [CrossRef]
- Ko, D.; Yoo, G.; Yun, S.T.; Jun, S.C.; Chung, H. Bacterial and fungal community composition across the soil depth profiles in a fallow field. J. Ecol. Environ. 2017, 41, 34. [Google Scholar] [CrossRef] [Green Version]
- De Leon, D.G.; Vahter, T.; Zobel, M.; Koppel, M.; Edesi, L.; Davison, J.; Al-Quraishy, S.; Hozzein, W.N.; Moora, M.; Oja, J.; et al. Different wheat cultivars exhibit variable responses to inoculation with arbuscular mycorrhizal fungi from organic and conventional farms. PLoS ONE 2020, 15, e0233878. [Google Scholar]
- Fawcett, J.K. The semi-micro Kjeldahl method for the determination of nitrogen. J. Med. Lab Technol. 1954, 12, 1–22. [Google Scholar]
- Li, Y.; Fang, F.; Wei, J.; Wu, X.; Cui, R.; Li, G.; Zheng, F.; Tan, D. Humic acid fertilizer improved soil properties and soil microbial diversity of continuous cropping peanut, a three-year experiment. Sci. Rep. 2019, 9, 12014. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Zhou, H.; Zhou, S.; Li, L.; Wei, C.; Yu, Y.; Hay, A.G. Fomesafen impacts bacterial communities and enzyme activities in the rhizosphere. Environ. Pollut. 2019, 253, 302–311. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH, fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UPARSE, highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project, improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R, A language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. 2020. Available online: https://www.R-project.org/ (accessed on 14 April 2022).
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG, integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG, Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Wan, W.; Tan, J.; Wang, Y.; Qin, Y.; He, H.; Wu, H.; Zuoa, W.; He, D. Responses of the rhizosphere bacterial community in acidic crop soil to pH: Changes in diversity, composition, interaction, and function. Sci. Total Environ. 2020, 700, 134418. [Google Scholar] [CrossRef]
- Rousk, J.; Baath, E.; Brookes, P.C.; Lauber, C.L.; Lozupone, C.; Caporaso, J.G.; Knight, R.; Fierer, N. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 2010, 4, 1340–1351. [Google Scholar] [CrossRef]
- Carrino-Kyker, S.R.; Coyle, K.P.; Kluber, L.A.; Burke, D.J. Fungal and bacterial communities exhibit consistent responses to reversal of soil acidification and phosphorus limitation over time. Microorganisms 2016, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, M.; Wani, S.P. Plant-growth-promoting rhizobacteria: Drought stress alleviators to ameliorate crop production in drylands. Ann. Microbiol. 2016, 66, 35–42. [Google Scholar] [CrossRef]
- Jian, S.; Li, J.; Chen, J.; Wang, G.; Mayes, M.A.; Dzantor, K.E.; Huie, D.; Luoc, Y. Soil extracellular enzyme activities, soil carbon and nitrogen storage under nitrogen fertilization, a meta-analysis. Soil Biol. Biochem. 2016, 101, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Li, S.; Zhang, W.; Ma, J.; Wang, J.; Cai, J.; Yang, G. Effects of the novel pyrimidynyloxybenzoic herbicide ZJ0273 on enzyme activities, microorganisms and its degradation in Chinese soils. Environ. Sci. Pollut. Res. 2015, 22, 4425–4433. [Google Scholar] [CrossRef] [PubMed]
- Benitez, M.S.; Osborne, S.L.; Lehman, R.M. Previous crop and rotation history effects on maize seedling health and associated rhizosphere microbiome. Sci. Rep. 2017, 7, 15709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokhorst, S.; Kardol, P.; Bellingham, P.J.; Kooyman, R.M.; Richardson, S.J.; Schmidt, S.; Wardle, D.A. Responses of communities of soil organisms and plants to soil aging at two contrasting long-term chronosequences. Soil Biol. Biochem. 2017, 106, 69–79. [Google Scholar] [CrossRef]
- Freedman, Z.; Zak, D.R. Soil bacterial communities are shaped by temporal and environmental filtering, evidence from a long-term chronosequence. Environ. Microbiol. 2015, 17, 3208–3218. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N. Embracing the unknown, disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef]
