
Self or Non-Self? It Is also a Matter of RNA Recognition and Editing by ADAR1



Abstract
:Simple Summary
Abstract
1. Introduction
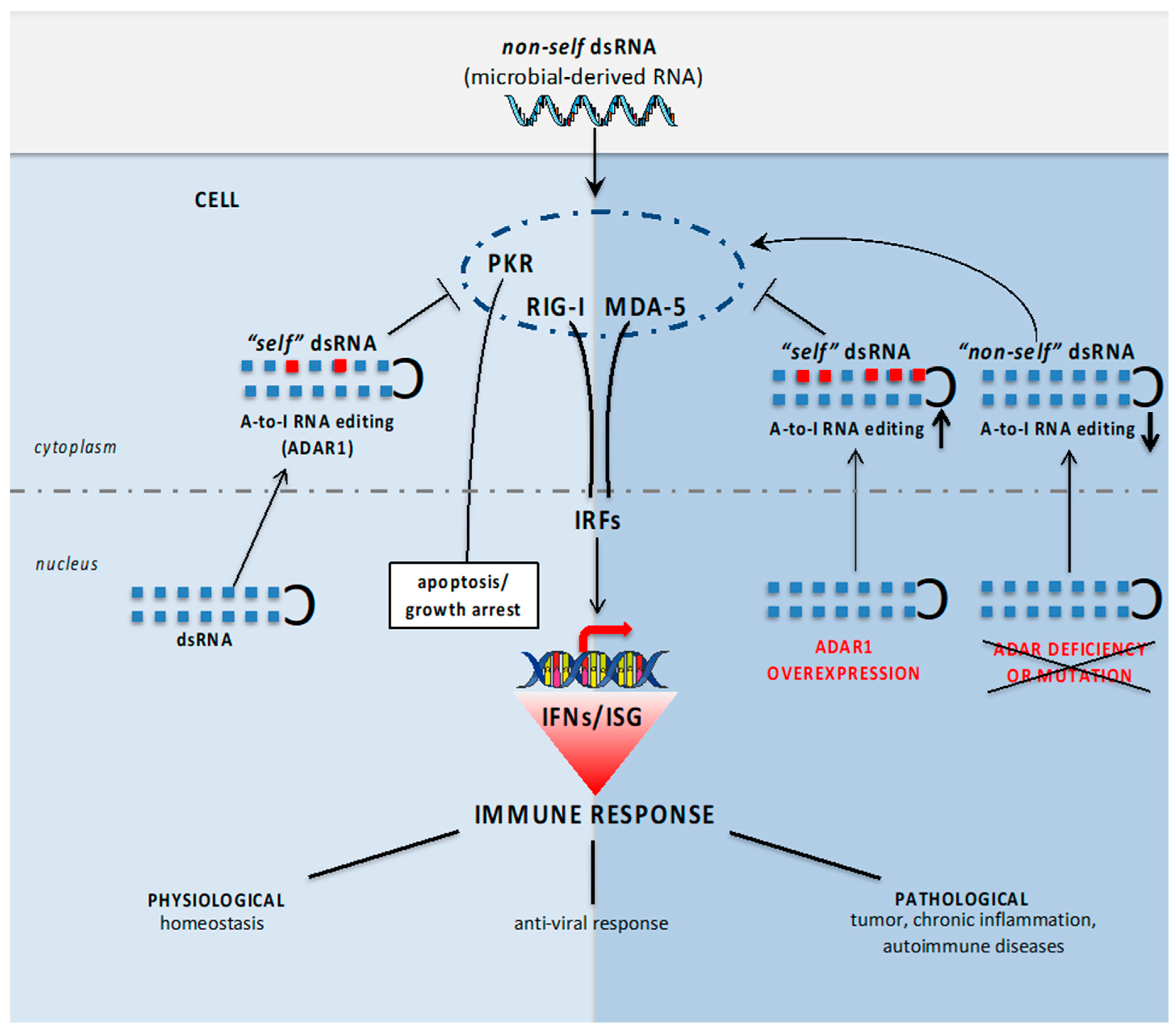
2. RNA Recognition: Self versus Non-Self RNA and Activation of the IFN Pathway
2.1. ADARs, A-to-I Editing and the Control of Innate Immune System Activation
2.2. Mutations in ADAR1
3. ADAR1 in the Immune System
4. ADAR1 and Its Role in Cancer
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zinkernagel, R.M. On ‘reactivity’ versus ‘tolerance’. Immunol. Cell Biol. 2004, 82, 343–352. [Google Scholar] [CrossRef]
- Barak, M.; Porath, H.T.; Finkelstein, G.; Knisbacher, B.A.; Buchumenski, I.; Roth, S.H.; Levanon, E.Y.; Eisenberg, E. Purifying selection of long dsRNA is the first line of defense against false activation of innate immunity. Genome Biol. 2020, 21, 26. [Google Scholar] [CrossRef] [PubMed]
- Mannion, N.M.; Greenwood, S.M.; Young, R.; Cox, S.; Brindle, J.; Read, D.; Nellaker, C.; Vesely, C.; Ponting, C.P.; McLaughlin, P.J.; et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 2014, 9, 1482–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crow, Y.J.; Chase, D.S.; Lowenstein Schmidt, J.; Szynkiewicz, M.; Forte, G.M.; Gornall, H.L.; Oojageer, A.; Anderson, B.; Pizzino, A.; Helman, G.; et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A 2015, 167, 296–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crow, Y.J.; Manel, N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat. Rev. Immunol. 2015, 15, 429–440. [Google Scholar] [CrossRef]
- Rice, G.I.; Forte, G.M.; Szynkiewicz, M.; Chase, D.S.; Aeby, A.; Abdel-Hamid, M.S.; Ackroyd, S.; Allcock, R.; Bailey, K.M.; Balottin, U.; et al. Assessment of interferon-related biomarkers in Aicardi-Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: A case-control study. Lancet. Neurol. 2013, 12, 1159–1169. [Google Scholar] [CrossRef] [Green Version]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat. Genet. 2012, 44, 1243–1248. [Google Scholar] [CrossRef] [Green Version]
- Baal, N.; Cunningham, S.; Obermann, H.L.; Thomas, J.; Lippitsch, A.; Dietert, K.; Gruber, A.D.; Kaufmann, A.; Michel, G.; Nist, A.; et al. ADAR1 Is Required for Dendritic Cell Subset Homeostasis and Alveolar Macrophage Function. J. Immunol. 2019, 202, 1099–1111. [Google Scholar] [CrossRef] [Green Version]
- Marcu-Malina, V.; Goldberg, S.; Vax, E.; Amariglio, N.; Goldstein, I.; Rechavi, G. ADAR1 is vital for B cell lineage development in the mouse bone marrow. Oncotarget 2016, 7, 54370–54379. [Google Scholar] [CrossRef] [Green Version]
- Nakahama, T.; Kato, Y.; Shibuya, T.; Inoue, M.; Kim, J.I.; Vongpipatana, T.; Todo, H.; Xing, Y.; Kawahara, Y. Mutations in the adenosine deaminase ADAR1 that prevent endogenous Z-RNA binding induce Aicardi-Goutieres-syndrome-like encephalopathy. Immunity 2021, 54, 1976–1988.e1977. [Google Scholar] [CrossRef]
