1. Introduction
Cytochromes P450, the heme-thiolate enzymes found in all domains of life, from
Eubacteria and
Archaea to
Eukarya, probably appeared about 3.5 billion years ago [
1], when the oxygen content in the atmosphere was negligible. It is suggested that ancient cytochromes P450 acted as reducing enzymes and could play the role of NO reductases [
2]. When green plants began to release oxygen into the atmosphere about 2 billion years ago, cytochromes P450 became involved in the synthesis and oxidative metabolism of fatty acids and steroids [
3]. Later, the catalytic oxidation of hydrophobic compounds became the primary function of cytochromes P450.
Nowadays, cytochromes P450 act as terminal oxidases in monooxygenase systems, oxidizing various exogenous and endogenous substrates. They are involved in the oxidative metabolism and detoxification of low molecular weight foreign lipophilic compounds (xenobiotics) as well as in the synthesis of pigments, hormones, second messengers, antibiotics, and toxins. To perform these functions better, the eukaryotic P450s became membrane incorporated. In most cases, they are associated with the membranes of the endoplasmic reticulum (ER), where they interact with their partner proteins via lateral diffusion and the formation of dissociative complexes.
All known eukaryotic cytochromes P450 and most bacterial analogs are not self-sufficient in their catalytic function. The monooxygenase reaction requires two electrons, which are usually transferred to cytochrome P450 from a protein partner. For most eukaryotic cytochromes P450, the role of electron donor is played by NADPH-cytochrome P450 reductase (CPR), a flavoprotein that contains two flavin cofactors, FAD and FMN. These flavins are situated in distinct protein domains termed FAD and FMN domains. These two domains are connected with a flexible connecting loop. The reducing equivalents from NADPH are first acquired by FAD and then transferred to FMN, which serves as an ultimate electron donor for P450.
While the majority of animal cytochrome P450 species are involved in xenobiotic metabolism, the predominant part of the plant P450s participates in biosynthetic pathways. They play a critical role in synthesizing lignin, UV protectants, pigments, defense compounds, fatty acids, hormones, and secondary messengers. In particular, cytochrome-P450-dependent cinnamate-4-hydroxylase (C4H), p-coumaroyl quinate/shikimate-3’-hydroxylase (C3′H), and ferulate-5-hydroxylase (F5H) are the critical branching points in the phenylpropanoid metabolizing pathway, which is required for the biosynthesis of monolignol and serves as a starting point for the production of flavonoids, coumarins, lignans, and lignin.
This investigation represents a part of our studies aimed at elucidating the mechanisms of function and regulation of phenylpropanoid-metabolizing monooxygenases from
Sorghum bicolor, a U.S. strategic plant for biofuel production [
4,
5,
6]. The detailed mechanistic knowledge of these enzymes will enable specific manipulation of lignin composition and content and thus economize the industrial cost of biofuel production. Furthermore, the high flexibility of the hinge region of SbCPR demonstrated in our previous report [
7] offers potential for manipulating the functional properties of the enzyme through rational engineering in this region. The present study explores the functional mechanisms of NADPH-cytochrome P450 reductase 2b, one of the three CPR enzymes in
Sorghum bicolor serving as electron donors for C4H, C3′H, and F5H P450 enzymes. This enzyme is referred to as SbCPR from now on.
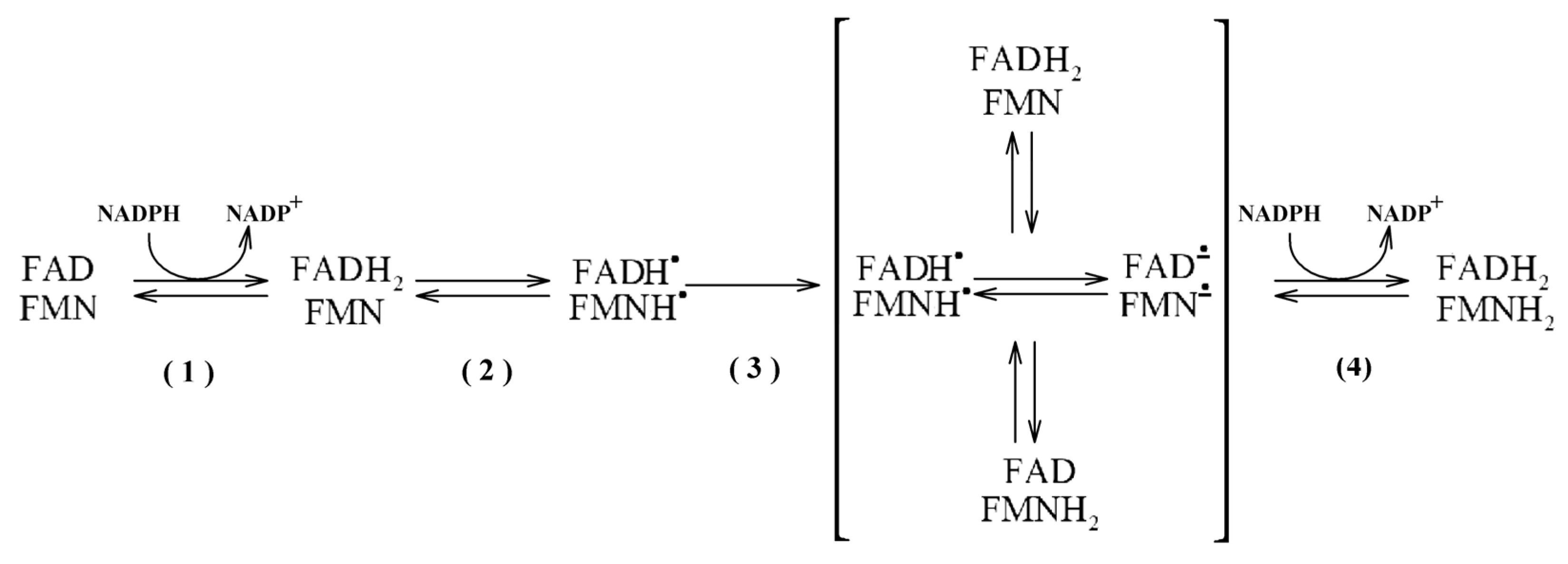
In general, the functional redox cycling in SbCPR enzymes follows the scheme common to all known CPRs. Their reduction from the completely oxidized (FAD, FMN) to the four-electron reduced state (FADH
2, FMNH
2) includes (1) hydride transfer from NADPH to FAD, resulting in a two-electron reduced (FADH
2, FMN) state; (2) inter-flavin electron transfer from FADH
2 to FMN with the formation of the neutral (blue) disemiquinone (FADH
•, FMNH
•); (3) establishing a transient equilibrium between the latter and the other two-electron reduced states (FAD, FMNH
2), (FADH
2, FMN) and anionic semiquinones (FAD
, FMN
); (4) supply of another pair of electrons from NADPH yielding the four-electron reduced (FADH
2, FMNH
2) state. This sequence of events is illustrated in
Scheme 1.
In most eukaryotic monooxygenases, this four-electron reduced state is believed to serve as the P450 electron donor so that FMN cofactor interchanges between the hydroquinone and semiquinone states. By contrast, the FMN moiety in the bacterial CPR-P450 chimera P450BM-3 shuttles between the semiquinone and oxidized states instead [
8]. However, in all cases, the transfer of electrons between the FAD and FMN domains remains an obligatory step in the CPR redox cycle.
In this perspective, understanding electron transfer mechanisms and the respective conformational rearrangements has become a challenge for researchers. In the first solved CPR structure [
9] and several subsequently published structures of CPR from various species [
10,
11,
12], the distance between FAD and FMN is around 4 Å, which is considered favorable for the inter-flavin electron transfer. However, this proximity of the two domains does not allow the electron acceptor protein to reach FMN. This circumstance and the largely disordered structure of the connecting loop brought forward a hypothesis of a large-scale opening-and-closing transition involved in the CPR electron transfer mechanism.