- Bardgett, R.D.; van der Putten, W.H. Belowground biodiversity and ecosystem functioning. Nature 2014, 515, 505–511. [Google Scholar] [CrossRef]
- van Elsas, J.D.; Chiurazzi, M.; Mallon, C.A.; Elhottova, D.; Kristufek, V.; Salles, J.F. Microbial diversity determines the invasion of soil by a bacterial pathogen. Proc. Natl. Acad. Sci. USA 2012, 109, 1159–1164. [Google Scholar] [CrossRef] [Green Version]
- Berg, G.; Koberl, M.; Rybakova, D.; Muller, H.; Grosch, R.; Smalla, K. Plant microbial diversity is suggested as the key to future biocontrol and health trends. FEMS Microbiol. Ecol. 2017, 93, 28430944. [Google Scholar] [CrossRef] [PubMed]
- Cong, W.F.; Hoffland, E.; Li, L.; Six, J.; Sun, J.H.; Bao, X.G.; Zhang, F.S.; Werf, W.V.D. Intercropping enhances soil carbon and nitrogen. Global Change Biol. 2015, 21, 1715–1726. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Yu, L.; Liu, Y.; Zhang, Y.; Yang, W.; Li, Z.; Wang, J. Effects of reduced nitrogen input on productivity and N2O emissions in a sugarcane/soybean intercropping system. Eur. J. Agron. 2016, 81, 78–85. [Google Scholar] [CrossRef]
- Jauri, P.V.; Altieri, N.; Pérez, C.A.; Kinkel, L. Cropping history effects on pathogen suppressive and signaling dynamics in streptomyces communities. Phytobiomes J. 2018, 2, 14–23. [Google Scholar] [CrossRef]
- Meyer, S.T.; Ptacnik, R.; Hillebrand, H.; Bessler, H.; Buchmann, N.; Ebeling, A.; Eisenhauer, N.; Engels, C.; Fischer, M.; Halle, S.; et al. Biodiversity–multifunctionality relationships depend on identity and number of measured functions. Nat. Ecol. Evol. 2018, 2, 44–49. [Google Scholar] [CrossRef]
- Steven, B.; Belnap, J.; Kuske, C.R. Chronic physical disturbance substantially alters the response of biological soil crusts to a wetting pulse, as characterized by metatranscriptomic sequencing. Front. Microbiol. 2018, 9, 2382. [Google Scholar] [CrossRef] [Green Version]
- Lugtenberg, B.; Kamilova, F. Plant-growth-promoting rhizobacteria. Annu. Rev. Microbiol. 2009, 63, 541–556. [Google Scholar] [CrossRef] [Green Version]
- Mendes, R.; Kruijt, M.; de Bruijn, I.; Dekkers, E.; van der Voort, M.; Schneider, J.H.M.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.H.M.; et al. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 2011, 27, 1097–1100. [Google Scholar] [CrossRef]
- Granzow, S.; Kaiser, K.; Wemheuer, B.; Pfeiffer, B.; Daniel, R.; Vidal, S.; Wemheuer, F. The effects of cropping regimes on fungal and bacterial communities of wheat and faba bean in a greenhouse pot experiment differ between plant species and compartment. Front. Microbiol. 2017, 8, 902. [Google Scholar] [CrossRef] [Green Version]
- Alami, M.M.; Xue, J.; Ma, Y.; Zhu, D.; Gong, Z.; Shu, S.; Wang, X. Structure, diversity, and composition of bacterial communities in rhizospheric soil of Coptis chinensis Franch under continuously cropped fields. Diversity 2020, 12, 57. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Lin, L.; Wang, X.; You, J.; Li, J.; Chen, X. Elevational is the main factor controlling the soil microbial community structure in alpine tundra of the Changbai Mountain. Sci. Rep. 2020, 10, 12442. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Valera, E.; Kyselkova, M.; Ahmed, E.; Sladecek, F.X.J.; Goberna, M.; Elhottova, D. Native soil microorganisms hinder the soil enrichment with antibiotic resistance genes following manure applications. Sci. Rep. 2019, 9, 6760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teplitski, M.; Warriner, K.; Bartz, J.; Schneider, K.R. Untangling metabolic and communication networks, interactions of enterics with phytobacteria and their implications in produce safety. Trends Microbiol. 2011, 19, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Stegen, J.C.; Lin, X.; Fredrickson, J.M.; Chen, X.; Kennedy, D.W.; Murray, C.J.; Rockhold, M.L.; Konopka, A. Quantifying community assembly processes and identifying features that impose them. ISME J. 2013, 7, 2069–2079. [Google Scholar] [CrossRef]
