
How Do Hexokinases Inhibit Receptor-Mediated Apoptosis?


Abstract
Simple Summary
Abstract
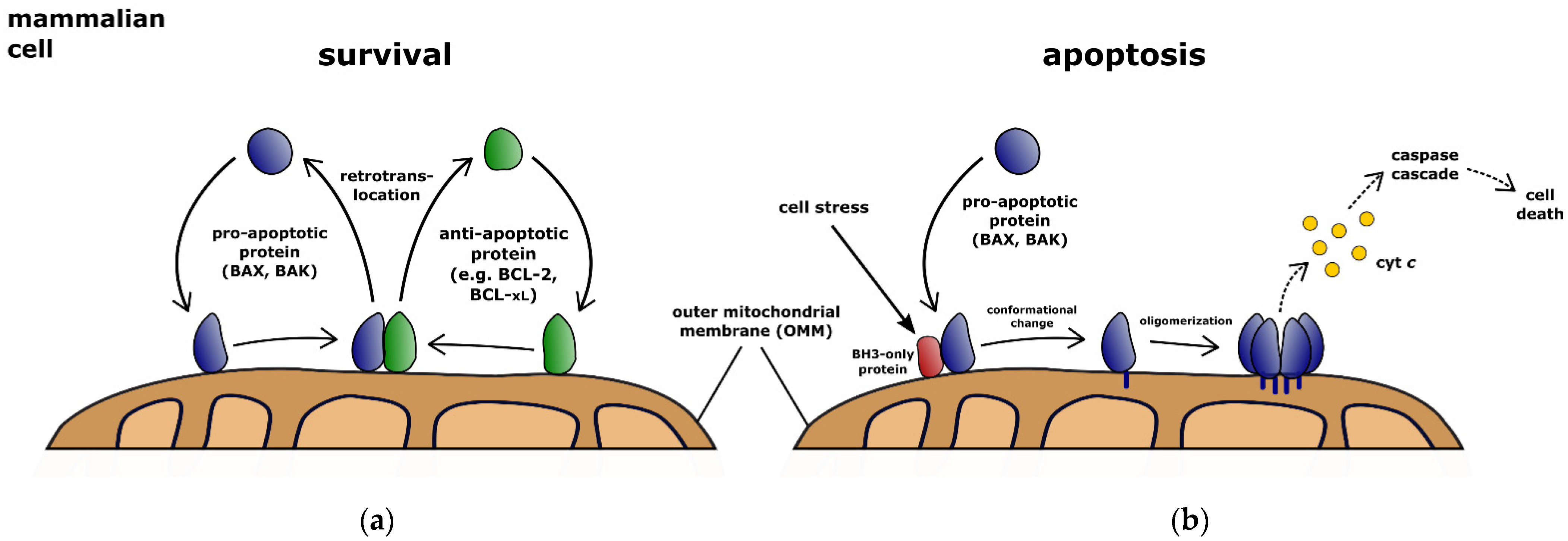
1. Pro-Apoptotic BCL-2 Activities Control the Molecular Decision to Apoptosis
2. Dynamic Retrotranslocation Determines the Effective BCL-2 Protein Pool
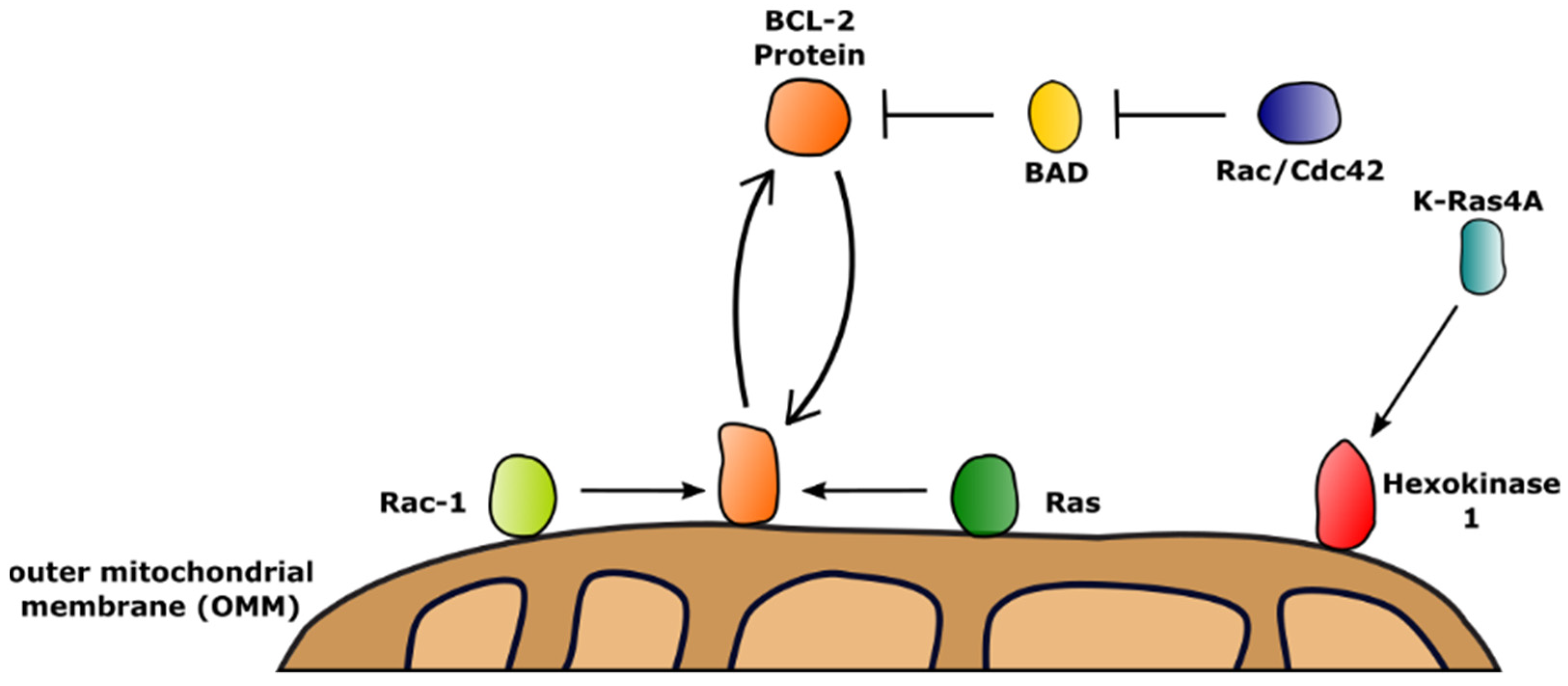
3. Membrane Receptors Guide the Function of BCL-2 Proteins by Activation of Downstream GTPases

{kind=link}
{kind=link}
{kind=link}
| GTPase Superfamily | Mode of Action |
|---|---|
| Ras | Raf-1-dependent phosphorylation of pro-apoptotic BAD/BIM [47,48,49] |
| Ras binding to BCL-2, increasing its anti-apoptotic effect [54] | |
| Activation of hexokinase I by K-Ras4A binding [55] | |
| Rho | Rac-1 binding to BCL-2, increasing its anti-apoptotic effect [50] |
| PAK-dependent phosphorylation of pro-apoptotic BAD by Rac/Cdc42 [50] |
4. Hexokinases: At the Crossroads between Glucose Metabolism and Apoptosis
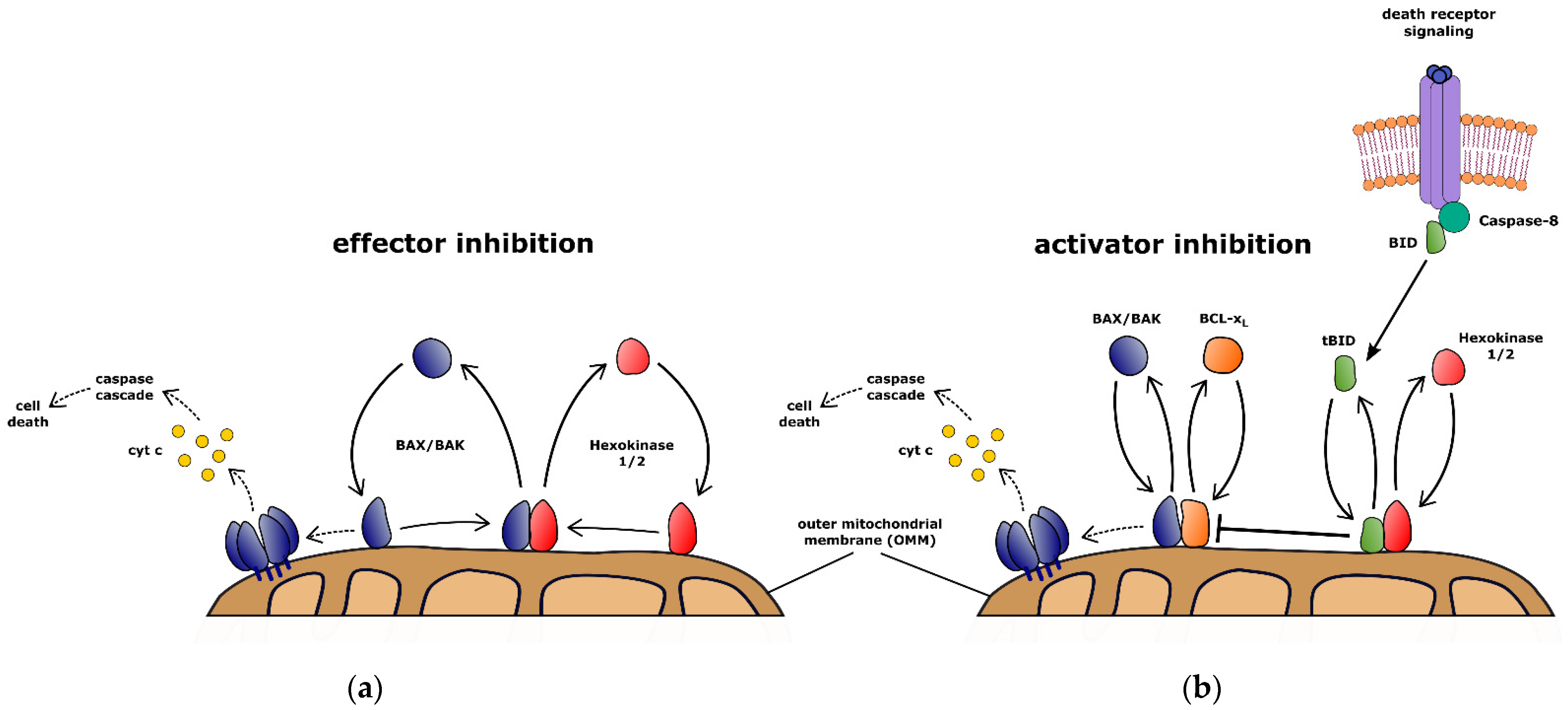
5. Hexokinase-Dependent Retrotranslocation Protects Cells against Extrinsic Apoptosis
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sulston, J.E.; Horvitz, H.R. Post-Embryonic Cell Lineages of the Nematode, Caenorhabditis Elegans. Dev. Biol. 1977, 56, 110–156. [Google Scholar] [CrossRef]
- Zychlinsky, A.; Prevost, M.C.; Sansonetti, P.J. Shigella Flexneri Induces Apoptosis in Infected Macrophages. Nature 1992, 358, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Clavería, C.; Giovinazzo, G.; Sierra, R.; Torres, M. Myc-Driven Endogenous Cell Competition in the Early Mammalian Embryo. Nature 2013, 500, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wide-Ranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Strasser, A.; McDunn, J.E.; Swanson, P.E. Cell Death. N. Engl. J. Med. 2009, 361, 1570–1583. [Google Scholar] [CrossRef]
- Oltvai, Z.N.; Milliman, C.L.; Korsmeyer, S.J. Bcl-2 Heterodimerizes in Vivo with a Conserved Homolog, Bax, That Accelerates Programmed Cell Death. Cell 1993, 74, 609–619. [Google Scholar] [CrossRef]
- Shimizu, S.; Narita, M.; Tsujimoto, Y. Bcl-2 Family Proteins Regulate the Release of Apoptogenic Cytochrome c by the Mitochondrial Channel VDAC. Nature 1999, 399, 483–487. [Google Scholar] [CrossRef]
- Eskes, R.; Antonsson, B.; Osen-Sand, A.; Montessuit, S.; Richter, C.; Sadoul, R.; Mazzei, G.; Nichols, A.; Martinou, J.C. Bax-Induced Cytochrome c Release from Mitochondria Is Independent of the Permeability Transition Pore but Highly Dependent on Mg2+ Ions. J. Cell Biol. 1998, 143, 217–224. [Google Scholar] [CrossRef]
- Martinou, I.; Desagher, S.; Eskes, R.; Antonsson, B.; André, E.; Fakan, S.; Martinou, J.C. The Release of Cytochrome c from Mitochondria during Apoptosis of NGF-Deprived Sympathetic Neurons Is a Reversible Event. J. Cell Biol. 1999, 144, 883–889. [Google Scholar] [CrossRef]
- Potts, M.B.; Vaughn, A.E.; McDonough, H.; Patterson, C.; Deshmukh, M. Reduced Apaf-1 Levels in Cardiomyocytes Engage Strict Regulation of Apoptosis by Endogenous XIAP. J. Cell Biol. 2005, 171, 925–930. [Google Scholar] [CrossRef]
- Ichim, G.; Lopez, J.; Ahmed, S.U.; Muthalagu, N.; Giampazolias, E.; Delgado, M.E.; Haller, M.; Riley, J.S.; Mason, S.M.; Athineos, D.; et al. Limited Mitochondrial Permeabilization Causes DNA Damage and Genomic Instability in the Absence of Cell Death. Mol. Cell 2015, 57, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Parsons, M.J.; Llambi, F.; Bouchier-Hayes, L.; Connell, S.; Muñoz-Pinedo, C.; Green, D.R. Resistance to Caspase-Independent Cell Death Requires Persistence of Intact Mitochondria. Dev. Cell 2010, 18, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; O’Neill, K.L.; Li, J.; Zhou, W.; Han, N.; Pang, X.; Wu, W.; Struble, L.; Borgstahl, G.; Liu, Z.; et al. BH3-Only Proteins Target BCL-XL/MCL-1, Not BAX/BAK, to Initiate Apoptosis. Cell Res. 2019, 29, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Llambi, F.; Moldoveanu, T.; Tait, S.W.G.; Bouchier-Hayes, L.; Temirov, J.; McCormick, L.L.; Dillon, C.P.; Green, D.R. A Unified Model of Mammalian BCL-2 Protein Family Interactions at the Mitochondria. Mol. Cell 2011, 44, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Roucou, X.; Montessuit, S.; Antonsson, B.; Martinou, J.-C. Bax Oligomerization in Mitochondrial Membranes Requires TBid (Caspase-8-Cleaved Bid) and a Mitochondrial Protein. Biochem. J. 2002, 368, 915–921. [Google Scholar] [CrossRef]