In further X-ray crystallographic studies, the CPR variant with a shortened connecting loop was found locked in the open state, which is flawed in terms of inter-flavin electron transfer, but effective in the transfer from FMN to the heme [
13]. In contrast, a variant of rat CPR where the two domains are interconnected with a disulfide bond [
14] was found locked in the closed conformation. These and other resolved CPR structures [
11,
12,
15,
16] demonstrated exceptional conformational flexibility of CPR and emphasized the pivotal functional role of the transitions between the enzyme’s closed and open states.
The conformational landscape of CPR and its relevance to the redox cycling of the enzyme was further explored with a wide variety of biophysical techniques ranging from NMR [
17] and small-angle neutron and X-ray scattering (SANS and SAX, [
18,
19,
20,
21]) to ion mobility mass spectrometry [
22] and single-molecule fluorescence resonance energy transfer (FRET [
23,
24,
25,
26]). Despite some contradictory observations in these studies, all of them are consistent in demonstrating a transition of the closed state of the enzyme into an open conformation upon the interdomain electron transfer event and the formation of the disemiquinone state (see [
27] for a review). At the same time, these studies also demonstrated that the initial two-state model is insufficient to adequately depict the conformational landscape of CPR.
According to the current concepts, instead of being represented by two discrete states, the enzyme exists in a dynamic equilibrium among multiple conformations differing in the relative positioning of the FMN and FAD domains [
27]. Most available data suggest that the closed conformations predominate in the completely oxidized CPR [
18,
19,
20,
21,
24,
26]. Some studies also suggest the further displacement of the conformational equilibrium towards the closed state upon the binding of NADPH to the oxidized enzyme [
25,
27]. There are also strong indications of the predominance of the closed conformations in the four-electron reduced enzyme [
20,
21,
23,
26].
Despite a foreseeable similarity of CPR enzymes from different organisms in general mechanisms of electron transport and the related protein choreography, the enzymes from different kingdoms of life may differ considerably in the kinetic and thermodynamic parameters of the individual steps of the redox cycle. Thus, the structure study of
Arabidopsis thaliana suggests that the oxidized state of plant CPR enzymes may have considerably more open conformation than that characteristic of their mammalian counterparts [
11]. Furthermore, the studies of the kinetics of electron transfer in plant CPR demonstrate that the rate of electron transfer from NADPH to FAD in these enzymes is over 50 times faster than in their mammalian counterparts [
28].
In the present study, we explore the conformational equilibrium in SbCPR and its modulation during the redox cycle of the enzyme with the use of a combination of the FRET-based detection of protein conformational rearrangements with the rapid scanning absorbance and fluorescence stop-flow technique and the pressure-perturbation approach. While the FRET-based methods and rapid scanning stop-flow techniques have already been applied in both mammalian [
24,
25,
27] and plant [
26] CPR studies, the present study represents the first attempt to explore the conformational landscape of CPR with pressure perturbation.
In pressure-perturbation studies, hydrostatic pressure is a variable parameter affecting the protein conformational landscapes. Along with the effects of temperature, varying pressure is indispensable for a detailed understanding of the mechanisms of protein conformational transitions. The basis of pressure effects is the change in system volume that accompanies biochemical processes [
29,
30,
31,
32]. According to Le Chatelier’s principle, increased pressure enhances processes accompanied by a decrease in system volume and, conversely, inhibits processes occurring with a volume increase. A prevalent part of the volume changes in protein transitions stems from the changes in interactions with solvents [
33,
34,
35,
36,
37,
38]. These include water penetration into the cavities and water constriction around solvent-exposed polar groups of the protein [
31,
38,
39,
40,
41,
42,
43]. Thus, the volume change resulting from the penetration of one water molecule into a protein cavity is equal to −18 mL/mol, while the solvation of a singly charged ion in water is characterized by Δ
V values of the order of −10 mL/mol [
31]. Generally speaking, pressure increase enhances protein hydration, which therefore constitutes the core of pressure-induced protein transitions [
33,
34,
37,
43,
44,
45,
46].
Ample conformational transitions necessary for CPR redox cycling are implied to be associated with significant changes in the protein–solvent interactions. The process of protein opening is reported to involve the breaking of several salt bridges [
11,
12,
24] and the subsequent hydration of the newly exposed charges on the protein surface. Therefore, pressure perturbation is the method of choice for exploring the CPR conformational landscape. It allows to judge the changes in protein hydration in the redox cycling of the enzyme and provides a simple means for determining the position of equilibria in the system of open and closed conformation at its different redox states.
To make the studies possible, we incorporate a FRET donor/acceptor pair into the FAD and FMN domains of SbCPR. Our study demonstrates that, although the closed conformation always predominates in the conformational landscape, the population of open state increases by order of magnitude upon the formation of the disemiquinone state. Our results are consistent with several open conformation sub-states differing in the opening transition volume change (ΔV0). The details of the SbCPR electron transfer mechanism revealed in this study will provide vital information for engineering the monolignol pathway and subsequent lignin polymerization in order to improve the use of Sorghum bicolor as a biofuel plant. In addition to elucidating the functional choreography of plant CPRs, our study demonstrates the high exploratory potential of a combination of the pressure-perturbation approach with the FRET-based monitoring of protein conformational dynamics.
2. Materials and Methods
2.1. Materials
DY-520XL and DY-731 were the products of Dyomics GMBH (Jena, Germany). Monobromobimane (MBBr) was obtained from Invitrogen/Molecular Probes (Eugene, OR, USA), now a part of ThermoFisher Scientific. Igepal CO-630, glucose oxidase, catalase, NADPH, glucose-6-phosphate, and glucose-6-phosphate-dehydrogenase were obtained from Sigma-Aldrich (St. Louis, MO, USA). 2′5′-ADP was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). All other chemicals were of the highest grade commercially available and were used without further purification.