- Gannon, H.S.; Zou, T.; Kiessling, M.K.; Gao, G.F.; Cai, D.; Choi, P.S.; Ivan, A.P.; Buchumenski, I.; Berger, A.C.; Goldstein, J.T.; et al. Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat. Commun. 2018, 9, 5450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizuka, J.J.; Manguso, R.T.; Cheruiyot, C.K.; Bi, K.; Panda, A.; Iracheta-Vellve, A.; Miller, B.C.; Du, P.P.; Yates, K.B.; Dubrot, J.; et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 2019, 565, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Golji, J.; Brodeur, L.K.; Chung, F.S.; Chen, J.T.; deBeaumont, R.S.; Bullock, C.P.; Jones, M.D.; Kerr, G.; Li, L.; et al. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat. Med. 2019, 25, 95–102. [Google Scholar] [CrossRef]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, J.; Franz, K.M.; Kagan, J.C. PRRs are watching you: Localization of innate sensing and signaling regulators. Virology 2015, 479–480, 104–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamers, M.M.; van den Hoogen, B.G.; Haagmans, B.L. ADAR1: “Editor-in-Chief” of Cytoplasmic Innate Immunity. Front. Immunol. 2019, 10, 1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, C.E. Adenosine deaminase acting on RNA (ADAR1), a suppressor of double-stranded RNA-triggered innate immune responses. J. Biol. Chem. 2019, 294, 1710–1720. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, M.J.; Purcell, C.A.; Pruijssers, A.J.; Zhao, Y.; Spurlock, C.F., 3rd; Sriram, S.; Ogden, K.M.; Dermody, T.S.; Scholz, M.B.; Crooke, P.S., 3rd; et al. Endogenous double-stranded Alu RNA elements stimulate IFN-responses in relapsing remitting multiple sclerosis. J. Autoimmun. 2019, 100, 40–51. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Ronnblom, L. The importance of the type I interferon system in autoimmunity. Clin. Exp. Rheumatol. 2016, 34, 21–24. [Google Scholar]
- Thompson, M.G.; Sacco, M.T.; Horner, S.M. How RNA modifications regulate the antiviral response. Immunol. Rev. 2021, 304, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Winkler, R.; Gillis, E.; Lasman, L.; Safra, M.; Geula, S.; Soyris, C.; Nachshon, A.; Tai-Schmiedel, J.; Friedman, N.; Le-Trilling, V.T.K.; et al. m(6)A modification controls the innate immune response to infection by targeting type I interferons. Nat. Immunol. 2019, 20, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Liddicoat, B.J.; Piskol, R.; Chalk, A.M.; Ramaswami, G.; Higuchi, M.; Hartner, J.C.; Li, J.B.; Seeburg, P.H.; Walkley, C.R. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 2015, 349, 1115–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pestal, K.; Funk, C.C.; Snyder, J.M.; Price, N.D.; Treuting, P.M.; Stetson, D.B. Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity 2015, 43, 933–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, J.F.; Yang, Q.; Liu, C.X.; Wu, M.; Chen, L.L.; Yang, L. N(6)-Methyladenosines Modulate A-to-I RNA Editing. Mol. Cell 2018, 69, 126–135.e126. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Xu, X.; Wang, Y.; Hawke, D.H.; Yu, S.; Han, L.; Zhou, Z.; Mojumdar, K.; Jeong, K.J.; Labrie, M.; et al. A-to-I RNA Editing Contributes to Proteomic Diversity in Cancer. Cancer Cell 2018, 33, 817–828.e817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz-Yaacov, N.; Bazak, L.; Buchumenski, I.; Porath, H.T.; Danan-Gotthold, M.; Knisbacher, B.A.; Eisenberg, E.; Levanon, E.Y. Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors. Cell Rep. 2015, 13, 267–276. [Google Scholar] [CrossRef] [Green Version]
