
Implementation of a Practical Teaching Course on Protein Engineering


Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Background Theory
1.2. Pedagogical Considerations
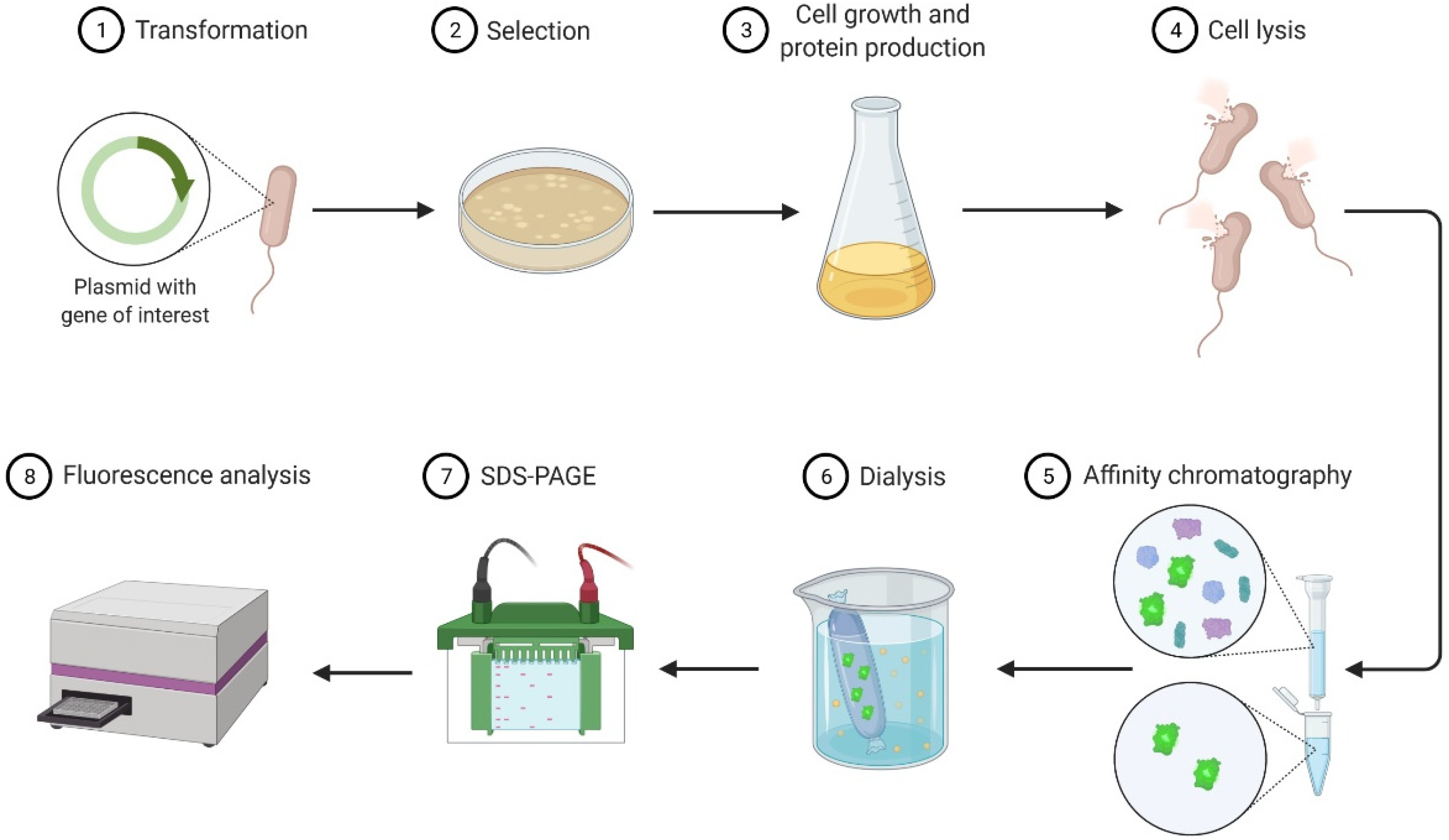
- To be proficient in carrying out the following procedures: bacterial growth, cell lysis, protein purification, protein quantification, and polyacrylamide gel electrophoresis.