- Wei, M.C.; Lindsten, T.; Mootha, V.K.; Weiler, S.; Gross, A.; Ashiya, M.; Thompson, C.B.; Korsmeyer, S.J. TBID, a Membrane-Targeted Death Ligand, Oligomerizes BAK to Release Cytochrome c. Genes Dev. 2000, 14, 2060–2071. [Google Scholar] [CrossRef]
- Schleicher, R.I.; Reichenbach, F.; Kraft, P.; Kumar, A.; Lescan, M.; Todt, F.; Göbel, K.; Hilgendorf, I.; Geisler, T.; Bauer, A.; et al. Platelets Induce Apoptosis via Membrane-Bound FasL. Blood 2015, 126, 1483–1493. [Google Scholar] [CrossRef]
- Hinds, M.G.; Lackmann, M.; Skea, G.L.; Harrison, P.J.; Huang, D.C.S.; Day, C.L. The Structure of Bcl-w Reveals a Role for the C-Terminal Residues in Modulating Biological Activity. EMBO J. 2003, 22, 1497–1507. [Google Scholar] [CrossRef]
- Suzuki, M.; Youle, R.J.; Tjandra, N. Structure of Bax: Coregulation of Dimer Formation and Intracellular Localization. Cell 2000, 103, 645–654. [Google Scholar] [CrossRef]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J.; et al. Structure of Bcl-XL-Bak Peptide Complex: Recognition between Regulators of Apoptosis. Science 1997, 275, 983–986. [Google Scholar] [CrossRef]
- Jeong, S.-Y.; Gaume, B.; Lee, Y.-J.; Hsu, Y.-T.; Ryu, S.-W.; Yoon, S.-H.; Youle, R.J. Bcl-x(L) Sequesters Its C-Terminal Membrane Anchor in Soluble, Cytosolic Homodimers. EMBO J. 2004, 23, 2146–2155. [Google Scholar] [CrossRef] [PubMed]
- Edlich, F.; Banerjee, S.; Suzuki, M.; Cleland, M.M.; Arnoult, D.; Wang, C.; Neutzner, A.; Tjandra, N.; Youle, R.J. Bcl-x(L) Retrotranslocates Bax from the Mitochondria into the Cytosol. Cell 2011, 145, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Edlich, F. BCL-2 Proteins and Apoptosis: Recent Insights and Unknowns. Biochem. Biophys. Res. Commun. 2018, 500, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Edlich, F. The Great Migration of Bax and Bak. Mol. Cell. Oncol. 2015, 2, e995029. [Google Scholar] [CrossRef]
- Todt, F.; Cakir, Z.; Reichenbach, F.; Emschermann, F.; Lauterwasser, J.; Kaiser, A.; Ichim, G.; Tait, S.W.G.; Frank, S.; Langer, H.F.; et al. Differential Retrotranslocation of Mitochondrial Bax and Bak. EMBO J. 2015, 34, 67–80. [Google Scholar] [CrossRef]
- Schellenberg, B.; Wang, P.; Keeble, J.A.; Rodriguez-Enriquez, R.; Walker, S.; Owens, T.W.; Foster, F.; Tanianis-Hughes, J.; Brennan, K.; Streuli, C.H.; et al. Bax Exists in a Dynamic Equilibrium between the Cytosol and Mitochondria to Control Apoptotic Priming. Mol. Cell 2013, 49, 959–971. [Google Scholar] [CrossRef]
- Edlich, F.; Martinou, J.-C. Bcl-2 Protein Interplay on the Outer Mitochondrial Membrane. In Mitochondria and Cell Death; Springer: New York, NY, USA, 2016; pp. 69–83. ISBN 978-1-4939-3612-0. [Google Scholar]
- Todt, F.; Cakir, Z.; Reichenbach, F.; Youle, R.J.; Edlich, F. The C-Terminal Helix of Bcl-x(L) Mediates Bax Retrotranslocation from the Mitochondria. Cell Death Differ. 2013, 20, 333–342. [Google Scholar] [CrossRef]
- Reichenbach, F.; Wiedenmann, C.; Schalk, E.; Becker, D.; Funk, K.; Scholz-Kreisel, P.; Todt, F.; Wolleschak, D.; Döhner, K.; Marquardt, J.U.; et al. Mitochondrial BAX Determines the Predisposition to Apoptosis in Human AML. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 4805–4816. [Google Scholar] [CrossRef]
- Funk, K.; Czauderna, C.; Klesse, R.; Becker, D.; Hajduk, J.; Oelgeklaus, A.; Reichenbach, F.; Fimm-Todt, F.; Lauterwasser, J.; Galle, P.R.; et al. BAX Redistribution Induces Apoptosis Resistance and Selective Stress Sensitivity in Human HCC. Cancers 2020, 12, 1437. [Google Scholar] [CrossRef]
- Marquardt, J.U.; Edlich, F. Predisposition to Apoptosis in Hepatocellular Carcinoma: From Mechanistic Insights to Therapeutic Strategies. Front. Oncol. 2019, 9, 1421. [Google Scholar] [CrossRef]