- George, C.X.; Gan, Z.; Liu, Y.; Samuel, C.E. Adenosine deaminases acting on RNA, RNA editing, and interferon action. J. Interferon Cytokine Res. 2011, 31, 99–117. [Google Scholar] [CrossRef] [Green Version]
- George, C.X.; Samuel, C.E. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc. Natl. Acad. Sci. USA 1999, 96, 4621–4626. [Google Scholar] [CrossRef] [Green Version]
- Desterro, J.M.; Keegan, L.P.; Lafarga, M.; Berciano, M.T.; O’Connell, M.; Carmo-Fonseca, M. Dynamic association of RNA-editing enzymes with the nucleolus. J. Cell Sci. 2003, 116, 1805–1818. [Google Scholar] [CrossRef] [Green Version]
- Nishikura, K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, E.; Levanon, E.Y. A-to-I RNA editing—Immune protector and transcriptome diversifier. Nat. Rev. Genet. 2018, 19, 473–490. [Google Scholar] [CrossRef] [PubMed]
- Sommer, B.; Kohler, M.; Sprengel, R.; Seeburg, P.H. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 1991, 67, 11–19. [Google Scholar] [CrossRef]
- Higuchi, M.; Single, F.N.; Kohler, M.; Sommer, B.; Sprengel, R.; Seeburg, P.H. RNA editing of AMPA receptor subunit GluR-B: A base-paired intron-exon structure determines position and efficiency. Cell 1993, 75, 1361–1370. [Google Scholar] [CrossRef]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 2000, 406, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Horsch, M.; Seeburg, P.H.; Adler, T.; Aguilar-Pimentel, J.A.; Becker, L.; Calzada-Wack, J.; Garrett, L.; Gotz, A.; Hans, W.; Higuchi, M.; et al. Requirement of the RNA-editing enzyme ADAR2 for normal physiology in mice. J. Biol. Chem. 2011, 286, 18614–18622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athanasiadis, A.; Rich, A.; Maas, S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004, 2, e391. [Google Scholar] [CrossRef]
- Bazak, L.; Haviv, A.; Barak, M.; Jacob-Hirsch, J.; Deng, P.; Zhang, R.; Isaacs, F.J.; Rechavi, G.; Li, J.B.; Eisenberg, E.; et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014, 24, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Blow, M.; Futreal, P.A.; Wooster, R.; Stratton, M.R. A survey of RNA editing in human brain. Genome Res. 2004, 14, 2379–2387. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.D.; Kim, T.T.; Walsh, T.; Kobayashi, Y.; Matise, T.C.; Buyske, S.; Gabriel, A. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004, 14, 1719–1725. [Google Scholar] [CrossRef] [Green Version]
- Levanon, E.Y.; Eisenberg, E.; Yelin, R.; Nemzer, S.; Hallegger, M.; Shemesh, R.; Fligelman, Z.Y.; Shoshan, A.; Pollock, S.R.; Sztybel, D.; et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 2004, 22, 1001–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.; Mu, X.; Yang, F.; Greenwald, E.; Park, J.W.; Jacob, E.; Zhang, C.Z.; Hur, S. Breaching Self-Tolerance to Alu Duplex RNA Underlies MDA5-Mediated Inflammation. Cell 2018, 172, 797–810.e713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, O.; Di Segni, A.; Cesarkas, K.; Porath, H.T.; Marcu-Malina, V.; Mizrahi, O.; Stern-Ginossar, N.; Kol, N.; Farage-Barhom, S.; Glick-Saar, E.; et al. RNA editing by ADAR1 leads to context-dependent transcriptome-wide changes in RNA secondary structure. Nat. Commun. 2017, 8, 1440. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Calis, J.J.A.; Wu, X.; Sun, T.; Yu, Y.; Sarbanes, S.L.; Dao Thi, V.L.; Shilvock, A.R.; Hoffmann, H.H.; Rosenberg, B.R.; et al. Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 2018, 172, 811–824.e814. [Google Scholar] [CrossRef] [Green Version]