- To reinforce understanding of the following topics: plasmid design, recombinant protein expression, and protein purification.
- To acquire skills to operate the following equipment: UV-Vis spectrophotometer, centrifuges, sonicator, microplate reader, and electrophoresis apparatus.
- To improve the ability in critical thinking, team organization, and scientific concepts exposition and writing skills.
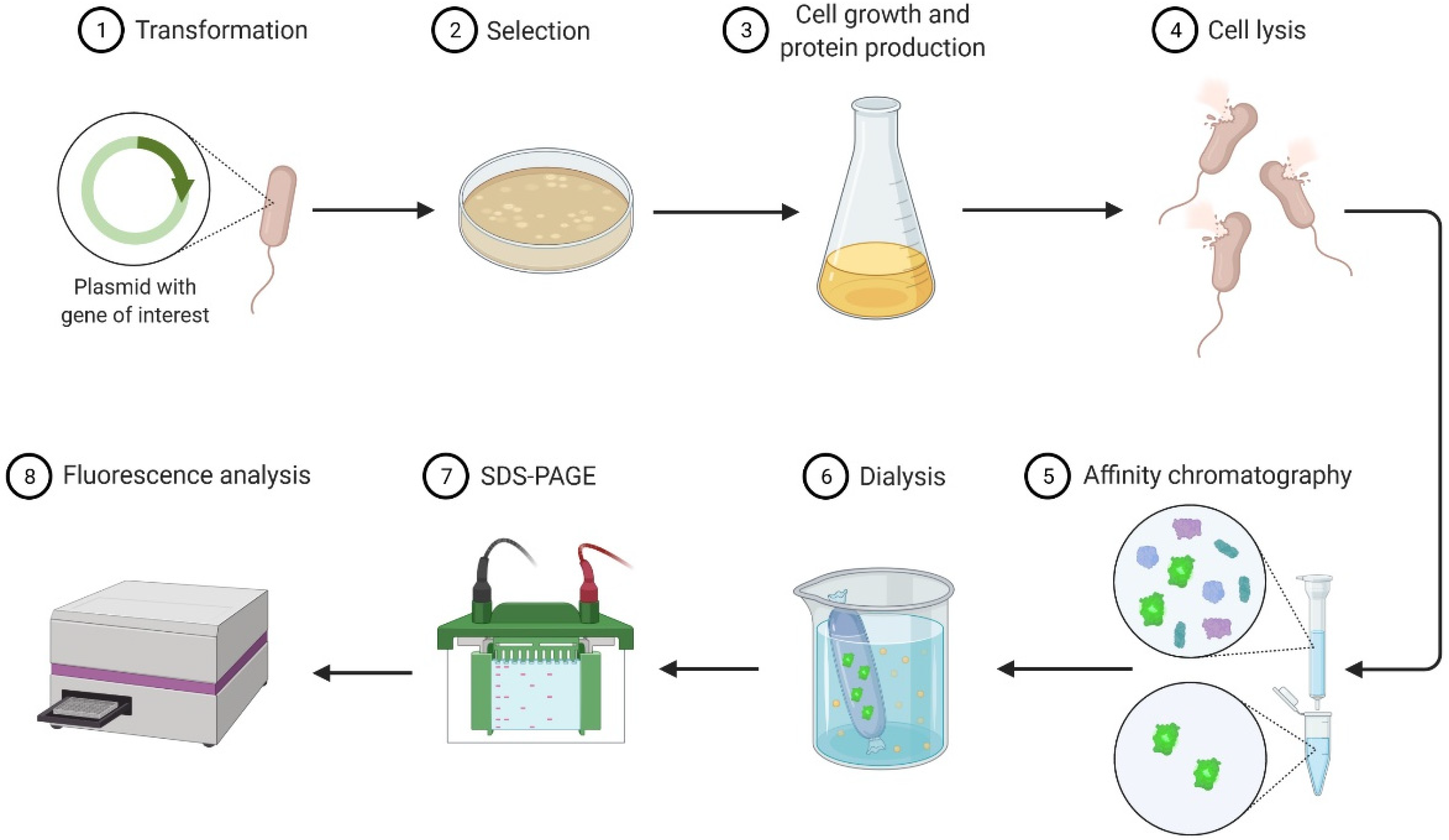
2. Materials and Methods
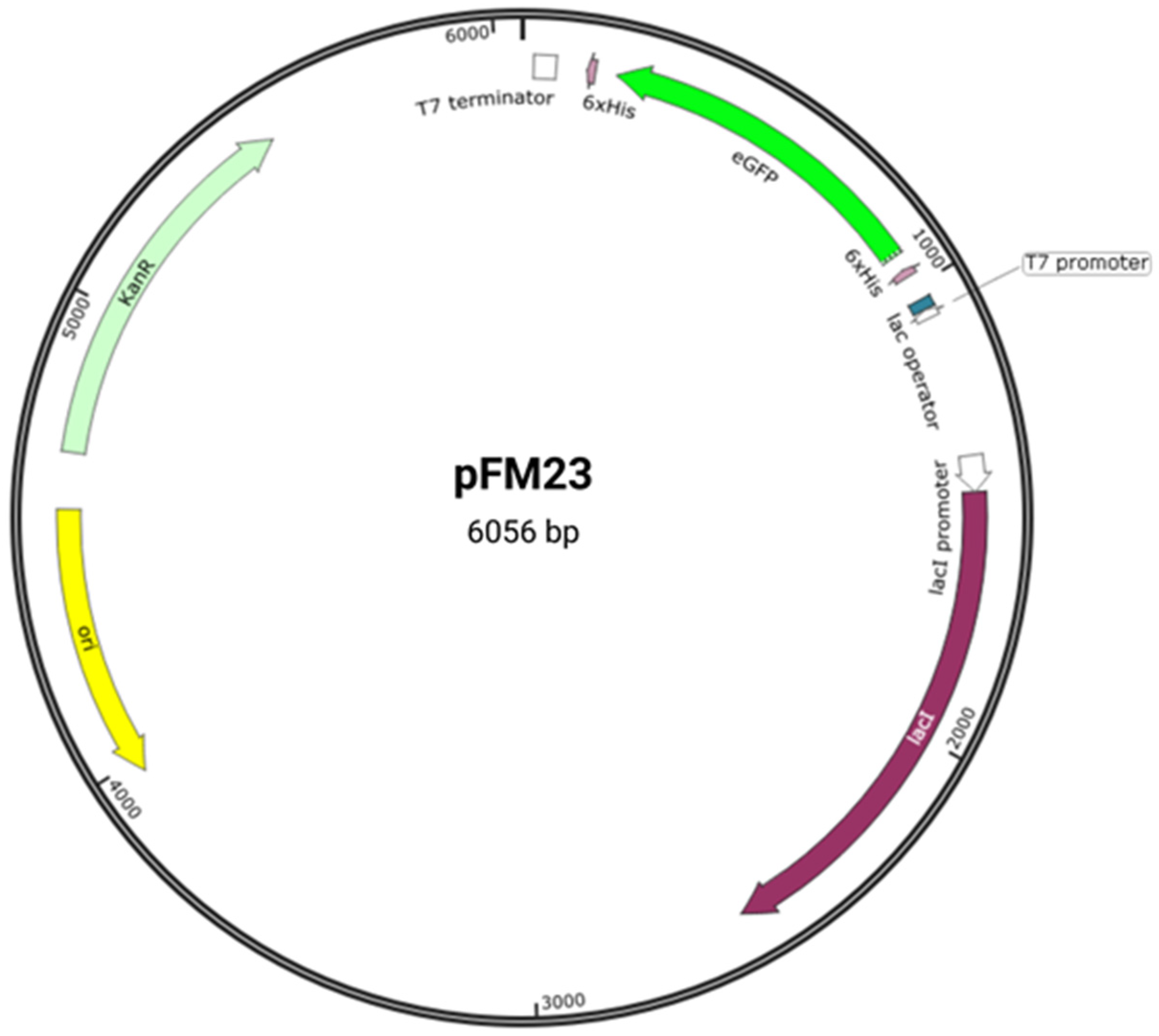
2.1. Bacterial Strain and Culture Medium
2.2. Reagents and Equipment
2.3. Session 1—Bacterial Growth Curve and Chemical Induction
2.3.1. Pre-Lab Preparation
2.3.2. Lab Session
2.3.3. Post-Lab Preparation
2.4. Session 2—Cell Disruption and Contact with the Chromatographic Resin
2.4.1. Pre-Lab Preparation
2.4.2. Lab Session
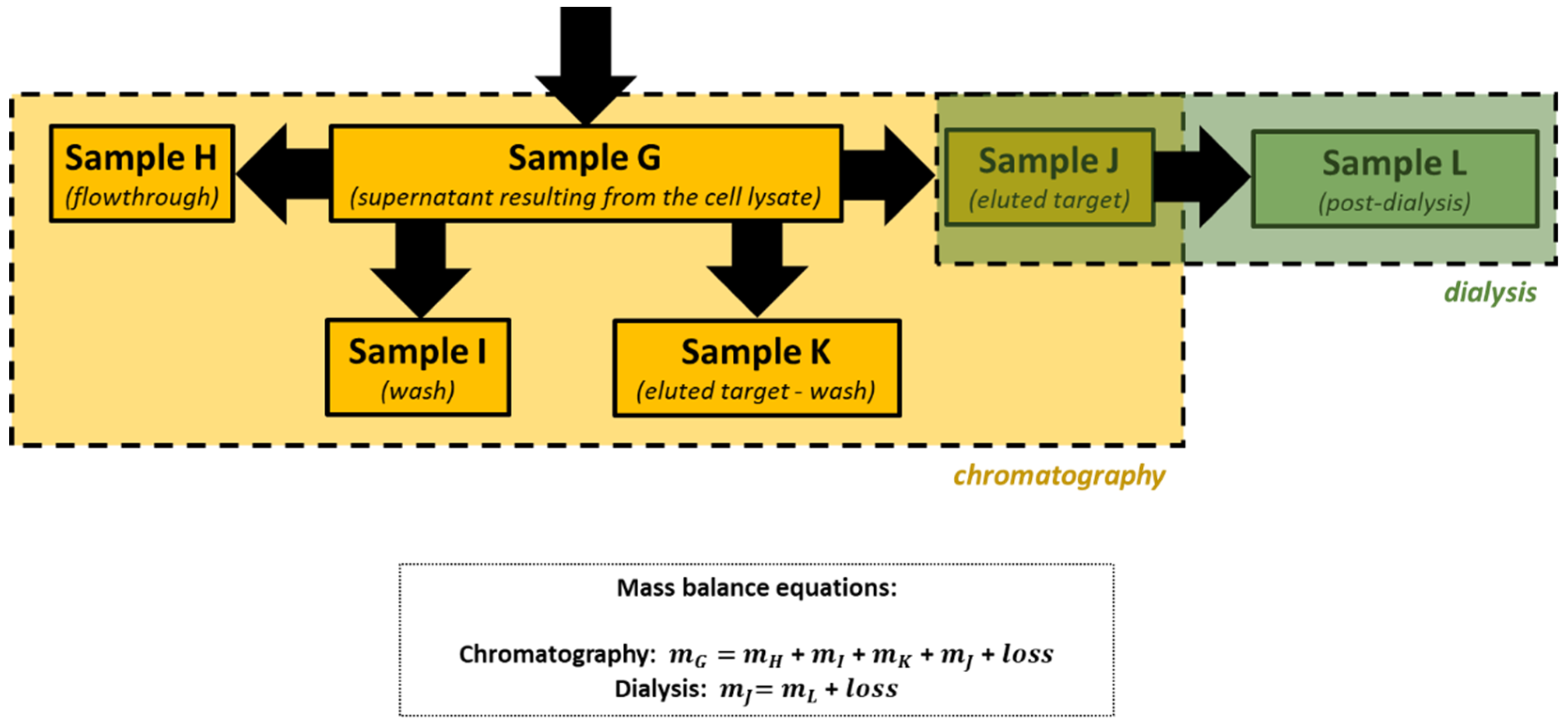
2.5. Session 3—Affinity Chromatography and Dialysis