2.2. Cloning, Protein Expression, and Purification
The SbCPR cDNA corresponding to the
Sorghum bicolor gene SORBI_3007G088000 was modified with a truncation of its N-terminal transmembrane sequence (Δ2–50) and the addition of the C-terminal hexahistidine tag. The resulting construct was cloned into a pET-30a (+) vector. For the SbCPR C596S mutant, site-directed mutations were created in the SbCPR coding region by PCR-based amplification using Phusion High-Fidelity DNA polymerase (New England Biolabs, Ipswich, MA, USA). The amplification was performed using the forward primer CTTCGGAAGCAGAAATAGCAAGATGGACT, and the reverse primer TATTTCTGCTTCCGAAGAAGAACACGGATG was followed by
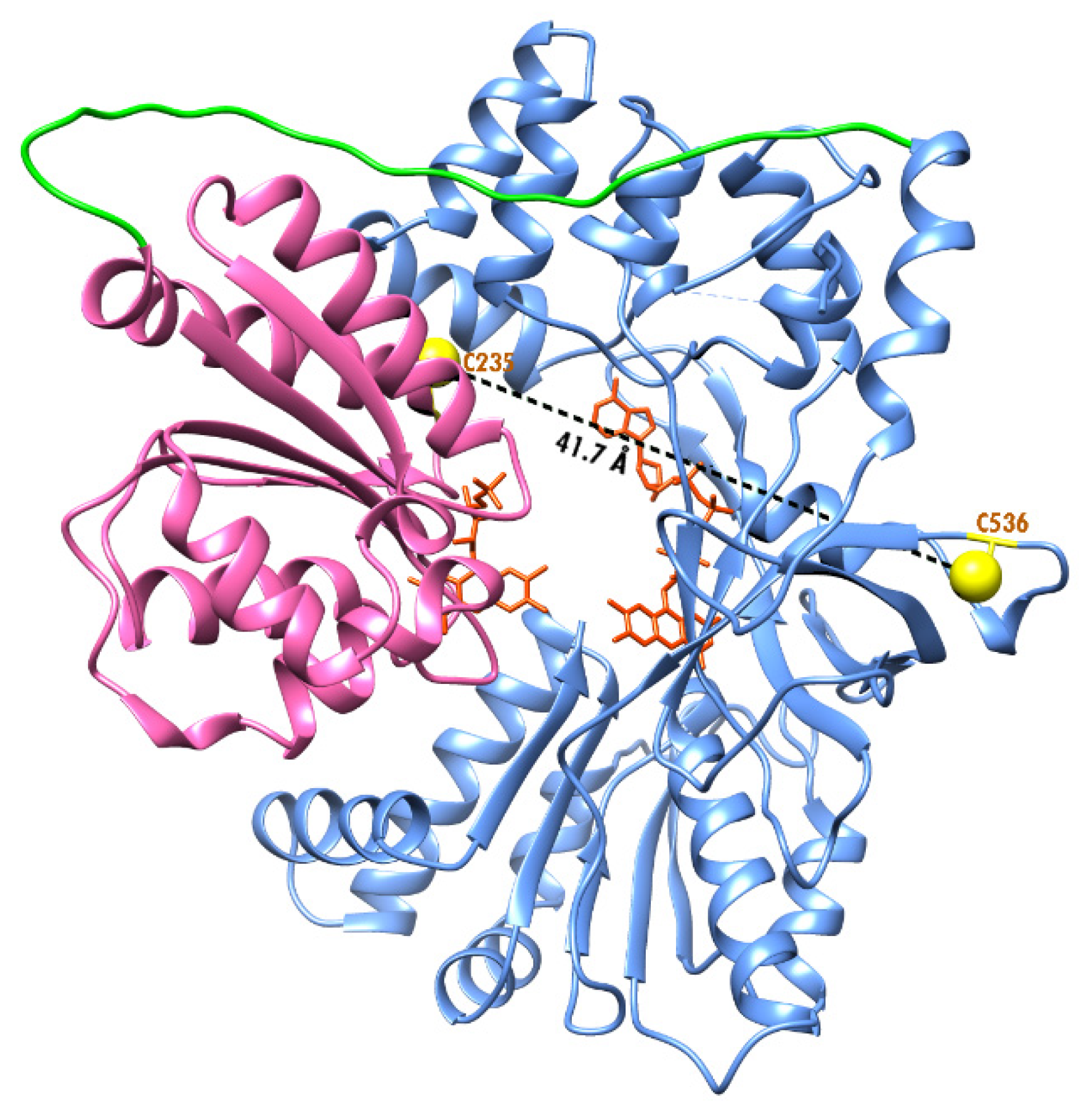
DpnI (New England Biolabs, Ipswich, MA, USA) digestion to remove the template strand prior to transformation to XL1-blue competent cell for amplification. C596S mutation was confirmed by DNA sequencing (Fisher Scientific, Waltham, MA, USA). The replacement was performed to limit the possible location of the incorporated fluorescent probes to Cys-235 and Cys-536 (see
Section 3.1). The purification methods were the same for the wild-type and C596S mutant. The vectors were transformed into
Escherichia coli Rosetta 2 (DE3) cells. Three liters of Lysogeny Broth medium complemented with 25 μg mL
−1 chloramphenicol and 50 μg mL
−1 kanamycin were inoculated with 20 mL from the culture. The cells were grown at 37 °C until the optical density of the culture at 600 nm reached 0.6~0.8. At this point, the temperature was set at 25 °C and 0.5 mM IPTG was added. After the incubation of the culture for 16 h, the cells were harvested by centrifugation at 5000 rpm for 20 min at 4 °C and resuspended in the Buffer A (50 mM Tris-HCl, 300 mM NaCl, pH 8.0) with 20 mM imidazole. After sonicating on ice for 30 min with a Model 450 sonicator (Branson Ultrasonics, Danbury, CT, USA), the cell debris was removed by ultracentrifugation. The clear lysate was loaded onto the column of Ni-NTA agarose (Qiagen, Germantown, MD, USA) and extensively washed with the same buffer. Modified CPR protein was eluted by Buffer A containing 250 mM imidazole, pH 8.0. After concentrating the protein to 2 mg mL
−1, its solution was dialyzed against 5 mM potassium phosphate buffer and applied onto a CHT ceramic hydroxyapatite column (Bio-Rad, Hercules, CA, USA). The fraction containing SbCPR was eluted by a linear phosphate gradient and then concentrated to ~30 mg/mL. Final purity was analyzed by SDS-PAGE, and the concentration was determined by Bradford assay (Bio-Rad).
2.3. Incorporation of Thiol-Reactive Fluorescent Probes
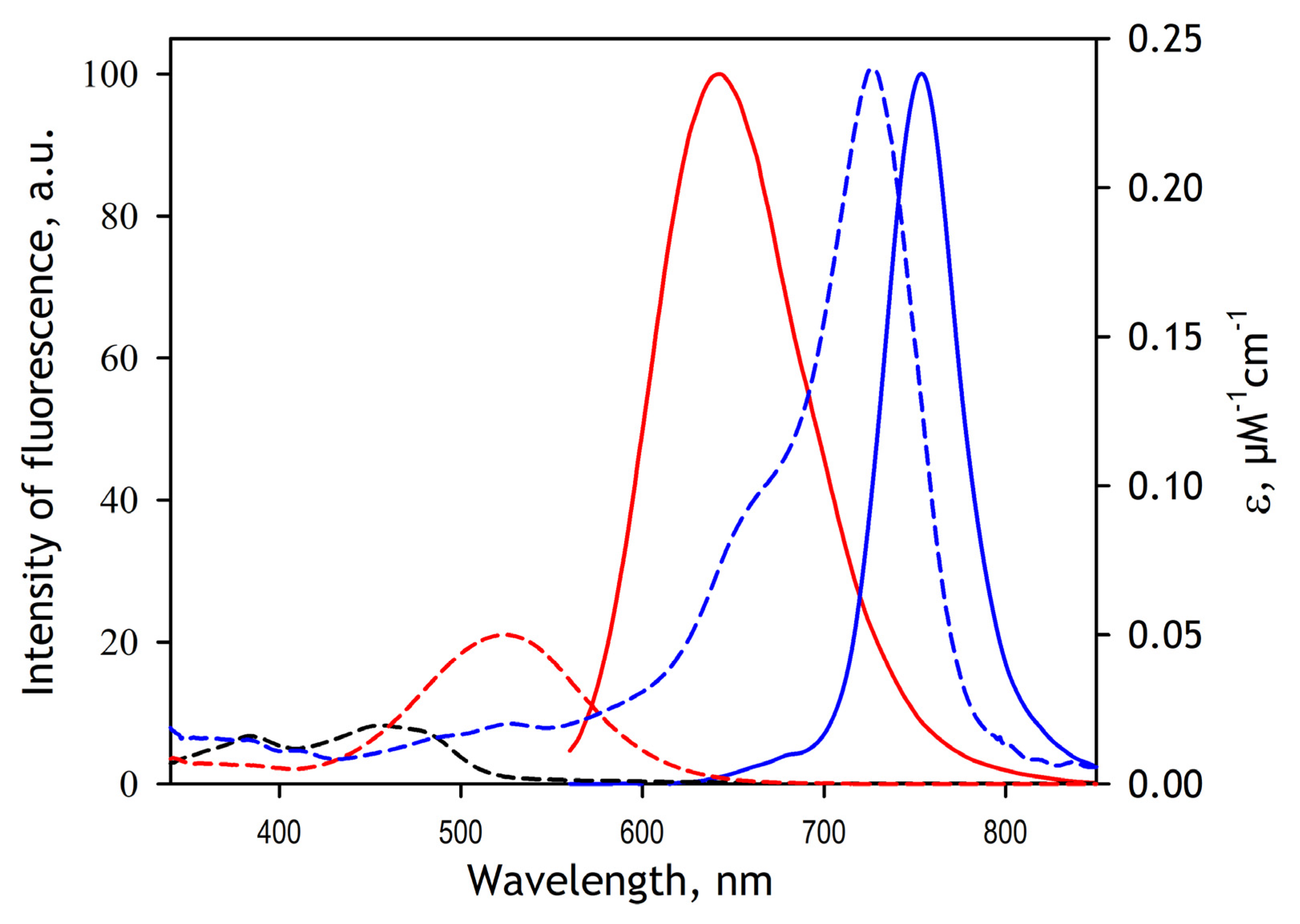
