
Diabetes Mellitus: A Path to Amnesia, Personality, and Behavior Change
 , ,
, ,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Study Population | Study Design | Study Period | Subgroup | Results |
|---|---|---|---|---|---|
| Espeland MA et al. 2013 [24] | N = 1366 Age range = 72–89 years at start of study; 698 women obtained a repeat MRI scan | Cohort study | 4.7 years | Women with DM and women without DM | 145 diabetic women had smaller brain volume (0.6% less; p = 0.05), gray matter volumes that was smaller (1.5% less; p = 0.01), ischemic lesion volumes which was larger (21.8% greater; p = 0.02), in white matter (18.8% greater; p = 0.02), both overall and in gray matter (27.5% greater; p = 0.06). |
| Moran et al. 2013 [25] | N = 713 Age = ≥55 years | Cross-sectional study | 2 years (2008–2010) | Subjects with Type 2 DM (T2DM) = 350 Subjects without Type 2 DM (T2DM) = 363 | An MRI scan revealed an association betweenT2DM and greater cerebral infarcts and lesser total white, gray, and hippocampal volumes (p < 0.05). T2DM was associated with poorer visuospatial construction, planning, speed, and visual memory (p ≤ 0.05) |
| Ball et al. 2011 [26] | N = 4 tissue culture and brain slices from streptozotocin (STZ)-diabetic rats | Experimental animal study | 2–3 weeks | Relative connexin (Cx) protein levels were assessed by Western blotting using extracts from cultured astrocytes grown in high (25 mmol/L) or low (5.5 mmol/L) | Astrocytic growth in high glucose reduced dye-labeled area by 75%; actin level rose by 24%. Oxidative stress and regionally-selective down-regulation of connexin protein content affect gap junctional communication in brain of STZ-diabetic rats |
| Janelidze et al. 2017 [27] | N = 1015 Age = ≥60 years | Cohort study | 5.7 yrs (3.0–9.6) | Alzheimer’s Disease, Vascular dementia, dementia with Lewy bodies (DLB), Parkinson’s disease with dementia (PDD), and frontotemporal dementia (FTD) | The CSF/plasma albumin ratio (an indicator of BBB and blood-CSF barrier permeability) was increased in individuals with diabetes (diagnosed with diabetes or taking antidiabetic medications) compared with those without diabetes (p = 0.015). Diabetes was linked to high CSF levels of ICAM-1 (p < 0.001), VCAM-1 (p = 0.007), and VEGF (p = 0.024). |