- Hartner, J.C.; Schmittwolf, C.; Kispert, A.; Muller, A.M.; Higuchi, M.; Seeburg, P.H. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J. Biol. Chem. 2004, 279, 4894–4902. [Google Scholar] [CrossRef] [Green Version]
- Hartner, J.C.; Walkley, C.R.; Lu, J.; Orkin, S.H. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat. Immunol. 2009, 10, 109–115. [Google Scholar] [CrossRef]
- Wang, Q.; Miyakoda, M.; Yang, W.; Khillan, J.; Stachura, D.L.; Weiss, M.J.; Nishikura, K. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J. Biol. Chem. 2004, 279, 4952–4961. [Google Scholar] [CrossRef] [Green Version]
- Heraud-Farlow, J.E.; Chalk, A.M.; Linder, S.E.; Li, Q.; Taylor, S.; White, J.M.; Pang, L.; Liddicoat, B.J.; Gupte, A.; Li, J.B.; et al. Protein recoding by ADAR1-mediated RNA editing is not essential for normal development and homeostasis. Genome Biol. 2017, 18, 166. [Google Scholar] [CrossRef]
- De Reuver, R.; Dierick, E.; Wiernicki, B.; Staes, K.; Seys, L.; De Meester, E.; Muyldermans, T.; Botzki, A.; Lambrecht, B.N.; Van Nieuwerburgh, F.; et al. ADAR1 interaction with Z-RNA promotes editing of endogenous double-stranded RNA and prevents MDA5-dependent immune activation. Cell Rep. 2021, 36, 109500. [Google Scholar] [CrossRef]
- Herbert, A. Mendelian disease caused by variants affecting recognition of Z-DNA and Z-RNA by the Zalpha domain of the double-stranded RNA editing enzyme ADAR. Eur. J. Hum. Genet. 2020, 28, 114–117. [Google Scholar] [CrossRef]
- Maurano, M.; Snyder, J.M.; Connelly, C.; Henao-Mejia, J.; Sidrauski, C.; Stetson, D.B. Protein kinase R and the integrated stress response drive immunopathology caused by mutations in the RNA deaminase ADAR1. Immunity 2021, 54, 1948–1960.e1945. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Rigby, R.E.; Young, G.R.; Hvidt, A.K.; Davis, T.; Tan, T.K.; Bridgeman, A.; Townsend, A.R.; Kassiotis, G.; Rehwinkel, J. Adenosine-to-inosine editing of endogenous Z-form RNA by the deaminase ADAR1 prevents spontaneous MAVS-dependent type I interferon responses. Immunity 2021, 54, 1961–1975.e1965. [Google Scholar] [CrossRef] [PubMed]
- Vongpipatana, T.; Nakahama, T.; Shibuya, T.; Kato, Y.; Kawahara, Y. ADAR1 Regulates Early T Cell Development via MDA5-Dependent and -Independent Pathways. J. Immunol. 2020, 204, 2156–2168. [Google Scholar] [CrossRef] [PubMed]
- Nakahama, T.; Kato, Y.; Kim, J.I.; Vongpipatana, T.; Suzuki, Y.; Walkley, C.R.; Kawahara, Y. ADAR1-mediated RNA editing is required for thymic self-tolerance and inhibition of autoimmunity. EMBO Rep. 2018, 19, e46303. [Google Scholar] [CrossRef] [PubMed]
- Crow, M.K. Type I interferon in the pathogenesis of lupus. J. Immunol. 2014, 192, 5459–5468. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, E.; Booiman, T.; van Hamme, J.L.; Jansen, M.H.; van Dort, K.A.; Vanderver, A.; Rice, G.I.; Crow, Y.J.; Kootstra, N.A.; Kuijpers, T.W. ADAR1 Facilitates HIV-1 Replication in Primary CD4+ T Cells. PLoS ONE 2015, 10, e0143613. [Google Scholar] [CrossRef]