In this study, we used DY-520XL and DY731 fluorescent dyes manufactured by Dyomics GMBH, Jena, Germany) as FRET donor and acceptor fluorophores. Both probes were used as maleimide derivatives (product numbers 520XL-03 and 731-03, respectively). Incorporating these probes into SbCPR involves their attachment to the thiol groups of the cysteine residues of the protein. Prior to modification, SbCPR was stored in 125 mM K-phosphate buffer, pH 7.4, containing 2 mM TCEP (Storage Buffer). TCEP was removed by passing the protein solution through a spin-out column of Bio-Gel P6 desalting resin (Bio-Rad, Hercules, CA, USA) equilibrated with 125 mM K-phosphate buffer, pH 7.4. After diluting the protein with the same buffer to the concentration of 10 µM, a 3 mM solution of DY-520XL maleimide in acetone was added to the final concentration of 10 µM, and the solution was incubated at 4 °C under continuous stirring. The increase in the fluorescence of DY-520XL at 630 nm (excitation at 520 nm) in the process of modification was monitored to ensure reaction completion. After the stabilization of fluorescence in approximately one hour of incubation, an acetone solution of the second probe (DY731 maleimide) was added to the final concentration of 10 µM. The reaction was followed by monitoring a decrease in the fluorescence of DY-520XL. The process of the second modification required 3–4 h for completion. Finally, the reaction was terminated by adding reduced glutathione to the concentration of 1 mM. The protein was concentrated to 100–200 µM and passed through a spin-out column of Bio-Gel P6 equilibrated with 125 mM potassium phosphate buffer, pH 7.4 to remove glutathione adducts unreacted dyes. The stoichiometry of labeling by DY-520XL and DY-731 was determined based on the spectrum of absorbance of the modified protein. This calculation used the extinction coefficients of 0.05 µM−1 cm−1 at 520 nm and 0.24 µM−1 cm−1 at 736 nm for DY-520XL and DY-731, respectively, as specified by the manufacturer.
2.4. Rapid Kinetic Studies with Absorbance and Fluorescence Spectroscopy
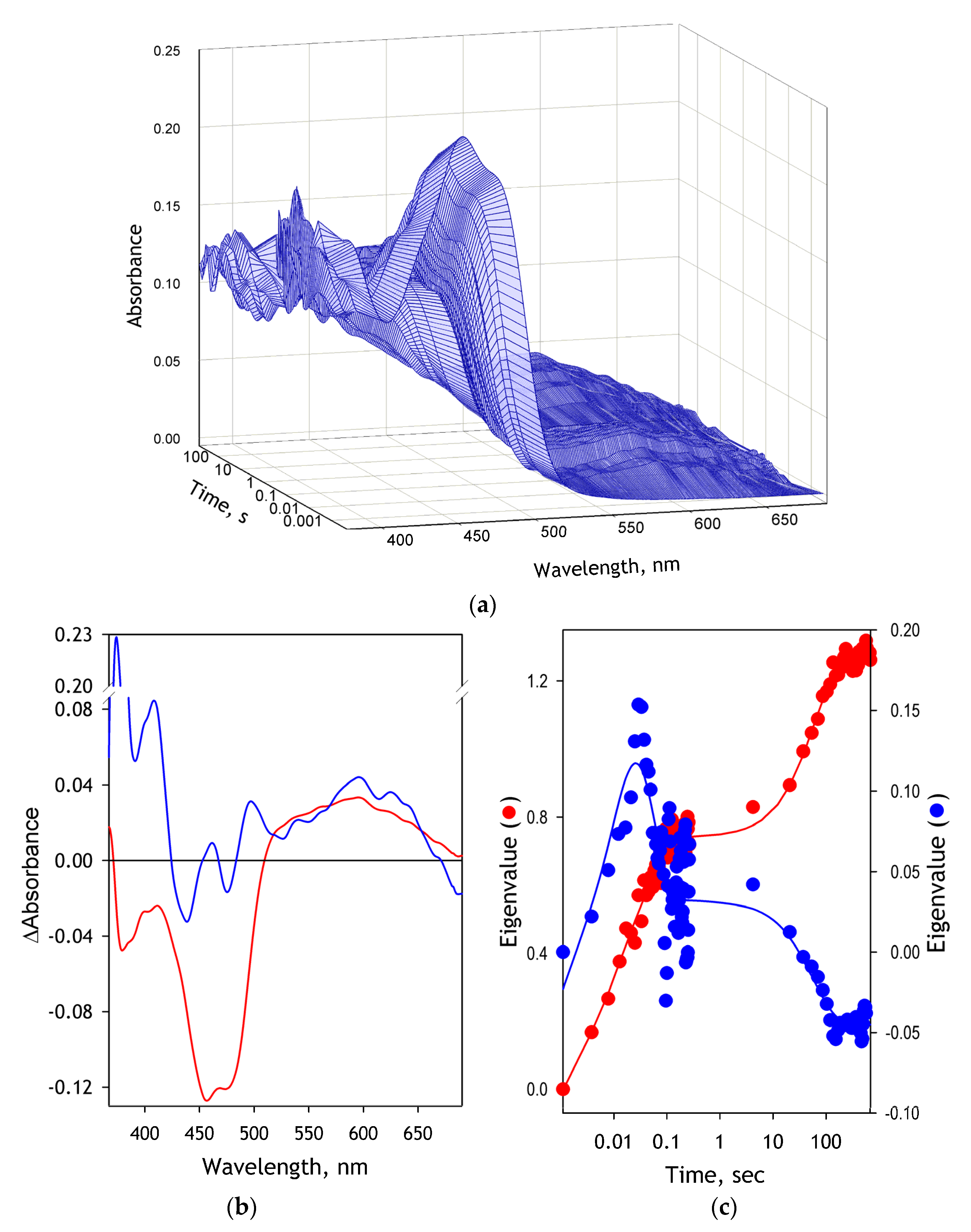
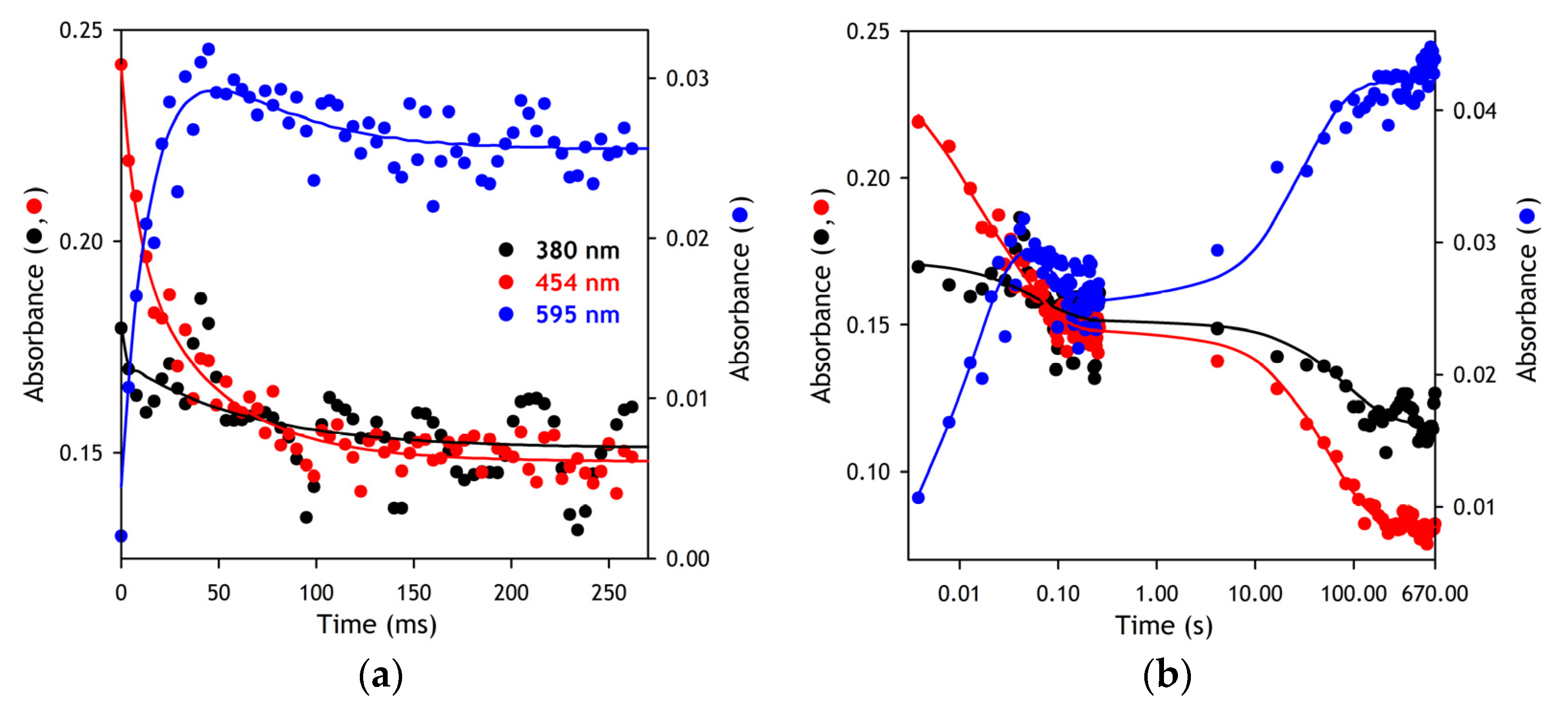
The kinetics of the NADPH-dependent reduction of SbCPR were studied with rapid-scanning stop-flow spectroscopy. The experiments were performed at 5 °C in 20 mM HEPES buffer, pH 7.4, containing an oxygen-scavenging system consisting of 60 mM glucose, 300 units/mL glucose oxidase, and 2000 units/mL catalase. The concentration of SbCPR and NADPH in the optical cell was equal to 20 μM and 200 μM, respectively. The solution of NADPH also contained 2 mM glucose-6-phosphate and 4 units/mL glucose-6-phosphate dehydrogenase, which were added to keep the concentration of NADPH constant. The stop-flow experiments were performed with the use of RX 2000 Rapid Mixing Stopped-flow Accessory manufactured by Applied Photophysics Ltd. (Leatherhead, Surrey, U.K.) connected to the master channel of an MC2000-2 two-channel CCD spectrometer (Ocean Optics, Inc., Dunedin, FL, USA) equipped with a custom-made thermostated cell holder and a PX-2 pulsed xenon lamp light source (Ocean Optics). The RX 2000 Accessory was custom modified to allow remote control of mixing from the data acquisition software. The absorbance spectra in the range of 320–700 nm were collected with the time intervals changing from 2 ms to 2 s per spectrum using custom data acquisition software.
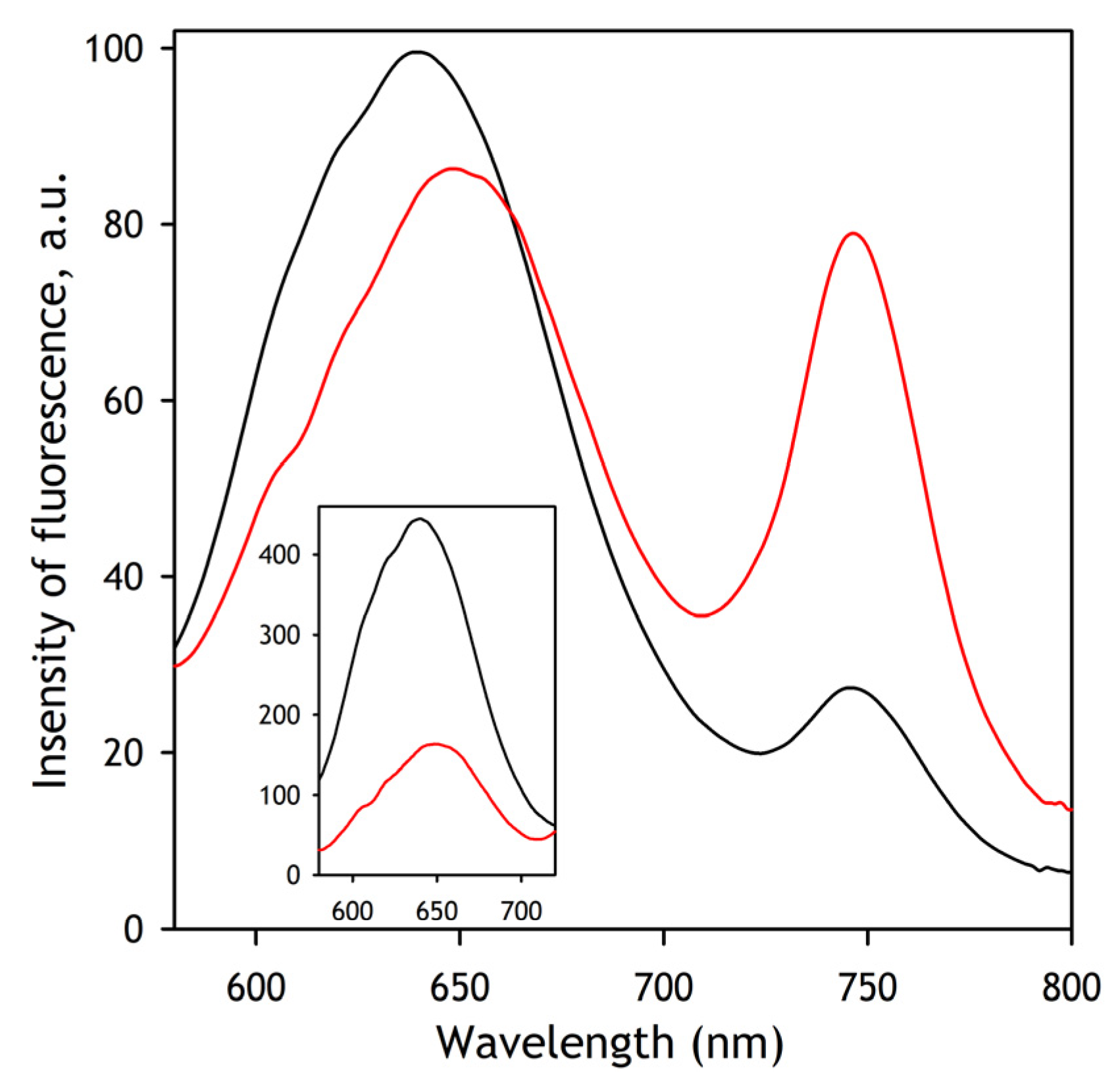
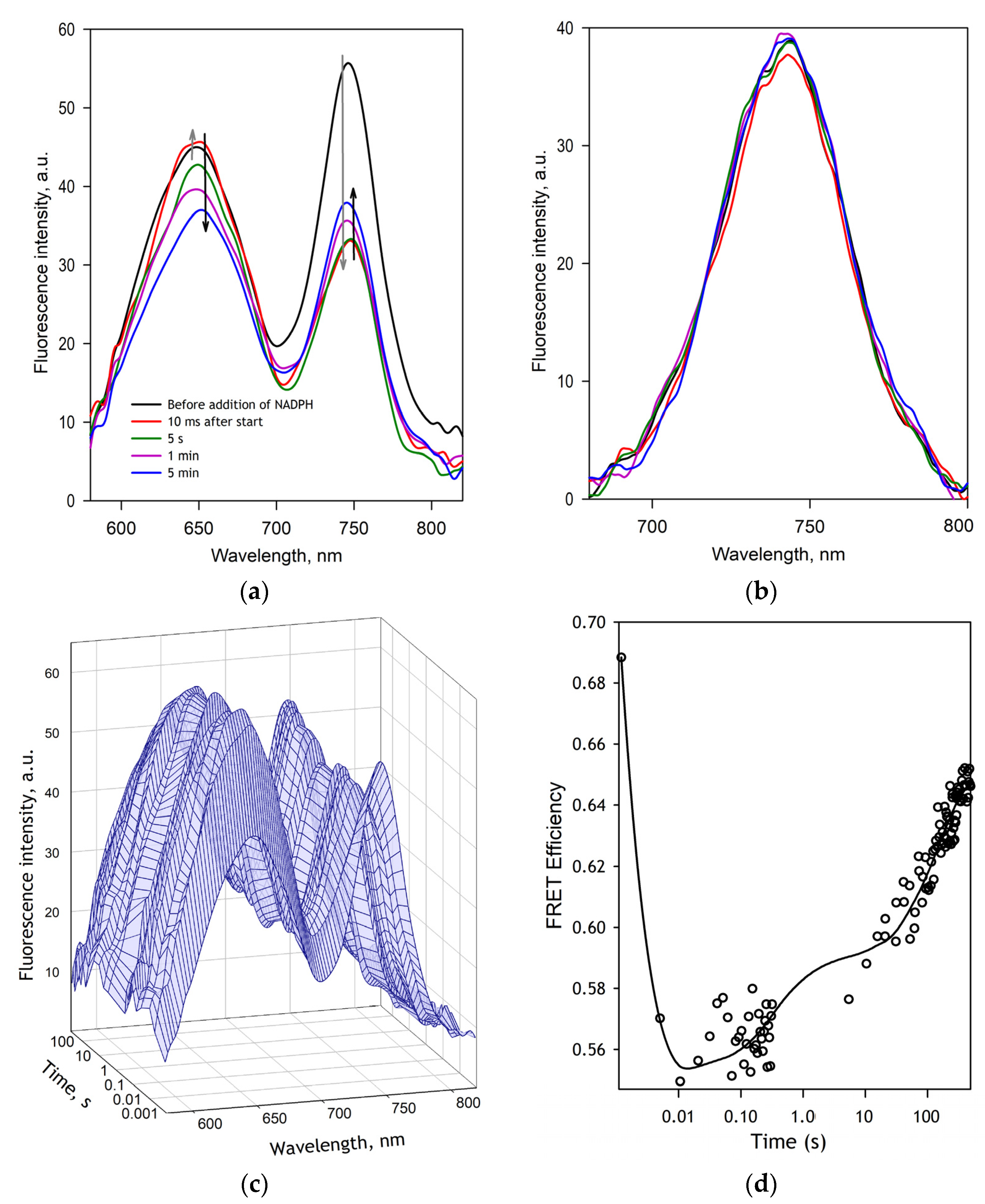
Rapid kinetics of the changes in SbCPR conformation during the reduction process were studied with FRET-based monitoring in a setup similar to that described above for the absorbance spectroscopy. In these experiments, the concentration of SbCPR-2DY and NADPH in the optical cell was equal to 5 and 100 µM, respectively, and the temperature was maintained at 5 °C. The composition of the other ingredients was the same as indicated above for the absorbance spectroscopy experiments. In these studies, the master channel of the MC2000-2 spectrometer was connected with a Vis-NIR 3 mm liquid light guide (Model 77635, Newport Corporation, Irvine, CA, USA) to the fluorescence window of the cell holder. The excitation light was provided with an M505F1 light-emitting diode (Thorlabs Inc., Newton, NJ, USA) emitting at 505 nm and functioning in a continuous wave mode. We used M617L3 light-emitting diode (Thorlabs Inc.) emitting at 617 nm as a light source in the experiments with direct excitation of the acceptor fluorophore. The fluorescence spectra were recorded in the range of 580–950 nm collected with the time intervals changing from 10 ms to 2 s per spectrum using custom data acquisition software.
2.5. Pressure Perturbation Experiments
Pressure-perturbation experiments were performed using a custom-built high-pressure optical cell [
47] connected to a manual pressure generator (High Pressure Equipment, Erie, PA, USA) capable of generating a pressure of up to 6000 bar. The emission spectra were recorded with an MC2000-2 spectrometer (Ocean Optics) connected with a Vis-NIR 3 mm liquid light guide (Model 77635, Newport Corporation, Irvine, CA, USA) to the fluorescence window of the high-pressure cell. The spectra were recorded in the 580–900 nm region with a step of 1 nm. All spectra were corrected for the changes in protein concentration due to pressure-dependent compression of water as described earlier [
48]. The excitation light was provided with an M505F1 light-emitting diode (Thorlabs Inc., Newton, NJ, USA) emitting at 505 nm in the continuous wave mode. The experiments were performed with 5 µM SbCPR-2DY at 25 °C in 20 mM Na-Hepes buffer. The experiments with the reduced SbCPR-2DY were carried out in the presence of an oxygen-scavenging system consisting of glucose oxidase (30 units/mL), 60 mM glucose, and 2000 units/mL catalase. The NADPH concentrations used in the experiments with the partially and fully reduced enzyme were equal to 5 and 100 µM, respectively. The experiments in the presence of 2′,5′-ADP were performed at 1 mM concentration of the latter.