2.6. Session 4—Total Protein Concentration
2.7. Session 5—SDS-PAGE (Sodium Dodecyl Sulfate—Polyacrylamide Gel Electrophoresis)
2.7.1. Pre-Lab Preparation
2.7.2. Lab Session
2.7.3. Post-Lab Preparation
2.8. Session 6: eGFP Concentration
3. Results and Discussion
3.1. Bacterial Growth
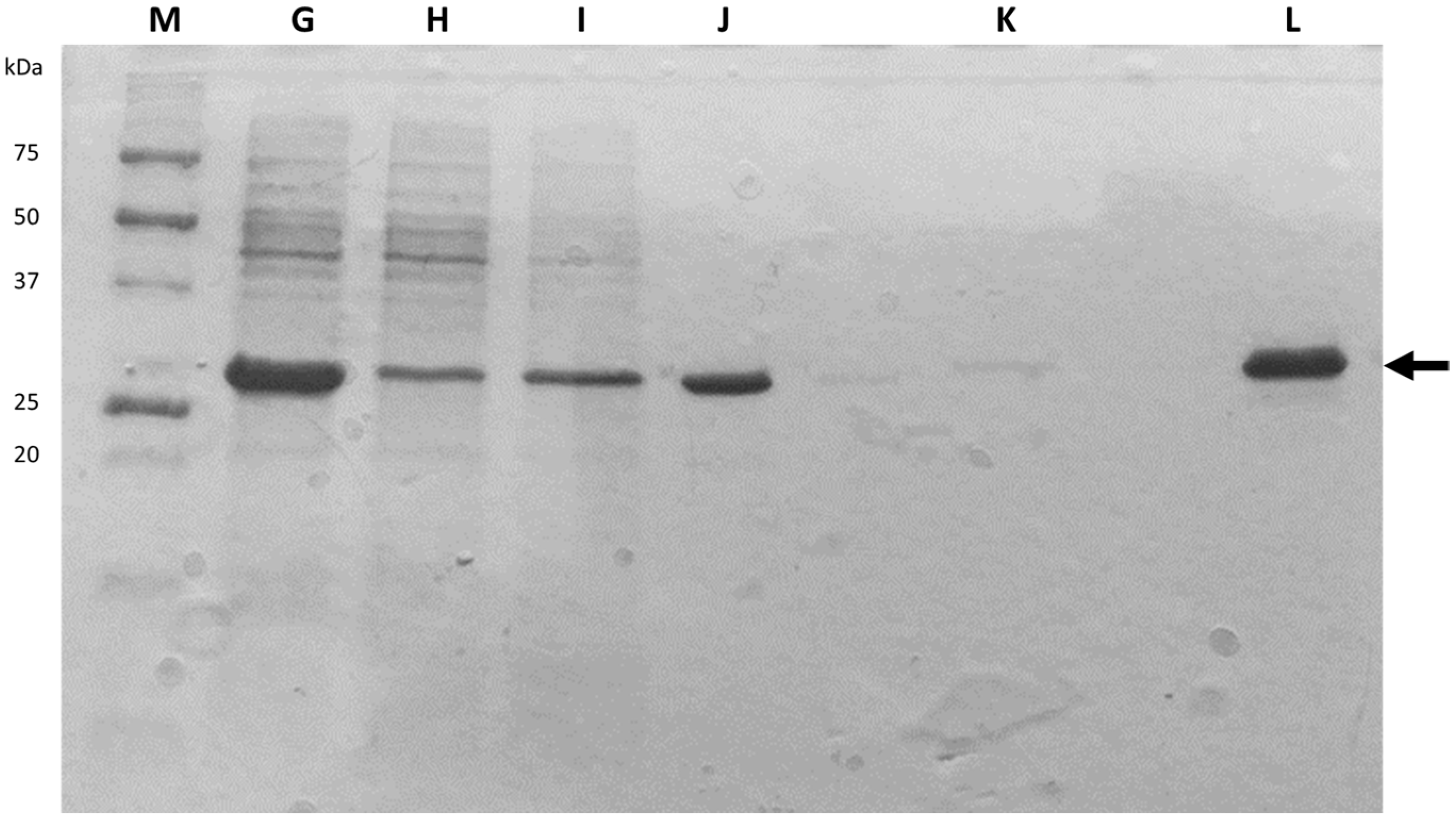
3.2. Protein Quantification and Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Poluri, K.M.; Gulati, K. Biotechnological and Biomedical Applications of Protein Engineering Methods. In Protein Engineering Techniques: Gateways to Synthetic Protein Universe; Springer: Singapore, 2017; pp. 103–134. [Google Scholar]
- Gomes, L.C.; Mergulhão, F.J. Production of Recombinant Proteins in Escherichia coli Biofilms: Challenges and Opportunities. In Advances in Medicine and Biology; Berhardt, L.V., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2019; Volume 152. [Google Scholar]