| Navaratna et al. 2013 [28] | Diabetes induced rats using streptozotocin | Experimental animal study | 6–12 weeks | Human brain microvascular endothelial cells with Advanced Glycation endproduct-BSA ((0–200 μg/mL) and treatment with non-glycated BSA (100 μg/mL) | Treatment with AGE-BSA (0–200 μg/mL) induced a dose-dependent increase in endothelial levels of MMP9. Study also showed neuronal TRKB trophic function is processed by MMP9-mediated degradation in the diabetic brain. |
| Vergoossen et al. 2020 [29] | N = 2302; age: 59 ± 8 years | Cohort study | 4 years (2013–2017) | 1361 subjects with normal glucose metabolism, 348 with prediabetes, and 510 with type 2 diabetes | Association of prediabetes and type 2 diabetes with white matter network organization was studied. Prediabetes and type 2 diabetes were linked with a lower node degree after full adjustment (standardized [st]βPrediabetes = −0.055 [95% CI −0.172, 0.062], stβType2diabetes = −0.256 [−0.379, −0.133], Ptrend < 0.001). Prediabetes was associated with lower local efficiency (stβ = −0.084 [95% CI −0.159, −0.008], p = 0.033) and lower clustering coefficient (stβ = −0.097 [95% CI −0.189, −0.005], p = 0.049). |
| Jackson et al. 2013 [30] | N = 56; Age = >70 years | Post mortem study | Brain tissues were divided in pathologically distinct groups: the group of brain tissues from patients with overt Type2DM and dementia (N = 15); brain samples from Alzheimer’s Disease patients without history of Type2DM (N = 14); brain specimens from age-matched healthy individuals as controls (N = 13) | Amylin oligomers and plaques were noted in the temporal lobe gray matter from patients with diabetes, but not found in controls. In addition, amylin deposit was observed in blood vessels and perivascular spaces. | |