2.6. Data Fitting
All data treatment and fitting, as well as the data acquisition in the absorbance and fluorescence spectroscopy experiments, were performed using our custom-designed SpectraLab software [
48]. The latest version of the software package is freely available on the author’s website [
49].
2.6.1. Interpretation of the Results of Rapid Scanning Absorbance Spectroscopy
The series of spectra obtained in the rapid scanning absorbance stop-flow experiments were subjected to the Principal Component Analysis (PCA), a linear algebra method commonly used to reduce the dimensionality of large datasets [
50]. It analyzes a set of M individual datasets (absorbance or fluorescence spectra in our case) of the dimensionality N (number data points in each spectrum). This dataset is used to construct the N × N covariance matrix, which is then transformed to find M-1 eigenvectors paired with M eigenvalues. The combination of each eigenvector with the corresponding set of eigenvalues is termed the Principal Component. The eigenvectors may be considered unified differences between the basis vector (the first spectrum in the series in our case) and other vectors (spectra) under analysis. The PCs are sorted based on their statistical significance. The first PC represents the most typical difference between the individual datasets (spectra), and the higher-order PCs contain the least significant deviations from the basis. This way, organizing information in PC allows reducing dimensionality without losing much information by discarding the components with low statistical significance. In practice, each dataset may be reconstituted with increasing accuracy by successive summarizing the basis vector and the eigenvectors multiplied by the respective eigenvalues. Thus, the set of eigenvalues deduced from the analysis of a spectral series reflects the changes in the amplitude of spectral alterations represented in the respective eigenvector, which might be considered a unified differential spectrum that characterizes the process under study. PCA is widely used for analyzing the results of rapid scanning kinetic experiments [
51,
52,
53,
54].
In our experiments, the first Principal Component yielded from this procedure typically covered over 98.5% of the total spectral changes. The time dependence of its eigenvalue was interpreted as reflecting the general kinetics of reduction. Approximation of these kinetics by a three-exponential equation was used to determine the kinetic constants of the individual phases of the reduction process.