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacharias, D.A.; Tsien, R.Y. Molecular biology and mutation of green fluorescent protein. Methods Biochem. Anal. 2006, 47, 83–120. [Google Scholar]
- Stepanenko, O.V.; Stepanenko, O.V.; Kuznetsova, I.M.; Verkhusha, V.V.; Turoverov, K.K. Beta-barrel scaffold of fluorescent proteins: Folding, stability and role in chromophore formation. Int. Rev. Cell. Mol. Biol. 2013, 302, 221–278. [Google Scholar] [PubMed] [Green Version]
- Stepanenko, O.V.; Verkhusha, V.V.; Kuznetsova, I.M.; Uversky, V.N.; Turoverov, K.K. Fluorescent proteins as biomarkers and biosensors: Throwing color lights on molecular and cellular processes. Curr. Protein Pept. Sci. 2008, 9, 338–369. [Google Scholar] [CrossRef] [Green Version]
- Cormack, B.P.; Valdivia, R.H.; Falkow, S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 1996, 173, 33–38. [Google Scholar] [CrossRef]
- Neylon, C. Chemical and biochemical strategies for the randomization of protein encoding DNA sequences: Library construction methods for directed evolution. Nucleic Acids Res. 2004, 32, 1448–1459. [Google Scholar] [CrossRef] [Green Version]
- Valdivia, R.H.; Hromockyj, A.E.; Monack, D.; Ramakrishnan, L.; Falkow, S. Applications for green fluorescent protein (GFP) in the study of hostpathogen interactions. Gene 1996, 173, 47–52. [Google Scholar] [CrossRef]
- Crameri, A.; Whitehorn, E.A.; Tate, E.; Stemmer, W.P.C. Improved Green Fluorescent Protein by Molecular Evolution Using DNA Shuffling. Nat. Biotechnol. 1996, 14, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, D.W.; Anderson, M.T.; Herzenberg, L.A. Flow Cytometric Analysis and FACS Sorting of Cells Based on GFP Accumulation. In Methods in Cell Biology; Sullivan, K.F., Kay, S.A., Eds.; Academic Press: Cambridge, MA, USA, 1998; Volume 58, pp. 315–341. [Google Scholar]
- Zacharias, D.A.; Baird, G.S.; Tsien, R.Y. Recent advances in technology for measuring and manipulating cell signals. Curr. Opin. Neurobiol. 2000, 10, 416–421. [Google Scholar] [CrossRef]
- Gomes, L.C.; Mergulhão, F.J. Applications of Green Fluorescent Protein in Biofilm Studies. In Advances in Medicine and Biology; Berhardt, L.V., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2018; Volume 132. [Google Scholar]
- Mergulhão, F.J.; Monteiro, G.A. Analysis of factors affecting the periplasmic production of recombinant proteins in Escherichia coli. J. Microbiol. Biotechnol. 2007, 17, 1236–1241. [Google Scholar] [PubMed]
- Gomes, L.C.; Monteiro, G.A.; Mergulhão, F.J. The Impact of IPTG Induction on Plasmid Stability and Heterologous Protein Expression by Escherichia coli Biofilms. Int. J. Mol. Sci. 2020, 21, 576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novagen. pET System Manual, 11th ed.; 2005; Available online: https://kirschner.med.harvard.edu/files/protocols/Novagen_petsystem.pdf (accessed on 20 February 2022).