| Currais et al. 2012 [31] | Type 1 Diabetes induced mice; mean age = 6 months | Experimental animal study | 4 months | Type1 DM induced senescence-accelerated prone 8 (SAMP8) and senescence-resistant 1 (SAMR1) mice. Age matched non-diabetic SAMP8 mice | Type 1 Diabetes Mellitus increased Aβ and glial fibrillary acidic protein (GFAP) immunoreactivity in the hippocampus of SAMP8 mice and in age-matched SAMR1 mice to a lesser extent. Analysis showed aggregation of Aβ within astrocyte processes surrounding vessels. Western blot analyses from Type 1 Diabetes Mellitus SAMP8 mice showed raised APP processing and protein glycation along with increased inflammation. Type 1 Diabetes Mellitus increased tau phosphorylation in the SAMR1 mice but did not further increase it in the SAMP8 mice |
| Willette et al. 2015 [32] | N = 186; age = 60.37 ± 5.63 | Cross-sectional study | Normoglycemic 135 Pre Diabetic 43 Diabetic 8 | Pittsburgh Compound B (PiB) Positron Emission Tomography revealed in participants with normoglycemia, higher insulin resistance corresponded to higher PiB uptake in frontal and temporal regions, suggesting raised amyloid deposition. |
2. Objectives of the Study
3. Material and Methods
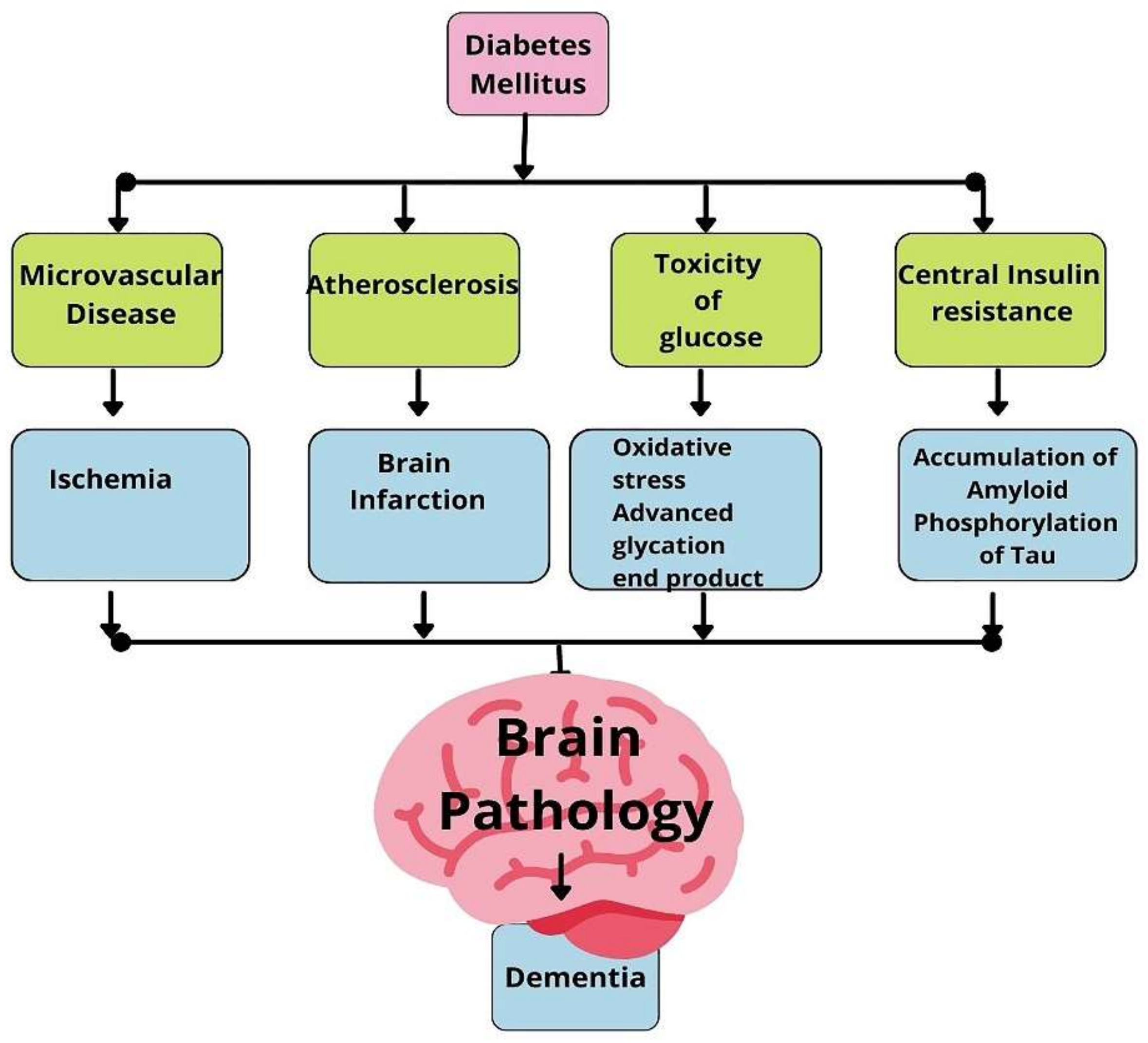
4. Vascular Dementia
5. Pathology of Vascular Dementia
6. Diabetes Mellitus as a Risk Factor for Vascular Dementia
7. Inflammation of Blood Vessels in Diabetes Mellitus
8. Inflammation, Atherosclerotic Change in the Blood Vessel of Diabetics
9. Altered Blood–Brain Barrier Integrity in Vascular Dementia
10. Inflammation Oxidative Stress in Diabetes Mellitus Linked to Vascular Dementia
11. Alzheimer’s Disease
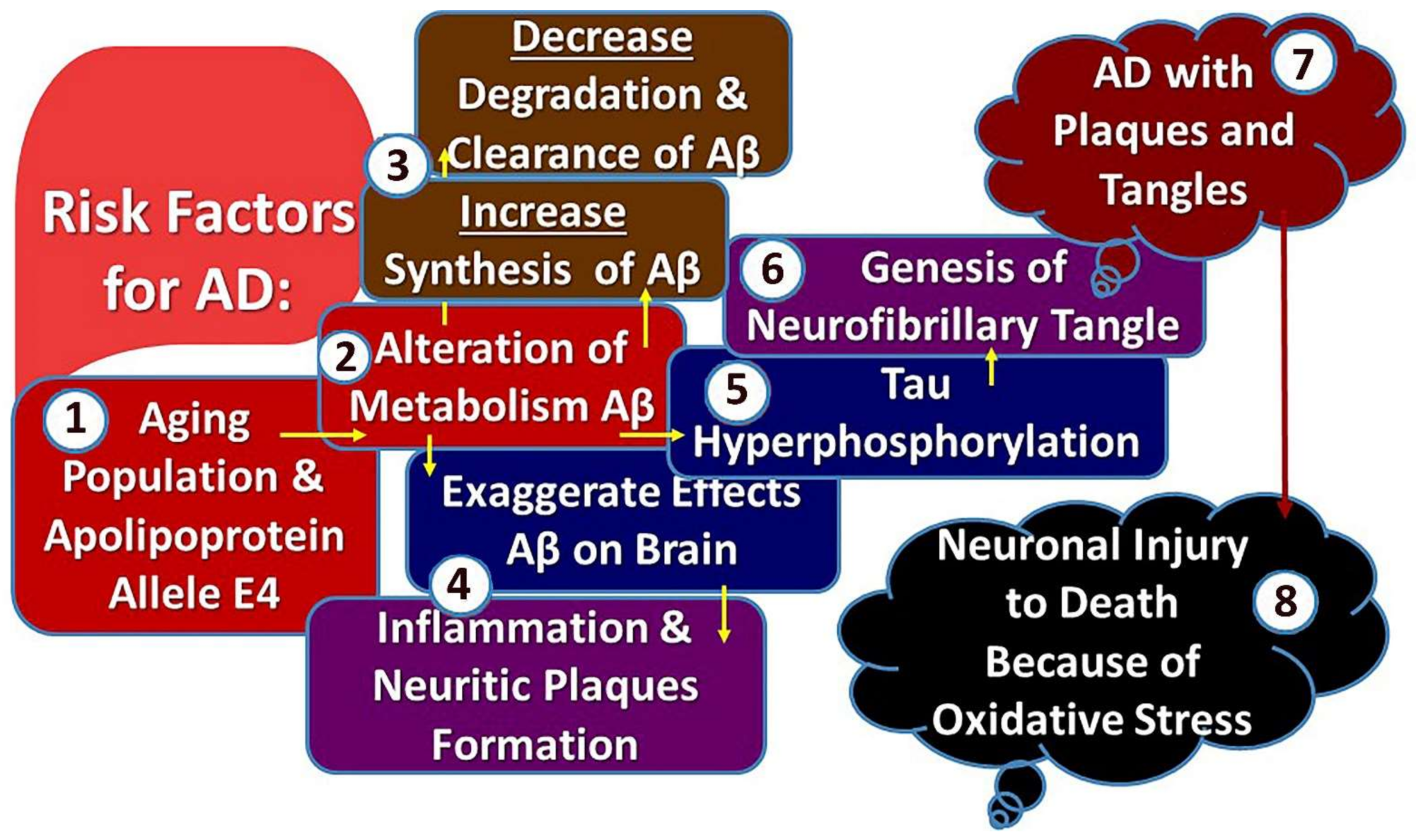
12. Pathogenesis of Alzheimer’s Disease
- (1)
- The formation of senile plaque, which is an extracellular lesion consisting of an accumulation of amyloid-β (Aβ) protein-42 (Aβ42) in its nucleus.
- (2)
- Neuro fibrillar tangles that are intraneuronal findings consisting of phosphorylated tau protein (P-tau) [11].
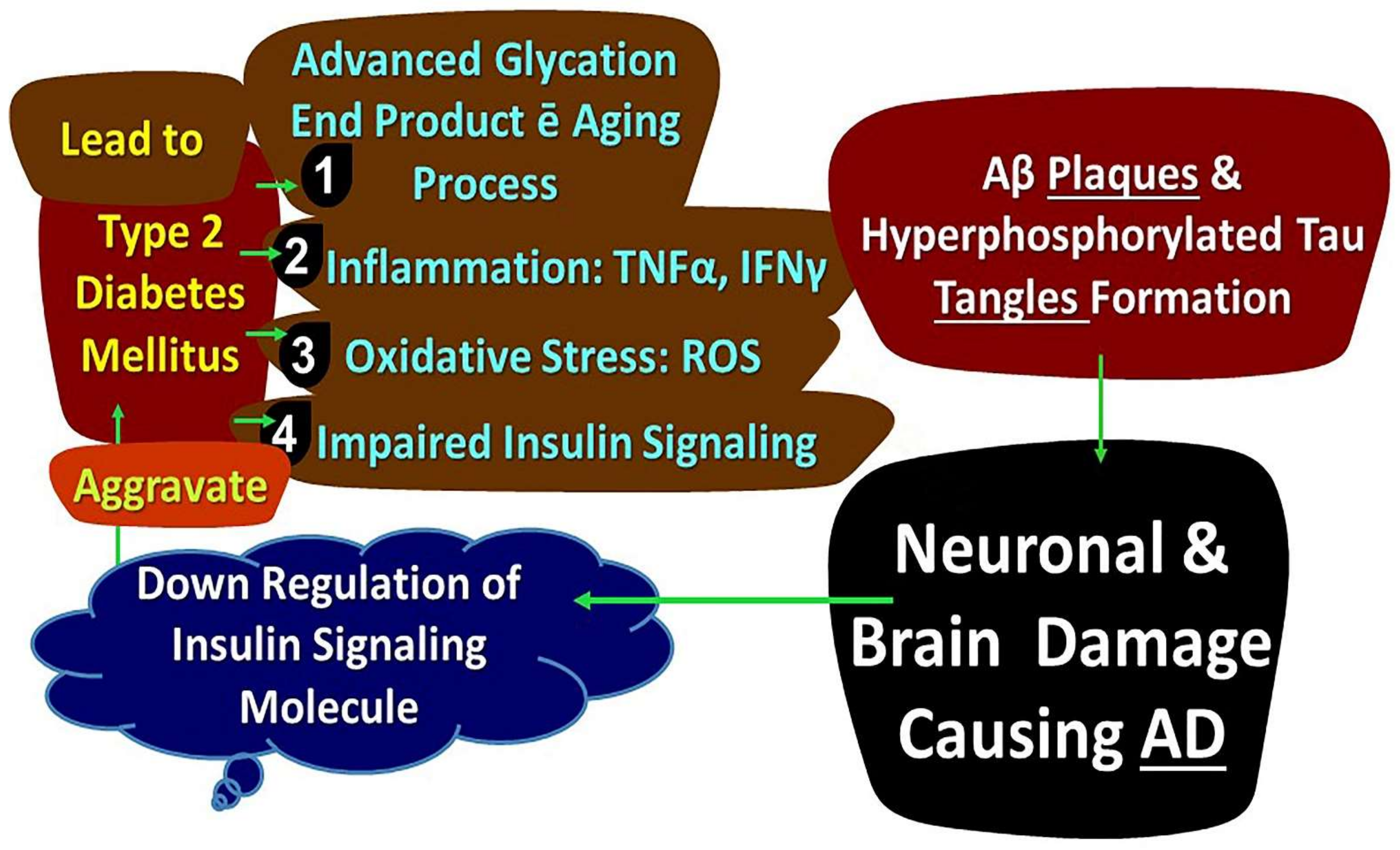
13. Diabetes Mellitus, Insulin Resistance, and Alzheimer’s Disease
14. Diabetes Mellitus, Inflammation, and Metabolism of Energy
15. Dysfunctional Adipose Tissue, Diabetes Mellitus, and Dementia
16. Glymphatic System Disruption and Dementia
17. APO Lipoprotein E: Type 2 Diabetes and Alzheimer’s Disease Relationship Modifier
18. Study Limitations
- This study is a narrative review, so a meta-analysis was not conducted.