2.6.2. Interpretation of the Results of Fluorescence Spectroscopy
The quantitative interpretation of FRET results was based on the application of PCA to a series of emission spectra recorded in rapid kinetics or pressure perturbation experiments. Approximation of the first principal vector (>98% of the observed changes) with a combination of the prototypical spectra of emission of DY-520XL and DY731 normalized proportionally to their quantum yields (see
Appendix A) allowed us to resolve the changes in the integral intensities of emission of each of the dyes and interpret them in terms of FRET efficiency. The quantum-yield-normalized intensities of the donor and acceptor fluorescence obtained in this way were used to determine the FRET efficiency according to the following equation:
where
Ia is the emission intensity of the acceptor,
Id is the residual donor emission in the presence of acceptor, and Φ
a and Φ
d are the quantum yields of the fluorescence of the acceptor and the donor, respectively.
Distances between the donor and acceptor fluorophores (
R) were calculated using the following equation:
where
R0 is the Förster distance calculated using the absorbance and emission spectra of the protein-bound donor and acceptor fluorophores. These calculations were performed using PhotoChemCad software [
55], assuming the values of the orientation factor (
κ) and the refractive index (
n) to be equal to 0.667 and 1.4, respectively.
2.6.3. Fitting of the Results of Pressure-Perturbation Experiments
The interpretation of the effect of pressure on protein equilibria in this article is based on the equation for the pressure dependence of the equilibrium constant ([
56], Equation (1)):
or in integral form, [
57] (p. 212, Equtation (9)):
where
Keq is the equilibrium constant of the reaction at pressure
P,
P½ is the pressure at which
Keq = 1 (“half pressure” of the conversion), Δ
V0 is the standard molar reaction volume, and
K0eq is the equilibrium constant extrapolated to zero pressure,
K0eq =
eP½ΔV0/RT. For the equilibrium
A ⇌
B and
Keq = [
B]/[
A], Equation (4) may be transformed into the following relationship:
where
C0 = [
A] + [
B]. To determine the Δ
V0 and
P½ parameters from the experimental datasets describing pressure-induced changes in the amplitude of a signal derived from the fluorescence spectra (
Ap), this equation was complemented with the offset (
A0) and scaling factor (
Amax) parameters and used in the following form:
Prior to the analysis, all spectra were corrected for the compression of the solvent [
48].
2.7. MALDI/TOF Analysis for the Fluorescence Dye Modified Peptides
Prior to MALDI/TOF analysis, the unmodified SbCPR-C596 and its adducts with monobromobimane (MBBr), DY-520XL and DY-731 (see
Appendix B) were subjected to SDS-PAGE. After staining the gel slabs, their fragments containing the SbCPR protein band were isolated and subjected to in-gel trypsinolysis, following the established protocol [
58]. The digested peptides were analyzed by MALDI/TOF MS. The peptide mass spectra were obtained using procedure and collection programs supplied by the manufacturer (Applied Biosystems, Waltham, MA, USA). The matrix, a-cyano-4-hydroxycinnamic acid, CHCA (Sigma-Aldrich, St. Louis, MO, USA), was prepared as a solution of 10 mg mL
−1 in 50% water/acetonitrile with 0.1% TFA. The matrix solution was mixed 1:1 with the trypsin digest, applied to the sample plate, and dried. Spectra were collected using a 4800 MALDI TOF/TOF Analyzer (Applied Biosystems, Waltham, MA, USA), using the data collection programs in the positive mode for MS spectra.
4. Discussion
Incorporating DY520-XL and DY731 fluorescence dyes into the FAD and FMN domains of CPR from Sorghum bicolor provided means for the direct observation of conformational rearrangements during the redox cycling of the enzyme. A close match of the kinetic constant derived from the absorbance and fluorescence spectroscopy assays in rapid scanning stop-flow experiments allowed us to track the changes in the average inter-probe distance during the flavoprotein reduction process. Furthermore, applying pressure-perturbation spectroscopy for portraying the conformational landscape of the enzyme, we were able to determine the positions of equilibrium between its open and closed conformations at different points of the electron-transfer pathway. These studies revealed the presence of several open protein sub-states that differ in the protein solvation pattern. Through unveiling the functional choreography of plant CPRs, our study provides the essential information for rational engineering these pivotal enzymes of the monolignol biosynthetic pathway.
The results of our rapid kinetics experiments suggest that the binding of NADPH to the enzyme is exceptionally fast and takes place during the dead time of the stop-flow device (~2 ms). Similar to that reported for human CPR [
62], the reduction of SbCPR by NADPH occurs without the intermittent occurrence of the detectable charge-transfer species. The first resolved kinetic phase is the formation of the blue (neutral) disemiquinone resulting from the interdomain electron transfer between FADH
2 and FMN immediately coupled to the charge-transfer step. However, the rate constant of this process (around 200 s
−1 at 5 °C, see
Table 1) is an order of magnitude higher than the values reported by Gutierez et al. for human CPR (20 s
−1 at 25 °C) [
62] and by Oprian and Coon for rabbit CPR (28 s
−1 at unspecified temperature) [
63]. Thus, the interdomain electron transfer in SbCPR occurs much faster than in the mammalian enzymes. This observation is consistent with the higher flexibility of the interdomain loop in plant CPRs as compared to the mammalian orthologs, which is suggested by their X-ray structures [
7,
11].
The second kinetic step presumably corresponds to establishing a transient equilibrium of the red and blue disemiquinones with the other two-electron reduced states, (FAD, FMNH
2) and (FADH
2, FMN). It has a rate constant of around 20 s
−1 (
Table 1), which is also much higher than those reported for the human (3.7 s
−1 [
62]) and the rabbit (5.4 s
−1 [
63]) enzymes. Notably, the amplitude of the 595 nm band and, respectively, the fractional content of the blue semiquinone species at the end of this phase is considerably higher in SbCPR than that observed with the mammalian reductases.
Interestingly, the kinetics of the reduction of SbCPR reveals a noticeable difference with that reported for another plant CPR, ATR2 enzyme from
Arabidopsis thaliana. Studying this enzyme, Whitelaw and co-authors reported a resolution of the hydride transfer step (425 s
−1 at 6 °C) from the interdomain electron transfer stage (49 s
−1) [
28]. Thus, according to our results, both of these electron transfer events appear to be considerably faster in SbCPR than in ATR2. Furthermore, the fractional content of the blue semiquinone state in the transient equilibrium mixture established after the second phase of reduction is markedly higher in SbCPR than in ATR2, similar to what is observed in the comparison of SbCPR with the mammalian reductases.
The third, extremely slow, kinetic phase of the reduction corresponds to a partial transition of the enzyme to the four-electron reduced state followed by a comproportionation between the two- and four-electron reduced molecules leading to the three-electron reduced enzyme ((FADH
2, FADH
•) ⇌ (FMNH
•, FMNH
2)), the apparent final state. This incomplete reducibility of CPR and the formation of a three-electron reduced state through a comproportionation reaction has already been reported for mammalian reductases [
64] and the flavoprotein domain of the bacterial P450BM-3 [
61].
The changes in FRET efficiency in SbCPR-2DY during its NADPH-dependent reduction are in good agreement with the kinetics of changes in the redox state of the enzyme. The kinetic curves registered in our fluorescence stop-flow experiments obey three-exponential kinetics with the rate constants closely similar to those obtained with absorbance spectroscopy. According to our results, the first resolved phase of reduction that leads to the disemiquinone state is associated with an increase in the averaged inter-probe distance by 4.1 Å. However, the subsequent equilibration between several two-electron reduced states and the further reduction of the enzyme results in the opposite direction of the changes so that the averaged distance observed in the three-electron reduced enzyme is only 1.7 Å larger than in the oxidized enzyme (
Table 1).