- Lauterwasser, J.; Todt, F.; Zerbes, R.M.; Nguyen, T.N.; Craigen, W.; Lazarou, M.; van der Laan, M.; Edlich, F. The Porin VDAC2 Is the Mitochondrial Platform for Bax Retrotranslocation. Sci. Rep. 2016, 6, 32994. [Google Scholar] [CrossRef] [PubMed]
- Andreu-Fernández, V.; García-Murria, M.J.; Bañó-Polo, M.; Martin, J.; Monticelli, L.; Orzáez, M.; Mingarro, I. The C-Terminal Domains of Apoptotic BH3-Only Proteins Mediate Their Insertion into Distinct Biological Membranes. J. Biol. Chem. 2016, 291, 25207–25216. [Google Scholar] [CrossRef] [PubMed]
- García-Murria, M.J.; Duart, G.; Grau, B.; Diaz-Beneitez, E.; Rodríguez, D.; Mingarro, I.; Martínez-Gil, L. Viral Bcl2s’ Transmembrane Domain Interact with Host Bcl2 Proteins to Control Cellular Apoptosis. Nat. Commun. 2020, 11, 6056. [Google Scholar] [CrossRef] [PubMed]
- Grunicke, H.H.; Maly, K. Role of GTPases and GTPase Regulatory Proteins in Oncogenesis. Crit. Rev. Oncog. 1993, 4, 389–402. [Google Scholar]
- Li, H.; Yao, X.-Q.; Grant, B.J. Comparative Structural Dynamic Analysis of GTPases. PLoS Comput. Biol. 2018, 14, e1006364. [Google Scholar] [CrossRef]
- Oldham, W.M.; Hamm, H.E. Heterotrimeric G Protein Activation by G-Protein-Coupled Receptors. Nat. Rev. Mol. Cell Biol. 2008, 9, 60–71. [Google Scholar] [CrossRef]
- Wingler, L.M.; Lefkowitz, R.J. Conformational Basis of G Protein-Coupled Receptor Signaling Versatility. Trends Cell Biol. 2020, 30, 736–747. [Google Scholar] [CrossRef]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.-G.; Schiöth, H.B. The G-Protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef]
- Yanamadala, V.; Negoro, H.; Denker, B.M. Heterotrimeric G Proteins and Apoptosis: Intersecting Signaling Pathways Leading to Context Dependent Phenotypes. Curr. Mol. Med. 2009, 9, 527–545. [Google Scholar] [CrossRef]
- Revankar, C.M.; Vines, C.M.; Cimino, D.F.; Prossnitz, E.R. Arrestins Block G Protein-Coupled Receptor-Mediated Apoptosis. J. Biol. Chem. 2004, 279, 24578–24584. [Google Scholar] [CrossRef]
- Kook, S.; Zhan, X.; Cleghorn, W.M.; Benovic, J.L.; Gurevich, V.V.; Gurevich, E.V. Caspase-Cleaved Arrestin-2 and BID Cooperatively Facilitate Cytochrome c Release and Cell Death. Cell Death Differ. 2014, 21, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Leloup, C.; Michaelson, D.M.; Fisher, A.; Hartmann, T.; Beyreuther, K.; Stein, R. M1 Muscarinic Receptors Block Caspase Activation by Phosphoinositide 3-Kinase- and MAPK/ERK-Independent Pathways. Cell Death Differ. 2000, 7, 825–833. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Graham, E.S.; Woo, K.K.; Aalderink, M.; Fry, S.; Greenwood, J.M.; Glass, M.; Dragunow, M. M1 Muscarinic Receptor Activation Mediates Cell Death in M1-HEK293 Cells. PLoS ONE 2013, 8, e72011. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.M.; Fuentes, G.; Rausell, A.; Valencia, A. The Ras Protein Superfamily: Evolutionary Tree and Role of Conserved Amino Acids. J. Cell Biol. 2012, 196, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Syrovatkina, V.; Alegre, K.O.; Dey, R.; Huang, X.-Y. Regulation, Signaling and Physiological Functions of G-Proteins. J. Mol. Biol. 2016, 428, 3850–3868. [Google Scholar] [CrossRef] [PubMed]
- Zha, J.; Harada, H.; Yang, E.; Jockel, J.; Korsmeyer, S.J. Serine Phosphorylation of Death Agonist BAD in Response to Survival Factor Results in Binding to 14-3-3 Not BCL-XL. Cell 1996, 87, 619–628. [Google Scholar] [CrossRef]
- Ley, R.; Balmanno, K.; Hadfield, K.; Weston, C.; Cook, S.J. Activation of the ERK1/2 Signaling Pathway Promotes Phosphorylation and Proteasome-Dependent Degradation of the BH3-Only Protein, Bim. J. Biol. Chem. 2003, 278, 18811–18816. [Google Scholar] [CrossRef]
- Jin, S.; Zhuo, Y.; Guo, W.; Field, J. P21-Activated Kinase 1 (Pak1)-Dependent Phosphorylation of Raf-1 Regulates Its Mitochondrial Localization, Phosphorylation of BAD, and Bcl-2 Association. J. Biol. Chem. 2005, 280, 24698–24705. [Google Scholar] [CrossRef]