- Studies in languages other than English could not be included.
- Articles that need to be accessed through institutional access could not be accessed.
19. Conclusions
20. Recommendations
21. Professional Explications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmad, R.; Haque, M. Oral Health Messiers: Diabetes Mellitus Relevance. Diabetes Metab. Syndr. Obes. 2021, 14, 3001–3015. [Google Scholar] [CrossRef]
- World Health Organization. Health Topics/Diabetes; WHO Press, World Health Organization: Geneva, Switzerland, 2021; Available online: https://www.who.int/health-topics/diabetes#tab=tab_1 (accessed on 25 October 2021).
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef]
- Albai, O.; Frandes, M.; Timar, R.; Roman, D.; Timar, B. Risk factors for developing dementia in type 2 diabetes mellitus patients with mild cognitive impairment. Neuropsychiatr. Dis. Treat. 2019, 15, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Candido, R.; Forbes, J.M.; Thomas, M.C.; Thallas, V.; Dean, R.G.; Burns, W.C.; Tikellis, C.; Ritchie, R.H.; Twigg, S.M.; Cooper, M.E.; et al. A breaker of advanced glycation end products attenuates diabetes-induced myocardial structural changes. Circ. Res. 2003, 92, 785–792. [Google Scholar] [CrossRef]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, D.; Taguchi, T.; Matsumura, T.; Pestell, R.; Edelstein, D.; Giardino, I.; Suske, G.; Rabbani, N.; Thornalley, P.; Sarthy, V.P.; et al. High glucose increases angiopoietin-2 transcription in microvascular endothelial cells through methylglyoxal modification of mSin3A. J. Biol. Chem. 2007, 282, 31038–31045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Yu, S.; Xu, W.; Xu, J. Enhancement of 26S Proteasome Functionality Connects Oxidative Stress and Vascular Endothelial Inflammatory Response in Diabetes Mellitus. Arter. Thromb. Vasc. Biol. 2012, 32, 2131–2140. [Google Scholar] [CrossRef] [Green Version]
- Arvanitakis, Z.; Wilson, R.S.; Li, Y.; Aggarwal, N.T.; Bennett, D.A. Diabetes and function in different cognitive systems in older individuals without dementia. Diabetes Care 2006, 29, 560–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef]
- Silva, M.V.F.; Loures, C.D.M.G.; Alves, L.C.V.; De Souza, L.C.; Borges, K.B.G.; Carvalho, M.D.G. Alzheimer’s disease: Risk factors and potentially protective measures. J. Biomed. Sci. 2019, 26, 33. [Google Scholar] [CrossRef] [Green Version]
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Global prevalence of dementia: A Delphi consensus study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef]
- Exalto, L.; Whitmer, R.; Kappele, L.; Biessels, G. An update on type 2 diabetes, vascular dementia, and Alzheimer’s disease. Exp. Gerontol. 2012, 47, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Karter, A.J.; Laiteerapong, N.; Chin, M.H.; Moffet, H.H.; Parker, M.M.; Sudore, R.; Adams, A.S.; Schillinger, D.; Adler, N.S.; Whitmer, R.A.; et al. Ethnic Differences in Geriatric Conditions and Diabetes Complications Among Older, Insured Adults with Diabetes: The Diabetes and Aging Study. J. Aging Health 2015, 27, 894–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meneilly, G.S.; Tessier, D.M. Diabetes, Dementia, and Hypoglycemia. Can. J. Diabetes 2016, 40, 73–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Román, G.C. Vascular dementia may be the most common form of dementia in the elderly. J. Neurol. Sci. 2002, 203–204, 7–10. [Google Scholar] [CrossRef]
- World Health Organization. Dementia. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 19 February 2022).