- Danan-Gotthold, M.; Guyon, C.; Giraud, M.; Levanon, E.Y.; Abramson, J. Extensive RNA editing and splicing increase immune self-representation diversity in medullary thymic epithelial cells. Genome Biol. 2016, 17, 219. [Google Scholar] [CrossRef] [Green Version]
- Kyewski, B.; Derbinski, J. Self-representation in the thymus: An extended view. Nat. Rev. Immunol. 2004, 4, 688–698. [Google Scholar] [CrossRef]
- Pujantell, M.; Riveira-Munoz, E.; Badia, R.; Castellvi, M.; Garcia-Vidal, E.; Sirera, G.; Puig, T.; Ramirez, C.; Clotet, B.; Este, J.A.; et al. RNA editing by ADAR1 regulates innate and antiviral immune functions in primary macrophages. Sci. Rep. 2017, 7, 13339. [Google Scholar] [CrossRef]
- Giacopuzzi, E.; Gennarelli, M.; Sacco, C.; Filippini, A.; Mingardi, J.; Magri, C.; Barbon, A. Genome-wide analysis of consistently RNA edited sites in human blood reveals interactions with mRNA processing genes and suggests correlations with cell types and biological variables. BMC Genom. 2018, 19, 963. [Google Scholar] [CrossRef] [Green Version]
- Cesarini, V.; Silvestris, D.A.; Tassinari, V.; Tomaselli, S.; Alon, S.; Eisenberg, E.; Locatelli, F.; Gallo, A. ADAR2/miR-589-3p axis controls glioblastoma cell migration/invasion. Nucleic Acids Res. 2018, 46, 2045–2059. [Google Scholar] [CrossRef] [PubMed]
- Fritzell, K.; Xu, L.D.; Lagergren, J.; Ohman, M. ADARs and editing: The role of A-to-I RNA modification in cancer progression. Semin. Cell Dev. Biol. 2018, 79, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Tassinari, V.; Cesarini, V.; Tomaselli, S.; Ianniello, Z.; Silvestris, D.A.; Ginistrelli, L.C.; Martini, M.; De Angelis, B.; De Luca, G.; Vitiani, L.R.; et al. ADAR1 is a new target of METTL3 and plays a pro-oncogenic role in glioblastoma by an editing-independent mechanism. Genome Biol. 2021, 22, 51. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; He, W.; Cai, H.; Hu, B.; Zheng, C.; Ke, X.; Xie, L.; Zheng, Z.; Wu, X.; Wang, H. A-to-I RNA editing of BLCAP lost the inhibition to STAT3 activation in cervical cancer. Oncotarget 2017, 8, 39417–39429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Wan, S.; Ou, Y.; Zhou, B.; Zhu, J.; Yi, X.; Guan, Y.; Jia, W.; Liu, X.; Wang, Q.; et al. RNA over-editing of BLCAP contributes to hepatocarcinogenesis identified by whole-genome and transcriptome sequencing. Cancer Lett. 2015, 357, 510–519. [Google Scholar] [CrossRef]
- Shigeyasu, K.; Okugawa, Y.; Toden, S.; Miyoshi, J.; Toiyama, Y.; Nagasaka, T.; Takahashi, N.; Kusunoki, M.; Takayama, T.; Yamada, Y.; et al. AZIN1 RNA editing confers cancer stemness and enhances oncogenic potential in colorectal cancer. JCI Insight. 2018, 3, e99976. [Google Scholar] [CrossRef]
- Teoh, P.J.; An, O.; Chung, T.H.; Chooi, J.Y.; Toh, S.H.M.; Fan, S.; Wang, W.; Koh, B.T.H.; Fullwood, M.J.; Ooi, M.G.; et al. Aberrant hyperediting of the myeloma transcriptome by ADAR1 confers oncogenicity and is a marker of poor prognosis. Blood 2018, 132, 1304–1317. [Google Scholar] [CrossRef]