- He, H.; Yim, M.; Liu, K.H.; Cody, S.C.; Shulkes, A.; Baldwin, G.S. Involvement of G Proteins of the Rho Family in the Regulation of Bcl-2-like Protein Expression and Caspase 3 Activation by Gastrins. Cell Signal. 2008, 20, 83–93. [Google Scholar] [CrossRef]
- Rebollo, A.; Pérez-Sala, D.; Martínez-A, C. Bcl-2 Differentially Targets K-, N-, and H-Ras to Mitochondria in IL-2 Supplemented or Deprived Cells: Implications in Prevention of Apoptosis. Oncogene 1999, 18, 4930–4939. [Google Scholar] [CrossRef]
- Ueda, S.; Kitazawa, S.; Ishida, K.; Nishikawa, Y.; Matsui, M.; Matsumoto, H.; Aoki, T.; Nozaki, S.; Takeda, T.; Tamori, Y.; et al. Crucial Role of the Small GTPase Rac1 in Insulin-Stimulated Translocation of Glucose Transporter 4 to the Mouse Skeletal Muscle Sarcolemma. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 2254–2261. [Google Scholar] [CrossRef] [PubMed]
- Velaithan, R.; Kang, J.; Hirpara, J.L.; Loh, T.; Goh, B.C.; Le Bras, M.; Brenner, C.; Clement, M.-V.; Pervaiz, S. The Small GTPase Rac1 Is a Novel Binding Partner of Bcl-2 and Stabilizes Its Antiapoptotic Activity. Blood 2011, 117, 6214–6226. [Google Scholar] [CrossRef] [PubMed]
- Denis, G.V.; Yu, Q.; Ma, P.; Deeds, L.; Faller, D.V.; Chen, C.-Y. Bcl-2, via Its BH4 Domain, Blocks Apoptotic Signaling Mediated by Mitochondrial Ras. J. Biol. Chem. 2003, 278, 5775–5785. [Google Scholar] [CrossRef]
- Amendola, C.R.; Mahaffey, J.P.; Parker, S.J.; Ahearn, I.M.; Chen, W.-C.; Zhou, M.; Court, H.; Shi, J.; Mendoza, S.L.; Morten, M.J.; et al. KRAS4A Directly Regulates Hexokinase 1. Nature 2019, 576, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Hoek, J.B.; Shulga, N. Activation of Glycogen Synthase Kinase 3β Disrupts the Binding of Hexokinase II to Mitochondria by Phosphorylating Voltage-Dependent Anion Channel and Potentiates Chemotherapy-Induced Cytotoxicity. Cancer Res. 2005, 65, 10545–10554. [Google Scholar] [CrossRef] [PubMed]
- Zaid, H.; Abu-Hamad, S.; Israelson, A.; Nathan, I.; Shoshan-Barmatz, V. The Voltage-Dependent Anion Channel-1 Modulates Apoptotic Cell Death. Cell Death Differ. 2005, 12, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 Is a Key Mediator of Aerobic Glycolysis and Promotes Tumor Growth in Human Glioblastoma Multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Arzoine, L.; Zilberberg, N.; Ben-Romano, R.; Shoshan-Barmatz, V. Voltage-Dependent Anion Channel 1-Based Peptides Interact with Hexokinase to Prevent Its Anti-Apoptotic Activity. J. Biol. Chem. 2009, 284, 3946–3955. [Google Scholar] [CrossRef]
- Xie, G.; Wilson, J.E. Rat Brain Hexokinase: The Hydrophobie N-Terminus of the Mitochondrially Bound Enzyme Is Inserted in the Lipid Bilayer. Arch. Biochem. Biophys. 1988, 267, 803–810. [Google Scholar] [CrossRef]
- Beurel, E.; Jope, R.S. The Paradoxical Pro- and Anti-Apoptotic Actions of GSK3 in the Intrinsic and Extrinsic Apoptosis Signaling Pathways. Prog. Neurobiol. 2006, 79, 173–189. [Google Scholar] [CrossRef]
- Arora, K.K.; Pedersen, P.L. Functional Significance of Mitochondrial Bound Hexokinase in Tumor Cell Metabolism. Evidence for Preferential Phosphorylation of Glucose by Intramitochondrially Generated ATP. J. Biol. Chem. 1988, 263, 17422–17428. [Google Scholar] [CrossRef]
- Pastorino, J.G.; Hoek, J.B. Regulation of Hexokinase Binding to VDAC. J. Bioenerg. Biomembr. 2008, 40, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Hájek, P.; Lin, C.; Shin, S.K.; Attardi, G.; Chomyn, A. Mitochondrial Outer Membrane Permeability Change and Hypersensitivity to Digitonin Early in Staurosporine-Induced Apoptosis. J. Biol. Chem. 2003, 278, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Srinivasula, S.M.; Druilhe, A.; Fernandes-Alnemri, T.; Alnemri, E.S. Caspase-2 Induces Apoptosis by Releasing Proapoptotic Proteins from Mitochondria. J. Biol. Chem. 2002, 277, 13430–13437. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.-P.; Austgen, K.; Nishino, M.; Coakley, K.M.; Hagen, A.; Han, D.; Papa, F.R.; Oakes, S.A. Caspase-2 Cleavage of BID Is a Critical Apoptotic Signal Downstream of Endoplasmic Reticulum Stress. Mol. Cell. Biol. 2008, 28, 3943–3951. [Google Scholar] [CrossRef]