- Smolina, K.; Wotton, C.J.; Goldacre, M.J. Risk of dementia in patients hospitalized with type 1 and type 2 diabetes in England, 1998–2011: A retrospective national record-linkage cohort study. Diabetologia 2015, 58, 942–950. [Google Scholar] [CrossRef] [Green Version]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and meta-analysis. Alzheimer’s Dement. 2013, 9, 63–75.e2. [Google Scholar] [CrossRef]
- Kadohara, K.; Sato, I.; Kawakami, K. Diabetes mellitus and risk of early-onset Alzheimer’s disease: A population-based case-control study. Eur. J. Neurol. 2017, 24, 944–949. [Google Scholar] [CrossRef]
- Heath, C.A.; Mercer, S.W.; Guthrie, B. Vascular comorbidities in younger people with dementia: A cross-sectional population-based study of 616 245 middle-aged people in Scotland. J. Neurol. Neurosurg. Psychiatry 2014, 86, 959–964. [Google Scholar] [CrossRef] [Green Version]
- Gudala, K.; Bansal, D.; Schifano, F.; Bhansali, A. Diabetes mellitus and risk of dementia: A meta-analysis of prospective observational studies. J. Diabetes Investig. 2013, 4, 640–650. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, C.; Hua, S.; Liao, H.; Wang, M.; Xiong, Y.; Cao, F. An updated meta-analysis of cohort studies: Diabetes and risk of Alzheimer’s disease. Diabetes Res. Clin. Pract. 2017, 124, 41–47. [Google Scholar] [CrossRef]
- Espeland, M.A.; Bryan, R.N.; Goveas, J.S.; Robinson, J.G.; Siddiqui, M.S.; Liu, S.; Hogan, P.E.; Casanova, R.; Coker, L.H.; Yaffe, K.; et al. Influence of type 2 diabetes on brain volumes and changes in brain volumes: Results from the Women’s Health Initiative Magnetic Resonance Imaging studies. Diabetes Care 2013, 36, 90–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, C.; Phan, T.G.; Chen, J.; Blizzard, L.; Beare, R.; Venn, A.; Münch, G.; Wood, A.G.; Forbes, J.; Greenaway, T.M.; et al. Brain atrophy in type 2 diabetes: Regional distribution and influence on cognition. Diabetes Care 2013, 36, 4036–4042. [Google Scholar] [CrossRef] [Green Version]
- Ball, K.K.; Harik, L.; Gandhi, G.K.; Cruz, N.F.; Dienel, G.A. Reduced gap junctional communication among astrocytes in experimental diabetes: Contributions of altered connexin protein levels and oxidative-nitrosative modifications. J. Neurosci. Res. 2011, 89, 2052–2067. [Google Scholar] [CrossRef] [Green Version]
- Janelidze, S.; Hertze, J.; Nägga, K.; Nilsson, K.; Nilsson, C.; Wennström, M.; van Westen, D.; Blennow, K.; Zetterberg, H.; Hansson, O. Increased blood-brain barrier permeability is associated with dementia and diabetes but not amyloid pathology or APOE genotype. Neurobiol. Aging 2017, 51, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Navaratna, D.; Fan, X.; Leung, W.; Lok, J.; Guo, S.; Xing, C.; Wang, X.; Lo, E.H. Cerebrovascular degradation of TRKB by MMP9 in the diabetic brain. J. Clin. Investig. 2013, 123, 3373–3377. [Google Scholar] [CrossRef]
- Vergoossen, L.W.; Schram, M.T.; de Jong, J.J.; Stehouwer, C.D.; Schaper, N.C.; Henry, R.M.; van der Kallen, C.J.; Dagnelie, P.C.; van Boxtel, M.P.; Eussen, S.J.; et al. White Matter Connectivity Abnormalities in Prediabetes and Type 2 Diabetes: The Maastricht Study. Diabetes Care 2020, 43, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.-W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Currais, A.; Prior, M.; Lo, D.; Jolivalt, C.; Schubert, D.; Maher, P. Diabetes exacerbates amyloid and neurovascular pathology in aging-accelerated mice. Aging Cell 2012, 11, 1017–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willette, A.A.; Johnson, S.C.; Birdsill, A.C.; Sager, M.A.; Christian, B.; Baker, L.D.; Craft, S.; Oh, J.; Statz, E.; Hermann, B.P.; et al. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimer’s Dement. 2015, 11, 504–510.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haroon, N.N.; Austin, P.C.; Shah, B.R.; Wu, J.; Gill, S.S.; Booth, G.L. Risk of dementia in seniors with newly diagnosed diabetes: A population-based study. Diabetes Care 2015, 38, 1868–1875. [Google Scholar] [CrossRef] [Green Version]
- Venkat, P.; Chopp, M.; Chen, J. Models and mechanisms of vascular dementia. Exp. Neurol. 2015, 272, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Plassman, B.; Langa, K.; Fisher, G.; Heeringa, S.; Weir, D.; Ofstedal, M.; Burke, J.; Hurd, M.; Potter, G.; Rodgers, W.; et al. Prevalence of dementia in the United States: The aging, demographics, and memory study. Neuroepidemiology 2007, 29, 125–132. [Google Scholar] [CrossRef]
- Xu, W.; Qiu, C.; Gatz, M.; Pedersen, N.L.; Johansson, B.; Fratiglioni, L. Mid- and late-life diabetes in relation to the risk of dementia: A population-based twin study. Diabetes 2009, 58, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Kimm, H.; Lee, P.H.; Shin, Y.J.; Park, K.; Jo, J.; Lee, Y.; Kang, H.C.; Jee, S. Mid-life and late-life vascular risk factors and dementia in Korean men and women. Arch. Gerontol. Geriatr. 2011, 52, e117–e122. [Google Scholar] [CrossRef]