- Mergulhão, F.J.M.; Monteiro, G.A.; Cabral, J.M.S.; Taipa, M.A. Design of bacterial vector systems for the production of recombinant proteins in Escherichia coli. J. Microbiol. Biotechnol. 2004, 14, 1–14. [Google Scholar]
- Sanchez-Garcia, L.; Martín, L.; Mangues, R.; Ferrer-Miralles, N.; Vázquez, E.; Villaverde, A. Recombinant pharmaceuticals from microbial cells: A 2015 update. Microb. Cell Factories 2016, 15, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baneyx, F. Recombinant protein expression in Escherichia coli. Curr. Opin. Biotechnol. 1999, 10, 411–421. [Google Scholar] [CrossRef]
- Pines, O.; Inouye, M. Expression and secretion of proteins in E. coli. Mol. Biotechnol. 1999, 12, 25–34. [Google Scholar] [CrossRef]
- Mergulhão, F.J.M.; Summers, D.K.; Monteiro, G.A. Recombinant protein secretion in Escherichia coli. Biotechnol. Adv. 2005, 23, 177–202. [Google Scholar] [CrossRef] [PubMed]
- Mergulhão, F.J.; Taipa, M.A.; Cabral, J.M.; Monteiro, G.A. Evaluation of bottlenecks in proinsulin secretion by Escherichia coli. J. Biotechnol. 2004, 109, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, H.P.; Mortensen, K.K. Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli. Microb. Cell Fact. 2005, 4, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urh, M.; Simpson, D.; Zhao, K. Affinity Chromatography: General Methods. In Methods in Enzymology; Burgess, R.R., Deutscher, M.P., Eds.; Academic Press: Cambridge, MA, USA, 2009; Volume 463, pp. 417–438. [Google Scholar]
- Spriestersbach, A.; Kubicek, J.; Schäfer, F.; Block, H.; Maertens, B. Purification of His-Tagged Proteins. In Methods in Enzymology; Lorsch, J.R., Ed.; Academic Press: Cambridge, MA, USA, 2015; Volume 559, pp. 1–15. [Google Scholar]
- Booth, W.T.; Schlachter, C.R.; Pote, S.; Ussin, N.; Mank, N.J.; Klapper, V.; Offermann, L.R.; Tang, C.; Hurlburt, B.K.; Chruszcz, M. Impact of an N-terminal Polyhistidine Tag on Protein Thermal Stability. ACS Omega 2018, 3, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Yanisch-Perron, C.; Vieira, J.; Messing, J. Improved M13 phage cloning vectors and host strains: Nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 1985, 33, 103–119. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Bentley, W.E.; Davis, R.H.; Kompala, D.S. Dynamics of induced CAT expression in E. coli. Biotechnol. Bioeng. 1991, 38, 749–760. [Google Scholar] [CrossRef]
- Einsfeldt, K.; Severo Júnior, J.B.; Corrêa Argondizzo, A.P.; Medeiros, M.A.; Alves, T.L.; Almeida, R.V.; Larentis, A.L. Cloning and expression of protease ClpP from Streptococcus pneumoniae in Escherichia coli: Study of the influence of kanamycin and IPTG concentration on cell growth, recombinant protein production and plasmid stability. Vaccine 2011, 29, 7136–7143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homem, V.; Alves, A.; Santos, L. Development and Validation of a Fast Procedure To Analyze Amoxicillin in River Waters by Direct-Injection LC–MS/MS. J. Chem. Educ. 2014, 91, 1961–1965. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, N.K.; Shrivastava, A. Recent Developments in Bioprocessing of Recombinant Proteins: Expression Hosts and Process Development. Front. Bioeng. Biotechnol. 2019, 7, 420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, J.M. SDS Polyacrylamide Gel Electrophoresis of Proteins. In The Protein Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 177–185. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Session Number | General Topic | Tasks |