- Rose, I.A.; Warms, J.V.B. Mitochondrial Hexokinase. J. Biol. Chem. 1967, 242, 1635–1645. [Google Scholar] [CrossRef]
- Sui, D.; Wilson, J.E. Structural Determinants for the Intracellular Localization of the Isozymes of Mammalian Hexokinase: Intracellular Localization of Fusion Constructs Incorporating Structural Elements from the Hexokinase Isozymes and the Green Fluorescent Protein. Arch. Biochem. Biophys. 1997, 345, 111–125. [Google Scholar] [CrossRef]
- John, S.; Weiss, J.N.; Ribalet, B. Subcellular Localization of Hexokinases I and II Directs the Metabolic Fate of Glucose. PLoS ONE 2011, 6, e17674. [Google Scholar] [CrossRef]
- Nawaz, M.H.; Ferreira, J.C.; Nedyalkova, L.; Zhu, H.; Carrasco-López, C.; Kirmizialtin, S.; Rabeh, W.M. The Catalytic Inactivation of the N-Half of Human Hexokinase 2 and Structural and Biochemical Characterization of Its Mitochondrial Conformation. Biosci. Rep. 2018, 38, BSR20171666. [Google Scholar] [CrossRef]
- Polakis, P.G.; Wilson, J.E. An Intact Hydrophobic N-Terminal Sequence Is Critical for Binding of Rat Brain Hexokinase to Mitochondria. Arch. Biochem. Biophys. 1985, 236, 328–337. [Google Scholar] [CrossRef]
- Sun, L.; Shukair, S.; Naik, T.J.; Moazed, F.; Ardehali, H. Glucose Phosphorylation and Mitochondrial Binding Are Required for the Protective Effects of Hexokinases I and II. Mol. Cell. Biol. 2008, 28, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Bryan, N.; Raisch, K.P. Identification of a Mitochondrial-Binding Site on the N-Terminal End of Hexokinase II. Biosci. Rep. 2015, 35, e00205. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Shulga, N.; Hoek, J.B. Mitochondrial Binding of Hexokinase II Inhibits Bax-Induced Cytochrome c Release and Apoptosis. J. Biol. Chem. 2002, 277, 7610–7618. [Google Scholar] [CrossRef] [PubMed]
- Abu-Hamad, S.; Zaid, H.; Israelson, A.; Nahon, E.; Shoshan-Barmatz, V. Hexokinase-I Protection against Apoptotic Cell Death Is Mediated via Interaction with the Voltage-Dependent Anion Channel-1. J. Biol. Chem. 2008, 283, 13482–13490. [Google Scholar] [CrossRef] [PubMed]
- Gall, J.M.; Wong, V.; Pimental, D.R.; Havasi, A.; Wang, Z.; Pastorino, J.G.; Bonegio, R.G.B.; Schwartz, J.H.; Borkan, S.C. Hexokinase Regulates Bax-Mediated Mitochondrial Membrane Injury Following Ischemic Stress. Kidney Int. 2011, 79, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s Double-Edged Sword Acting as Both Facilitator and Gatekeeper of Malignancy When Bound to Mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef]
- Ahn, K.J.; Hwang, H.S.; Park, J.H.; Bang, S.H.; Kang, W.J.; Yun, M.; Lee, J.D. Evaluation of the Role of Hexokinase Type II in Cellular Proliferation and Apoptosis Using Human Hepatocellular Carcinoma Cell Lines. J. Nucl. Med. 2009, 50, 1525–1532. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Holmuhamedov, E. Voltage-Dependent Anion Channel (VDAC) as Mitochondrial Governator—Thinking Outside the Box. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2006, 1762, 181–190. [Google Scholar] [CrossRef]
- Gu, J.J.; Singh, A.; Xue, K.; Mavis, C.; Barth, M.; Yanamadala, V.; Lenz, P.; Grau, M.; Lenz, G.; Czuczman, M.S.; et al. Up-Regulation of Hexokinase II Contributes to Rituximab-Chemotherapy Resistance and Is a Clinically Relevant Target for Therapeutic Development. Oncotarget 2018, 9, 4020–4033. [Google Scholar] [CrossRef]
- Schindler, A.; Foley, E. Hexokinase 1 Blocks Apoptotic Signals at the Mitochondria. Cell. Signal. 2013, 25, 2685–2692. [Google Scholar] [CrossRef]
- Liu, C.; Wang, X.; Zhang, Y. The Roles of HK2 on Tumorigenesis of Cervical Cancer. Technol. Cancer Res. Treat. 2019, 18, 153303381987130. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.; Fan, Z.; Cheng, H.; Huang, Q.; Yang, C.; Jin, K.; Luo, G.; Yu, X.; Liu, C. Hexokinase 2 Dimerization and Interaction with Voltage-dependent Anion Channel Promoted Resistance to Cell Apoptosis Induced by Gemcitabine in Pancreatic Cancer. Cancer Med. 2019, 8, 5903–5915. [Google Scholar] [CrossRef] [PubMed]