- Cheng, G.; Huang, C.; Deng, H.; Wang, H. Diabetes as a risk factor for dementia and mild cognitive impairment: A meta-analysis of longitudinal studies. Intern. Med. J. 2012, 42, 484–491. [Google Scholar] [CrossRef]
- Yu, J.H.; Han, K.; Park, S.; Cho, H.; Kim, J.W.; Seo, J.A.; Seo, J.A.; Kim, S.G.; Baik, S.H.; Park, Y.G.; et al. Incidence and Risk Factors for Dementia in Type 2 Diabetes Mellitus: A Nationwide Population-Based Study in Korea. Diabetes Metab. J. 2020, 44, 113–124. [Google Scholar] [CrossRef]
- Bir, S.C.; Khan, M.W.; Javalkar, V.; Toledo, E.G.; Kelley, R.E. Emerging Concepts in Vascular Dementia: A Review. J. Stroke Cerebrovasc. Dis. 2021, 30, 105864. [Google Scholar] [CrossRef]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.; A Schneider, J.; Wardlaw, J.M.; Greenberg, S.M. Cerebral microinfarcts: The invisible lesions. Lancet Neurol. 2012, 11, 272–282. [Google Scholar] [CrossRef] [Green Version]
- van Norden, A.G.; van Uden, I.W.; de Laat, K.F.; Gons, R.A.; Kessels, R.P.; van Dijk, E.J.; de Leeuw, F.E. Cerebral microbleeds are related to subjective cognitive failures: The RUN DMC study. Neurobiol. Aging 2013, 34, 2225–2230. [Google Scholar] [CrossRef]
- Thal, D.R.; Grinberg, L.T.; Attems, J. Vascular dementia: Different forms of vessel disorders contribute to the development of dementia in the elderly brain. Exp. Gerontol. 2012, 47, 816–824. [Google Scholar] [CrossRef] [Green Version]
- De Reuck, J.L. Histopathological stainings and definitions of vascular disruptions in the elderly brain. Exp. Gerontol. 2012, 47, 834–837. [Google Scholar] [CrossRef]
- Park, L.; Zhou, J.; Zhou, P.; Pistick, R.; El Jamal, S.; Younkin, L.; Pierce, J.; Arreguin, A.; Anrather, J.; Younkin, S.G.; et al. Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA 2013, 110, 3089–3094. [Google Scholar] [CrossRef] [Green Version]
- Brown, W.R.; Thore, C.R. Review: Cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol. Appl. Neurobiol. 2011, 37, 56–74. [Google Scholar] [CrossRef] [Green Version]
- Gouw, A.A.; Seewann, A.; van der Flier, W.M.; Barkhof, F.; Rozemuller, A.M.; Scheltens, P.; Geurts, J.J. Heterogeneity of small vessel disease: A systematic review of MRI and histopathology correlations. J. Neurol. Neurosurg. Psychiatry 2011, 82, 126–135. [Google Scholar] [CrossRef]
- Jokinen, H.; Gouw, A.A.; Madureira, S.; Ylikoski, R.; van Straaten, E.C.W.; van der Flier, W.M.; Barkhof, F.; Scheltens, P.; Fazekas, F.; Schmidt, R.; et al. Incident lacunes influence cognitive decline: The LADIS study. Neurology 2011, 76, 1872–1878. [Google Scholar] [CrossRef]
- Umemura, T.; Kawamura, T.; Hotta, N. Pathogenesis and neuroimaging of cerebral large and small vessel disease in type 2 diabetes: A possible link between cerebral and retinal microvascular abnormalities. J. Diabetes Investig. 2017, 8, 134–148. [Google Scholar] [CrossRef] [Green Version]
- Pantoni, L. Cerebral small vessel disease: From pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010, 9, 689–701. [Google Scholar] [CrossRef]
- McCarty, J.L.; Leung, L.Y.; Peterson, R.B.; Sitton, C.W.; Sarraj, A.; Riascos, R.F.; Brinjikji, W. Ischemic Infarction in Young Adults: A Review for Radiologists. RadioGraphics 2019, 39, 1629–1648. [Google Scholar] [CrossRef]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef]
- Widlansky, M.E.; Jensen, D.M.; Wang, J.; Liu, Y.; Geurts, A.M.; Kriegel, A.J.; Liu, P.; Ying, R.; Zhang, G.; Casati, M.; et al. miR-29 contributes to normal endothelial function and can restore it in cardiometabolic disorders. EMBO Mol. Med. 2018, 10, e8046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanleenuwat, P.; Iwanowski, P.; Kozubski, W. Alzheimer’s dementia: Pathogenesis and impact of cardiovascular risk factors on cognitive decline. Postgrad. Med. 2019, 131, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Duan, W.; Alfaro, F.J.; Gavrieli, A.; Kourtelidis, F.; Novak, V. The resting perfusion pattern associates with functional decline in type 2 diabetes. Neurobiol. Aging 2017, 60, 192–202. [Google Scholar] [CrossRef]
- Bangen, K.J.; Werhane, M.L.; Weigand, A.J.; Edmonds, E.C.; Delano-Wood, L.; Thomas, K.; Nation, D.A.; Evangelista, N.D.; Clark, A.L.; Liu, T.T.; et al. Reduced Regional Cerebral Blood Flow Relates to Poorer Cognition in Older Adults with Type 2 Diabetes. Front. Aging Neurosci. 2018, 10, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, W.; Rao, H.; Spaeth, A.M.; Huang, R.; Tian, S.; Cai, R.; Sun, J.; Wang, S. Blood Pressure is Associated with Cerebral Blood Flow Alterations in Patients with T2DM as Revealed by Perfusion Functional MRI. Medicine 2015, 94, e2231. [Google Scholar] [CrossRef] [PubMed]