These results agree with multiple previous reports that demonstrated a transition from the closed conformation to a more open state upon the interdomain electron transfer event and the formation of the disemiquinone state (see [
27] for a review). Furthermore, there are strong indications of the predominance of the closed conformations in the three- and four-electron reduced enzyme [
20,
21,
23,
26], which is also consistent with our results.
It must be noted that the inter-probe distances estimated in our FRET kinetics experiments do not reflect the distances characteristic of any discrete protein conformations. They rather represent the averages over multiple conformational states, and their changes reflect the redox-state-dependent displacement of equilibrium between these various conformations. To explore better the conformational landscape of SbCPR and characterize its changes in the redox cycling of the enzyme, we used hydrostatic pressure as a tool for displacing the protein equilibria. When combined with such tools for detecting protein structural rearrangements as FRET, the pressure perturbation strategy allows determining the positions of its conformational equilibria in different redox states, assessing the changes in the protein interactions with solvent in its redox cycling, and estimating the inter-probe distances characteristic to the end-states of its conformational breathing.
According to our results, in all four studied states of the enzyme—oxidized ligand-free, oxidized ADP-bound, two-electron, and four-electron reduced—the enzyme exists in the equilibrium between its more closed and more open states. The inter-probe distance in the closed state is not affected by either reduction or the interactions with 2′,5′-ADP (the analog of the nucleotide cofactor). It is estimated to be around 38–40 Å in all four states of the enzyme (
Table 2). In contrast, the preferential conformation of the open state exhibits a pronounced change during the redox cycling. While the changes in the inter-probe distance associated with the conformational breathing of the oxidized enzyme are as small as 2 Å, the binding of ADP to SbCPR and its subsequent two-electron reduction increases the distance between the probes in the open conformation to 46–50 Å (
Table 2). In agreement with the previous observations [
20,
21,
23,
26], the further reduction of the enzyme partially reverts this change and decreases the amplitude of the conformational motion to 4 Å.
A unique feature of the pressure-perturbation approach is its ability to reveal conformational equilibria’s endpoints. Suppose that the inter-probe distances observed at any particular pressure represent weighted averages over a population of molecules existing in equilibrium between the low-pressure and pressure-promoted conformational states. Hence, the inter-probe distances estimated by extrapolating pressure dependencies to infinitely high and infinitely low pressures correspond to those characterizing the definite states (or ensembles of states with pressure-insensitive interconversion) representing the endpoints of conformational equilibrium. Therefore, comparing the end states of pressure-dependent transitions of SbCPR at different points of its redox cycle allows probing if the interactions of the enzyme with the nucleotide cofactor or reduction of its flavins results in an emergence of a new conformational state not present in the oxidized enzyme. The remarkable difference of the inter-probe-distance in the pressure-promoted end-state of the oxidized enzyme with those observed in SbCPR-ADP complex and two-electron reduced enzyme suggests that the binding of the nucleotide cofactor is a necessary prerequisite for the wide opening of the enzyme. This inference is consistent with a dramatic difference of the ΔV0 characteristic to the oxidized enzyme (89 mL/mol) with the ΔV0 values exhibited by SbCPR in all other probed states. According to this analysis, the nature of the low-amplitude conformational breathing observed in oxidized SbCPR is entirely different from the transitions between the closed and widely open conformations, which become possible only after binding the nucleotide cofactor.
However, it should be taken into account that the changes in the distance between the two protein-incorporated probes monitored in our experiments do not reflect the complete picture of the structural rearrangements, which may involve complex rotational and translational motions of different parts of the protein molecule. Therefore, the inter-probe distance may not be considered a full-fledged measure of the “degree of openness” of the enzyme molecule. Consequently, the actual variations in the accessibility of the FMN domain for interactions with the heme protein acceptor may not be exactly proportional to the variations in the inter-probe distances detected in our experiments.
The most notable and nontrivial conclusion derived from the fitting of the pressure dependencies of the FRET efficiency to Equation (6) is that the closed conformation of the enzyme heavily predominates in all four studied states of the enzyme at ambient pressure. According to the constant of equilibrium determined for the oxidized enzyme, the fraction of the closed form accounts for 99.6% of its total content. Despite a significant increase in the amplitude of the conformational breathing upon the binding of the nucleotide cofactor, its effect on the position of the conformation equilibrium is insignificant (99% of the closed state at 1 bar). However, the reduction of the enzyme with the formation of the disemiquinone displaces the equilibrium appreciably, and the fraction of the closed state decreases to 96%. Surprisingly, despite decreasing the amplitude of motions, the further reduction of SbCPR provokes the further opening of the enzyme so that the fraction of its closed form decreases to 89%.
It has to be noted that the high abundance of the closed conformation of the enzyme in our experiments is furthered by the low ionic strength of the buffer used in our studies (20 mM Na-HEPES,
I = 11.6 mM). The conformational equilibrium in CPR enzymes is known to be critically affected by ionic strength, and the abundance of open conformation is considerably increased at high salt concentrations [
19,
24,
25]. This strong ionic strength dependence is caused by the predominant role of charge pairing contacts in the inter-domain interactions [
12,
65]. The use of the low-ionic-strength media in our experiments was dictated by an objective to accurately determine FRET efficiency in the low-pressure (closed) end-state by displacing the
P½ of the opening transition to higher pressures. It is worth noting that the cytoplasmic ionic strength in cells of plants grown at normal soil salinity varies within 100–200 mM limits [
66], which is much higher than the ionic strength of our buffer (11.6 mM). Therefore, the actual position of the SbCPR conformational equilibrium in vivo must be more shifted towards the open state than observed in our experiments.
Another remarkable observation is a dramatic change in the Δ
V0 of the opening transition upon the binding of 2′,5′-ADP and the enzyme reduction. If in the oxidized SbCPR the volume change is as large as −88 mL/mol, the binding of the nucleotide cofactor decreases it (by absolute value) to −33 mL/mol. The subsequent electron transfer to the flavins and formation of the disemiquinone state further decreases Δ
V0 to −18 mL/mol. This observation suggests that the binding of the nucleotide cofactor and two-electron reduction results in some additional hydration of the enzyme, which decreases the changes in protein interactions with solvent necessary for the transition to the open conformation. Suppose we hypothesize that the predominant part of the volume change is originated from the electrostriction of water on the newly opened charges after breaking salt bridges. In that case, we can assume that the opening of the oxidized enzyme involves the dissociation of four salt links. That is calculated from the assumption that the solvation of a single-charged ion incurs the volume change of −10 mL/mol [
31]. In contrast, there is only one salt bridge to break for the opening transition of the two-electron reduced enzyme (Δ
V0 = −18 mL/mol).
The analysis of the recently resolved X-ray structure of CPR from the
Candida tropicalis yeast identified four salt-bridges connecting the FAD and FMN domains in the closed conformation of the enzyme [
12]. Comparing this structure with our structural model of SbCPR, we found that three of these charge pairs—D125/R515, E157/K669, and E193/R367—are retained in the sorghum enzyme, where they correspond to the pairs D168/R544, E203/K693, and D238/K414. The fourth one (K57/D338) has no analog in SbCPR despite the conservation of K57 residue, which corresponds to K96 in SbCPR. However, instead, this residue may be a part of a switching salt-link fork between D260 in the connecting loop and K96 and K126 in the FMN domain. Dissociation or switching these salt links may affect the flexibility of the connecting loop and thus modulate the interdomain interactions. Hypothetically, the dissociation of these salt bridges may be involved in the pressure-induced opening of the ligand-free oxidized enzyme. A decrease in the Δ
V0 of the opening transition upon the binding of the nucleotide cofactor and NADPH-dependent reduction may indicate that the interactions of the enzyme with 2′5′-ADP or NADPH promote the dissociation of some of these salt links. Further investigation of this system of molecular tethering and its linkage to the enzyme redox-state by a combination of site-directed mutagenesis and pressure-perturbation FRET spectroscopy may give more insight into the mechanisms controlling the inter-domain interactions in SbCPR.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}