- Ciscato, F.; Filadi, R.; Masgras, I.; Pizzi, M.; Marin, O.; Damiano, N.; Pizzo, P.; Gori, A.; Frezzato, F.; Chiara, F.; et al. Hexokinase 2 Displacement from Mitochondria-associated Membranes Prompts Ca2+ -dependent Death of Cancer Cells. EMBO Rep. 2020, 21, e49117. [Google Scholar] [CrossRef] [PubMed]
- Felgner, P.L.; Messer, J.L.; Wilson, J.E. Purification of a Hexokinase-Binding Protein from the Outer Mitochondrial Membrane. J. Biol. Chem. 1979, 254, 4946–4949. [Google Scholar] [CrossRef]
- Lindén, M.; Gellerfors, P.; Dean Nelson, B. Pore Protein and the Hexokinase-Binding Protein from the Outer Membrane of Rat Liver Mitochondria Are Identical. FEBS Lett. 1982, 141, 189–192. [Google Scholar] [CrossRef]
- Liu, M.Y.; Colombini, M. Regulation of Mitochondrial Respiration by Controlling the Permeability of the Outer Membrane through the Mitochondrial Channel, VDAC. Biochim. Biophys. Acta BBA-Bioenerg. 1992, 1098, 255–260. [Google Scholar] [CrossRef]
- Ujwal, R.; Cascio, D.; Colletier, J.-P.; Faham, S.; Zhang, J.; Toro, L.; Ping, P.; Abramson, J. The Crystal Structure of Mouse VDAC1 at 2.3 Å Resolution Reveals Mechanistic Insights into Metabolite Gating. Proc. Natl. Acad. Sci. USA 2008, 105, 17742–17747. [Google Scholar] [CrossRef]
- de Cerqueira Cesar, M.; Wilson, J.E. All Three Isoforms of the Voltage-Dependent Anion Channel (VDAC1, VDAC2, and VDAC3) Are Present in Mitochondria from Bovine, Rabbit, and Rat Brain. Arch. Biochem. Biophys. 2004, 422, 191–196. [Google Scholar] [CrossRef]
- Neumann, D.; Bückers, J.; Kastrup, L.; Hell, S.W.; Jakobs, S. Two-Color STED Microscopy Reveals Different Degrees of Colocalization between Hexokinase-I and the Three Human VDAC Isoforms. PMC Biophys. 2010, 3, 4. [Google Scholar] [CrossRef]
- Miyamoto, S.; Murphy, A.; Brown, J. Akt Mediates Mitochondrial Protection in Cardiomyocytes through Phosphorylation of Mitochondrial Hexokinase-II. Cell Death Differ. 2008, 15, 521–529. [Google Scholar] [CrossRef]
- Haloi, N.; Wen, P.-C.; Cheng, Q.; Yang, M.; Natarajan, G.; Camara, A.K.S.; Kwok, W.-M.; Tajkhorshid, E. Structural Basis of Complex Formation between Mitochondrial Anion Channel VDAC1 and Hexokinase-II. Commun. Biol. 2021, 4, 667. [Google Scholar] [CrossRef] [PubMed]
- Abu-Hamad, S.; Arbel, N.; Calo, D.; Arzoine, L.; Israelson, A.; Keinan, N.; Ben-Romano, R.; Friedman, O.; Shoshan-Barmatz, V. The VDAC1 N-Terminus Is Essential Both for Apoptosis and the Protective Effect of Anti-Apoptotic Proteins. J. Cell Sci. 2009, 122, 1906–1916. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Zakar, M.; Rosenthal, K.; Abu-Hamad, S. Key Regions of VDAC1 Functioning in Apoptosis Induction and Regulation by Hexokinase. Biochim. Biophys. Acta BBA-Bioenerg. 2009, 1787, 421–430. [Google Scholar] [CrossRef]
- Azoulay-Zohar, H.; Israelson, A.; Abu-Hamad, S.; Shoshan-Barmatz, V. In Self-Defence: Hexokinase Promotes Voltage-Dependent Anion Channel Closure and Prevents Mitochondria-Mediated Apoptotic Cell Death. Biochem. J. 2004, 377, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, P.L. Warburg, Me and Hexokinase 2: Multiple Discoveries of Key Molecular Events Underlying One of Cancers’ Most Common Phenotypes, the “Warburg Effect”, i.e., Elevated Glycolysis in the Presence of Oxygen. J. Bioenerg. Biomembr. 2007, 39, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N. Inhibition of Early Apoptotic Events by Akt/PKB Is Dependent on the First Committed Step of Glycolysis and Mitochondrial Hexokinase. Genes Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef]
- Majewski, N.; Nogueira, V.; Robey, R.B.; Hay, N. Akt Inhibits Apoptosis Downstream of BID Cleavage via a Glucose-Dependent Mechanism Involving Mitochondrial Hexokinases. Mol. Cell. Biol. 2004, 24, 730–740. [Google Scholar] [CrossRef]
- Robey, R.B.; Hay, N. Mitochondrial Hexokinases, Novel Mediators of the Antiapoptotic Effects of Growth Factors and Akt. Oncogene 2006, 25, 4683–4696. [Google Scholar] [CrossRef]