- Chau, A.C.M.; Cheung, E.Y.W.; Chan, K.H.; Chow, W.S.; Shea, Y.F.; Chiu, P.K.; Mak, H.K. Impaired cerebral blood flow in type 2 diabetes mellitus—A comparative study with subjective cognitive decline, vascular dementia, and Alzheimer’s disease subjects. Neuroimage Clin. 2020, 27, 102302. [Google Scholar] [CrossRef]
- Meerwaldt, R.; Zeebregts, C.J.; Navis, G.; Hillebrands, J.-L.; Lefrandt, J.D.; Smit, A.J. Accumulation of Advanced Glycation End Products and Chronic Complications in ESRD Treated by Dialysis. Am. J. Kidney Dis. 2009, 53, 138–150. [Google Scholar] [CrossRef]
- Senatus, L.M.; Schmidt, A.M. The AGE-RAGE Axis: Implications for Age-Associated Arterial Diseases. Front. Genet. 2017, 8, 187. [Google Scholar] [CrossRef] [Green Version]
- Piga, R.; Naito, Y.; Kokura, S.; Handa, O.; Yoshikawa, T. Short-term high glucose exposure induces monocyte-endothelial cells adhesion and transmigration by increasing VCAM-1 and MCP-1 expression in human aortic endothelial cells. Atherosclerosis 2007, 193, 328–334. [Google Scholar] [CrossRef]
- Koushki, K.; Shahbaz, S.K.; Mashayekhi, K.; Sadeghi, M.; Zayeri, Z.D.; Taba, M.Y.; Banach, M.; Al-Rasadi, K.; Johnston, T.P.; Sahebkar, A. Anti-inflammatory Action of Statins in Cardiovascular Disease: The Role of Inflammasome and Toll-Like Receptor Pathways. Clin. Rev. Allergy Immunol. 2021, 60, 175–199. [Google Scholar] [CrossRef]
- Beverly, J.K.; Budoff, M.J. Atherosclerosis: Pathophysiology of insulin resistance, hyperglycemia, hyperlipidemia, and inflammation. J. Diabetes 2020, 12, 102–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sada, K.; Nishikawa, T.; Kukidome, D.; Yoshinaga, T.; Kajihara, N.; Sonoda, K.; Senokuchi, T.; Motoshima, H.; Matsumura, T.; Araki, E. Hyperglycemia Induces Cellular Hypoxia through Production of Mitochondrial ROS Followed by Suppression of Aquaporin-1. PLoS ONE 2016, 11, e0158619. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Kim, S.; Nagar, H.; Choi, S.-J.; Jeon, B.H.; Piao, S.; Kim, C.-S. CR6-interacting factor 1 deficiency reduces endothelial nitric oxide synthase activity by inhibiting biosynthesis of tetrahydrobiopterin. Sci. Rep. 2020, 10, 842. [Google Scholar] [CrossRef]
- Sun, H.-J.; Wu, Z.-Y.; Nie, X.-W.; Bian, J.-S. Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link Between Inflammation and Hydrogen Sulfide. Front. Pharmacol. 2020, 10, 1568. [Google Scholar] [CrossRef] [Green Version]
- Di Marco, E.; Jha, J.C.; Sharma, A.; Wilkinson-Berka, J.L.; Jandeleit-Dahm, K.A.; de Haan, J.B. Are reactive oxygen species still the basis for diabetic complications? Clin. Sci. 2015, 129, 199–216. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ Res. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Tabit, C.E.; Shenouda, S.M.; Holbrook, M.; Fetterman, J.L.; Kiani, S.; Frame, A.A.; Kluge, M.A.; Held, A.; Dohadwala, M.M.; Gokce, N.; et al. Protein kinase C-β contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation 2013, 127, 86–95. [Google Scholar] [CrossRef]
- Katakami, N. Mechanism of Development of Atherosclerosis and Cardiovascular Disease in Diabetes Mellitus. J. Atheroscler. Thromb. 2018, 25, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.I.; Pichna, B.A.; Shi, Y.; Bowes, A.J.; Werstuck, G. Evidence supporting a role for endoplasmic reticulum stress in the development of atherosclerosis in a hyperglycaemic mouse model. Antioxid. Redox. Signal. 2009, 11, 2289–2298. [Google Scholar] [CrossRef]
- Zeadin, M.G.; Petlura, C.I.; Werstuck, G.H. Molecular mechanisms linking diabetes to the accelerated development of atherosclerosis. Can. J. Diabetes. 2013, 37, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Chew, P.; Yuen, D.Y.; Stefanovic, N.; Pete, J.; Coughlan, M.T.; Jandeleit-Dahm, K.A.; Thomas, M.C.; Rosenfeldt, F.; Cooper, M.E.; de Haan, J.B. Antiatherosclerotic and renoprotective effects of ebselen in the diabetic apolipoprotein E/GPx1-double knockout mouse. Diabetes 2010, 59, 3198–3207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, M.; Borja, A.J.; Hancin, E.C.; Auslander, T.; Revheim, M.-E.; Moghbel, M.C.; Werner, T.J.; Alavi, A.; Rajapakse, C.S. Imaging Atherosclerosis by PET, With Emphasis on the Role of FDG and NaF as Potential Biomarkers for This Disorder. Front. Physiol. 2020, 11, 511391. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, J.M.; Smith, C.; Dichgans, M. Mechanisms of sporadic cerebral small vessel disease: Insights from neuroimaging. Lancet Neurol. 2013, 12, 483–497. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Rosenberg, G.A. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 2011, 42, 3323–3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candelario-Jalil, E.; Thompson, J.; Taheri, S.; Grossetete, M.; Adair, J.C.; Edmonds, E.; Prestopnik, J.; Wills, J.; Rosenberg, G.A. Matrix Metalloproteinases Are Associated with Increased Blood–Brain Barrier Opening in Vascular Cognitive Impairment. Stroke 2011, 42, 1345–1350. [Google Scholar] [CrossRef] [Green Version]