- Nakano, M.; Fukami, T.; Gotoh, S.; Nakajima, M. A-to-I RNA Editing Up-regulates Human Dihydrofolate Reductase in Breast Cancer. J. Biol. Chem. 2017, 292, 4873–4884. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Crews, L.A.; Barrett, C.L.; Chun, H.J.; Court, A.C.; Isquith, J.M.; Zipeto, M.A.; Goff, D.J.; Minden, M.; Sadarangani, A.; et al. ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc. Natl. Acad. Sci. USA 2013, 110, 1041–1046. [Google Scholar] [CrossRef] [Green Version]
- Zipeto, M.A.; Court, A.C.; Sadarangani, A.; Delos Santos, N.P.; Balaian, L.; Chun, H.J.; Pineda, G.; Morris, S.R.; Mason, C.N.; Geron, I.; et al. ADAR1 Activation Drives Leukemia Stem Cell Self-Renewal by Impairing Let-7 Biogenesis. Cell Stem Cell 2016, 19, 177–191. [Google Scholar] [CrossRef]
- Mehdipour, P.; Marhon, S.A.; Ettayebi, I.; Chakravarthy, A.; Hosseini, A.; Wang, Y.; de Castro, F.A.; Loo Yau, H.; Ishak, C.; Abelson, S.; et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature 2020, 588, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Ishak, C.A.; De Carvalho, D.D. Endogenous Retroelements and the Viral Mimicry Response in Cancer Therapy and Cellular Homeostasis. Cancer Discov. 2021, 11, 2707–2725. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Cell Type/Organ | Mouse/ Human | Mutation/Gene Deficiency | Functional Effects | Reference |
|---|---|---|---|---|
| Single positive thymocytes | Mouse | Adar1 deletion in CD4+ T cells | Impaired T cell maturation, excessive ISG expression, defective TCR signaling, induction of autoimmunity (spontaneous colitis) | [54] |
| Primary CD4+ T lymphocytes | Human | Adar1-deficient AGS patients | Upregulated ISG expression, resistance to HIV-1 infection | [7,56] |
| mTECs | Mouse | None | Demonstration of high rates of editing events, acquisition of self-tolerance against edited-self, prevention of autoimmunity | [57] |
| B lymphocytes | Mouse | Adar1 deletion in CD19+ B cells | Impaired B cell maturation, reduced numbers of peripheral blood and splenic B cells, enhanced excessive ISG expression and apoptosis | [9] |
| Primary macrophages | Human | siRNA ADAR1 silencing | Enhanced production of IFN-I, chemokines and cytokines, inhibition of HIV-1 infection in bystander cells | [59] |
| DCs | Mouse | Adar1 deletion in CD11c+ cells | Peripheral systemic loss of CD103+/CD8+ DCs, reduced number and dysfunction of AM, loss of RNA editing in AM | [8] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tassinari, V.; Cerboni, C.; Soriani, A. Self or Non-Self? It Is also a Matter of RNA Recognition and Editing by ADAR1. Biology 2022, 11, 568. https://doi.org/10.3390/biology11040568
Tassinari V, Cerboni C, Soriani A. Self or Non-Self? It Is also a Matter of RNA Recognition and Editing by ADAR1. Biology. 2022; 11(4):568. https://doi.org/10.3390/biology11040568
Chicago/Turabian StyleTassinari, Valentina, Cristina Cerboni, and Alessandra Soriani. 2022. "Self or Non-Self? It Is also a Matter of RNA Recognition and Editing by ADAR1" Biology 11, no. 4: 568. https://doi.org/10.3390/biology11040568
APA StyleTassinari, V., Cerboni, C., & Soriani, A. (2022). Self or Non-Self? It Is also a Matter of RNA Recognition and Editing by ADAR1. Biology, 11(4), 568. https://doi.org/10.3390/biology11040568