|---|---|---|
| 1 | Protein expression | Bacterial growth curve and chemical induction |
| 2 | Protein extraction | Cell disruption and contact with the chromatographic resin |
| 3 | Protein purification | Affinity chromatography and dialysis |
| 4 | Protein quantification | Total protein concentration |
| 5 | Protein analysis | SDS-PAGE |
| 6 | Protein quantification | eGFP concentration |
| Group | R2 * | p-Value ** | µmax (min−1) | td (min) |
|---|---|---|---|---|
| G1 | 0.9430 | 0.0289 | 0.00634 | 109.4 |
| G2 | 0.9270 | 0.0372 | 0.00660 | 105.0 |
| G3 | 0.9401 | 0.0304 | 0.00673 | 103.0 |
| G4 | 0.9349 | 0.0331 | 0.00631 | 109.8 |
| Sample Identification | Session | Content |
|---|---|---|
| A | 1 | Cell culture in the exponential phase |
| B | 1 | Grown cell culture |
| C | 1 | Supernatant resulting from the centrifugation of the cell culture |
| D | 1 | Cell pellet resuspended in Buffer I |
| E | 2 | Cell lysate |
| F | 2 | Cell debris resulting from the centrifugation of the cell lysate |
| G | 2 | Supernatant resulting from the centrifugation of the cell lysate |
| H | 3 | Flowthrough (unbound material) |
| I | 3 | Wash |
| J | 3 | Eluted target |
| K | 3 | Eluted target (wash) |
| L | 3 | Post-dialysis |
| Group | Sample | Total Protein Mass (mg) | eGFP Mass (mg) | Purity (%) |
|---|---|---|---|---|
| G1 | G | 39.475 | 8.056 | 20.4 |
| H | 29.709 | 1.176 | 4.0 | |
| I | 3.759 | 1.207 | 32.1 | |
| J | 5.146 | 5.325 | 103.5 | |
| K | 0.248 | 0.051 | 20.6 | |
| L | 2.686 | 3.737 | 139.1 | |
| G2 | G | 68.138 | 9.323 | 13.7 |
| H | 54.898 | 1.282 | 2.3 | |
| I | 5.746 | 4.867 | 84.71 | |
| J | 6.868 | 3.169 | 46.14 | |
| K | 0.625 | 0.005 | 0.73 | |
| L | 2.672 | 2.745 | 102.73 | |
| G3 | G | 55.630 | 13.361 | 24.0 |
| H | 51.235 | 1.358 | 2.7 | |
| I | 1.993 | 1.067 | 53.5 | |
| J | 0.580 | 0.227 | 39.1 | |
| K | 0.191 | 0.007 | 3.7 | |
| L | 1.621 | 2.743 | 169.2 | |
| G4 | G | 23.586 | 13.530 | 57.36 |
| H | 7.107 | 2.664 | 37.48 | |
| I | 2.404 | 1.676 | 69.70 | |
| J | 13.856 | 9.791 | 70.66 | |
| K | 0.218 | 0.013 | 6.06 | |
| L | 1.088 | 1.207 | 110.9 |
| Group | Chromatography Yield (%) | Dialysis Yield (%) |
|---|---|---|
| G1 | 66.1 | 70.2 |
| G2 | 34.0 | 86.6 |
| G3 | 1.7 | 1208.4 * |
| G4 | 72.4 | 12.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, L.C.; Ferreira, C.; Mergulhão, F.J. Implementation of a Practical Teaching Course on Protein Engineering. Biology 2022, 11, 387. https://doi.org/10.3390/biology11030387
Gomes LC, Ferreira C, Mergulhão FJ. Implementation of a Practical Teaching Course on Protein Engineering. Biology. 2022; 11(3):387. https://doi.org/10.3390/biology11030387
Chicago/Turabian StyleGomes, Luciana C., Carla Ferreira, and Filipe J. Mergulhão. 2022. "Implementation of a Practical Teaching Course on Protein Engineering" Biology 11, no. 3: 387. https://doi.org/10.3390/biology11030387
APA StyleGomes, L. C., Ferreira, C., & Mergulhão, F. J. (2022). Implementation of a Practical Teaching Course on Protein Engineering. Biology, 11(3), 387. https://doi.org/10.3390/biology11030387