- Rodriguez, P.A.; Rothballer, M.; Chowdhury, S.P.; Nussbaumer, T.; Gutjahr, C.; Falter-Braun, P. Systems biology of plant-microbiome interactions. Mol. Plant 2019, 12, 804–821. [Google Scholar] [CrossRef] [Green Version]
- Kamutando, C.N.; Vikram, S.; Kamgan-Nkuekam, G.; Makhalanyane, T.P.; Greve, M.; Le Roux, J.J.; Richardson, D.M.; Cowan, D.A.; Valverde, A. The functional potential of the rhizospheric microbiome of an invasive tree species, Acacia Dealbata. Microb. Ecol. 2019, 77, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Richard, P.J.; Anna, K.; Stanislav, K. Pinpointing secondary metabolites that shape the composition and function of the plant microbiome. J. Exp. Bot. 2020, 424, 32995888. [Google Scholar]
- Ren, M.; Zhang, Z.; Wang, X.; Zhou, Z.; Chen, D.; Zeng, H.; Zhao, S.; Chen, L.; Hu, Y.; Zhang, C.; et al. Diversity and contributions to nitrogen cycling and carbon fixation of soil salinity shaped microbial communities in Tarim Basin. Front. Microbiol. 2018, 9, 431. [Google Scholar] [CrossRef] [Green Version]
- Roy, N.; Choi, K.; Khan, R.; Lee, S.W. Culturing simpler and bacterial wilt suppressive microbial communities from tomato rhizosphere. Plant Pathol. J. 2019, 35, 362–371. [Google Scholar] [CrossRef]
- Macias-Benitez, S.; Garcia-Martinez, A.M.; Jimenez, P.C.; Gonzalez, J.M.; Moral, M.T.; Rubio, J.P. Rhizospheric organic acids as biostimulants, monitoring feedbacks on soil microorganisms and biochemical properties. Front. Plant Sci. 2020, 11, 633. [Google Scholar] [CrossRef]
- Bubici, G.; Kaushal, M.; Prigigallo, M.I.; Gómez-Lama Cabanás, C.; Mercado-Blanco, J. Biological control agents against Fusarium wilt of banana. Front. Microbiol. 2019, 10, 616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushal, M.; Mahuku, G.; Swennen, R. Comparative transcriptome and expression profiling of resistant and susceptible banana cultivars during infection by Fusarium oxysporum. Int. J. Mol. Sci. 2021, 22, 3002. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Parameters | Tanzania | Uganda | ||||
|---|---|---|---|---|---|---|---|
| Meru | Rombo | Rungwe | Fort Portal | Luweero | Kyenjojo | ||
| 1. | Sampling time | May 2019 | May 2019 | May 2019 | June 2019 | June 2019 | June 2019 |
| 2. | Average annual temperature (°C) | Max. 25.8 ± 0.5 Min. 16.3 ± 0.7 | Max. 23.5 ± 0.5 Min. 14.6 ± 0.9 | Max. 21.6 ± 0.7 Min. 12.8 ± 0.8 | Max. 25.0 ± 0.5 Min. 15.2 ± 0.7 | Max. 26.8 ± 0.6 Min. 19.2 ± 0.7 | Max. 25.8 ± 0.7 Min. 15.9 ± 0.5 |
| 3. | Soil temperature at the time of sampling (°C) | 21 ± 0.5 | 17.7 ± 0.5 | 16.4 ± 0.5 | 20.2 ± 0.5 | 23.4 ± 0.5 | 21.9 ± 0.5 |
| 4. | Average annual precipitation (mm) | 233.3 | 110.3 | 955.0 | 160.5 | 125.3 | 158.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaushal, M.; Tumuhairwe, J.B.; Kaingo, J.; Richard, M.; Nakamanya, F.; Taulya, G.; Coyne, D. Compositional Shifts in Microbial Diversity under Traditional Banana Cropping Systems of Sub-Saharan Africa. Biology 2022, 11, 756. https://doi.org/10.3390/biology11050756
Kaushal M, Tumuhairwe JB, Kaingo J, Richard M, Nakamanya F, Taulya G, Coyne D. Compositional Shifts in Microbial Diversity under Traditional Banana Cropping Systems of Sub-Saharan Africa. Biology. 2022; 11(5):756. https://doi.org/10.3390/biology11050756
Chicago/Turabian StyleKaushal, Manoj, John Baptist Tumuhairwe, Jacob Kaingo, Malingumu Richard, Florence Nakamanya, Godfrey Taulya, and Danny Coyne. 2022. "Compositional Shifts in Microbial Diversity under Traditional Banana Cropping Systems of Sub-Saharan Africa" Biology 11, no. 5: 756. https://doi.org/10.3390/biology11050756
APA StyleKaushal, M., Tumuhairwe, J. B., Kaingo, J., Richard, M., Nakamanya, F., Taulya, G., & Coyne, D. (2022). Compositional Shifts in Microbial Diversity under Traditional Banana Cropping Systems of Sub-Saharan Africa. Biology, 11(5), 756. https://doi.org/10.3390/biology11050756