- Perevoshchikova, I.V.; Zorov, S.D.; Kotova, E.A.; Zorov, D.B.; Antonenko, Y.N. Hexokinase Inhibits Flux of Fluorescently Labeled ATP through Mitochondrial Outer Membrane Porin. FEBS Lett. 2010, 584, 2397–2402. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Chandel, N.S.; Li, X.X.; Schumacker, P.T.; Colombini, M.; Thompson, C.B. Outer Mitochondrial Membrane Permeability Can Regulate Coupled Respiration and Cell Survival. Proc. Natl. Acad. Sci. USA 2000, 97, 4666–4671. [Google Scholar] [CrossRef]
- Bobba, A.; Amadoro, G.; La Piana, G.; Petragallo, V.A.; Calissano, P.; Atlante, A. Glucose-6-Phosphate Tips the Balance in Modulating Apoptosis in Cerebellar Granule Cells. FEBS Lett. 2015, 589, 651–658. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, J.; Guo, Y.; Ge, W.; Zhou, X.; Pan, M. High Glucose Induces Apoptosis of HUVECs in a Mitochondria-dependent Manner by Suppressing Hexokinase 2 Expression. Exp. Ther. Med. 2019, 18, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Ludwig, R.L.; Vousden, K.H. Mitochondrial Localization of TIGAR under Hypoxia Stimulates HK2 and Lowers ROS and Cell Death. Proc. Natl. Acad. Sci. USA 2012, 109, 20491–20496. [Google Scholar] [CrossRef] [PubMed]
- Mergenthaler, P.; Kahl, A.; Kamitz, A.; van Laak, V.; Stohlmann, K.; Thomsen, S.; Klawitter, H.; Przesdzing, I.; Neeb, L.; Freyer, D.; et al. Mitochondrial Hexokinase II (HKII) and Phosphoprotein Enriched in Astrocytes (PEA15) Form a Molecular Switch Governing Cellular Fate Depending on the Metabolic State. Proc. Natl. Acad. Sci. USA 2012, 109, 1518–1523. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Cassina, A.; Lim, F.; Cerrato, T.; Palomo, G.M.; Diaz-Nido, J. Mitochondrial Hexokinase II Promotes Neuronal Survival and Acts Downstream of Glycogen Synthase Kinase-3. J. Biol. Chem. 2009, 284, 3001–3011. [Google Scholar] [CrossRef]
- Baines, C.P.; Song, C.-X.; Zheng, Y.-T.; Wang, G.-W.; Zhang, J.; Wang, O.-L.; Guo, Y.; Bolli, R.; Cardwell, E.M.; Ping, P. Protein Kinase Cε Interacts With and Inhibits the Permeability Transition Pore in Cardiac Mitochondria. Circ. Res. 2003, 92, 873–880. [Google Scholar] [CrossRef]
- Lauterwasser, J.; Fimm-Todt, F.; Oelgeklaus, A.; Schreiner, A.; Funk, K.; Falquez-Medina, H.; Klesse, R.; Jahreis, G.; Zerbes, R.M.; O’Neill, K.; et al. Hexokinases Inhibit Death Receptor-Dependent Apoptosis on the Mitochondria. Proc. Natl. Acad. Sci. USA 2021, 118, e2021175118. [Google Scholar] [CrossRef]
- Lauterwasser, J.; Fimm-Todt, F.; Edlich, F. Assessment of Dynamic BCL-2 Protein Shuttling Between Outer Mitochondrial Membrane and Cytosol. Methods Mol. Biol. 2019, 1877, 151–161. [Google Scholar] [CrossRef]
- Bogner, C.; Leber, B.; Andrews, D.W. Apoptosis: Embedded in Membranes. Curr. Opin. Cell Biol. 2010, 22, 845–851. [Google Scholar] [CrossRef]
- Lovell, J.F.; Billen, L.P.; Bindner, S.; Shamas-Din, A.; Fradin, C.; Leber, B.; Andrews, D.W. Membrane Binding by TBid Initiates an Ordered Series of Events Culminating in Membrane Permeabilization by Bax. Cell 2008, 135, 1074–1084. [Google Scholar] [CrossRef]
- Kalezic, A.; Udicki, M.; Srdic Galic, B.; Aleksic, M.; Korac, A.; Jankovic, A.; Korac, B. Tissue-Specific Warburg Effect in Breast Cancer and Cancer-Associated Adipose Tissue-Relationship between AMPK and Glycolysis. Cancers 2021, 13, 2731. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cheng, H.; Zhou, Y.; Zhu, Y.; Bian, R.; Chen, Y.; Li, C.; Ma, Q.; Zheng, Q.; Zhang, Y.; et al. Myostatin Induces Mitochondrial Metabolic Alteration and Typical Apoptosis in Cancer Cells. Cell Death Dis. 2013, 4, e494. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.L.; Gaudet, S.; Albeck, J.G.; Burke, J.M.; Sorger, P.K. Non-Genetic Origins of Cell-to-Cell Variability in TRAIL-Induced Apoptosis. Nature 2009, 459, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Flores-Romero, H.; Hohorst, L.; John, M.; Albert, M.-C.; King, L.E.; Beckmann, L.; Szabo, T.; Hertlein, V.; Luo, X.; Villunger, A.; et al. BCL-2-Family Protein TBID Can Act as a BAX-like Effector of Apoptosis. EMBO J. 2022, 41, e108690. [Google Scholar] [CrossRef]
- Lopez, J.; Bessou, M.; Riley, J.S.; Giampazolias, E.; Todt, F.; Rochegüe, T.; Oberst, A.; Green, D.R.; Edlich, F.; Ichim, G.; et al. Mito-Priming as a Method to Engineer Bcl-2 Addiction. Nat. Commun. 2016, 7, 10538. [Google Scholar] [CrossRef]
- Sarosiek, K.A.; Chi, X.; Bachman, J.A.; Sims, J.J.; Montero, J.; Patel, L.; Flanagan, A.; Andrews, D.W.; Sorger, P.; Letai, A. BID Preferentially Activates BAK While BIM Preferentially Activates BAX, Affecting Chemotherapy Response. Mol. Cell 2013, 51, 751–765. [Google Scholar] [CrossRef]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed Anuclear Cell Death Delimits Platelet Life Span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef]
- Certo, M.; Del Gaizo Moore, V.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria Primed by Death Signals Determine Cellular Addiction to Antiapoptotic BCL-2 Family Members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef]
- Ni Chonghaile, T.; Sarosiek, K.A.; Vo, T.-T.; Ryan, J.A.; Tammareddi, A.; Moore, V.D.G.; Deng, J.; Anderson, K.C.; Richardson, P.; Tai, Y.-T.; et al. Pretreatment Mitochondrial Priming Correlates with Clinical Response to Cytotoxic Chemotherapy. Science 2011, 334, 1129–1133. [Google Scholar] [CrossRef]



| BCL-2 Family Member | Gene Name | Activity | Associated Diseases | Interacting BCL-2 Family Proteins in Cancer |
|---|---|---|---|---|
| BCL-2 | BCL2 | Anti-apoptotic | Follicular lymphoma 1, | BAX, BAD, BIM, tBID, PUMA |
| high-grade B-cell lymphoma | ||||
| BCL-xL | BCL2L1 | Anti-apoptotic | Absolute glaucoma, | BAX, BAK, BAD, BIM, tBID, PUMA |
| tongue carcinoma | ||||
| MCL-1 | MCL1 | Anti-apoptotic | Myeloid leukemia, | BAX, BAK, BIM, tBID, NOXA, PUMA |
| chlamydia | ||||
| BAX | BAX | Pro-apoptotic | T-cell acute lymphoblastic leukemia, | MCL-1, BFL-1, BCL-xL, BCL-2, BCL-w, |
| colorectal cancer | BCL-B, PUMA, BIM, tBID | |||
| BAK | BAK1 | Pro-apoptotic | Absolute glaucoma, | MCL-1, BFL-1, BCL-xL, PUMA, BIM, tBID |
| keratoacanthoma | ||||
| BID | BID | Pro-apoptotic | Bladder transitional cell papilloma, | MCL-1, BFL-1, BCL-xL, BCL-2, BCL-w, |
| colon adenocarcinoma | BCL-B, BAX, BAK | |||
| BIM | BCL2L11 | Pro-apoptotic | Interleukin-7 receptor alpha deficiency, | MCL-1, BFL-1, BCL-xL, BCL-2, BCL-w, |
| lymphoproliferative syndrome | BCL-B, BAX, BAK | |||
| BAD | BAD | Pro-apoptotic | B-cell lymphoma, | BCL-2, BCL-xL, BCL-w |
| transient cerebral ischemia |
| HK | Tissue Distribution | Subcellular Localization | Functions | Suggested Interactions in Cell Death Signaling | References |
|---|---|---|---|---|---|
| I | All mammalian tissues, | OMM, cytosol | Glucose catabolism, | BCL-xL | [56] |
| main isoform in the brain | apoptosis regulator | BID | [56,57] * | ||
| BIM | [56] | ||||
| BAX | [56] | ||||
| BAK | [56] | ||||
| VDAC | [58,59] | ||||
| II | Heart, skeletal muscle, | OMM, cytosol | Glucose catabolism, | BAX | [56,60] * |
| adipose tissue | glycogen synthesis, | BAK | [56] | ||
| apoptosis regulator | VDAC | [58,59] | |||
| PKCε | [61] | ||||
| AKT | [62] | ||||
| PEA15 | [63] | ||||
| TIGAR | [64] | ||||
| III | Ubiquitously expressed at low levels, | Perinuclear | Glucose catabolism | ||
| highest expression in lung, kidney | compartment | ||||
| and liver | |||||
| IV | Liver, pancreatic islets, certain | Cytosol | Glucose catabolism, | BAD | [65] |
| parts of the brain and gut | intracellular glucose sensor | VDAC | [66] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schoeniger, A.; Wolf, P.; Edlich, F. How Do Hexokinases Inhibit Receptor-Mediated Apoptosis? Biology 2022, 11, 412. https://doi.org/10.3390/biology11030412
Schoeniger A, Wolf P, Edlich F. How Do Hexokinases Inhibit Receptor-Mediated Apoptosis? Biology. 2022; 11(3):412. https://doi.org/10.3390/biology11030412
Chicago/Turabian StyleSchoeniger, Axel, Philipp Wolf, and Frank Edlich. 2022. "How Do Hexokinases Inhibit Receptor-Mediated Apoptosis?" Biology 11, no. 3: 412. https://doi.org/10.3390/biology11030412
APA StyleSchoeniger, A., Wolf, P., & Edlich, F. (2022). How Do Hexokinases Inhibit Receptor-Mediated Apoptosis? Biology, 11(3), 412. https://doi.org/10.3390/biology11030412