- Nag, S.; Venugopalan, R.; Stewart, D.J. Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood–brain barrier breakdown. Acta Neuropathol. 2007, 114, 459–469. [Google Scholar] [CrossRef]
- Nag, S.; Manias, J.L.; Stewart, D.J. Expression of endothelial phosphorylated caveolin-1 is increased in brain injury. Neuropathol. Appl. Neurobiol. 2009, 35, 417–426. [Google Scholar] [CrossRef]
- Higashida, T.; Kreipke, C.W.; Rafols, J.A.; Peng, C.; Schafer, S.; Schafer, P.; Ding, J.Y.; Dornbos, D., III; Li, X.; Guthikonda, M.; et al. The role of hypoxia-inducible factor-1α, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J. Neurosurg. 2011, 114, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Kanekiyo, T. Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1965. [Google Scholar] [CrossRef] [Green Version]
- Marceau, F.; Regoli, D. Bradykinin receptor ligands: Therapeutic perspectives. Nat. Rev. Drug Discov. 2004, 3, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Tan, J.; Wan, L.; Chen, C.; Wu, B.; Ke, X.; Wu, R.; Ran, X. Increase in Blood–Brain Barrier Permeability is Modulated by Tissue Kallikrein via Activation of Bradykinin B1 and B2 Receptor-Mediated Signaling. J. Inflamm. Res. 2021, 14, 4283–4297. [Google Scholar] [CrossRef] [PubMed]
- Leeb-Lundberg, L.M. Bradykinin specificity and signaling at GPR100 and B2 kinin receptors. Br. J. Pharmacol. 2004, 143, 931–932. [Google Scholar] [CrossRef] [Green Version]
- Verhoog, Q.P.; Holtman, L.; Aronica, E.; van Vliet, E.A. Astrocytes as Guardians of Neuronal Excitability: Mechanisms Underlying Epileptogenesis. Front. Neurol. 2020, 11, 591690. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, S.; Sonobe, Y.; Cheng, Y.; Horiuchi, H.; Parajuli, B.; Kawanokuchi, J.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Interleukin-1β induces blood-brain barrier disruption by downregulating Sonic hedgehog in astrocytes. PLoS ONE 2014, 9, e110024. [Google Scholar] [CrossRef] [Green Version]
- da Fonseca, A.C.C.; Matias, D.; Garcia, C.; Amaral, R.; Geraldo, L.H.; Freitas, C.; Lima, F.R.S. The impact of microglial activation on blood-brain barrier in brain diseases. Front. Cell. Neurosci. 2014, 8, 362.e. [Google Scholar] [CrossRef] [Green Version]
- Burhans, M.S.; Hagman, D.K.; Kuzma, J.N.; Schmidt, K.A.; Kratz, M. Contribution of Adipose Tissue Inflammation to the Development of Type 2 Diabetes Mellitus. Compr. Physiol. 2018, 9, 1–58.e. [Google Scholar]
- Serlin, Y.; Levy, J.; Shalev, H. Vascular Pathology and Blood-Brain Barrier Disruption in Cognitive and Psychiatric Complications of Type 2 Diabetes Mellitus. Cardiovasc. Psychiatry Neurol. 2011, 2011, 609202. [Google Scholar]
- Wang, J.; Li, G.; Wang, Z.; Zhang, X.; Yao, L.; Wang, F.; Liu, S.; Yin, J.; Ling, E.-A.; Wang, L.; et al. High glucose-induced expression of inflammatory cytokines and reactive oxygen species in cultured astrocytes. Neuroscience 2012, 202, 58–68. [Google Scholar] [CrossRef]
- Prasad, S.; Sajja, R.K.; Naik, P.; Cucullo, L. Diabetes Mellitus and Blood-Brain Barrier Dysfunction: An Overview. J. Pharmacovigil. 2014, 2, 125. [Google Scholar]
- Shimizu, F.; Sano, Y.; Tominaga, O.; Maeda, T.; Abe, M.-A.; Kanda, T. Advanced glycation end-products disrupt the blood–brain barrier by stimulating the release of transforming growth factor–β by pericytes and vascular endothelial growth factor and matrix metalloproteinase–2 by endothelial cells in vitro. Neurobiol. Aging 2013, 34, 1902–1912. [Google Scholar] [CrossRef]
- Rochfort, K.; Collins, L.E.; Murphy, R.P.; Cummins, P.M. Downregulation of Blood-Brain Barrier Phenotype by Proinflammatory Cytokines Involves NADPH Oxidase-Dependent ROS Generation: Consequences for Interendothelial Adherens and Tight Junctions. PLoS ONE 2014, 9, e101815. [Google Scholar] [CrossRef] [Green Version]
- Bogush, M.; Heldt, N.A.; Persidsky, Y. Blood Brain Barrier Injury in Diabetes: Unrecognized Effects on Brain and Cognition. J. Neuroimmune Pharmacol. 2017, 12, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Papachristoforou, E.; Lambadiari, V.; Maratou, E.; Makrilakis, K. Association of Glycemic Indices (Hyperglycemia, Glucose Variability, and Hypoglycemia) with Oxidative Stress and Diabetic Complications. J. Diabetes Res. 2020, 2020, 7489795. [Google Scholar] [CrossRef]
- Cipolla, M.J.; Huang, Q.; Sweet, J.G. Inhibition of Protein Kinase Cβ Reverses Increased Blood–Brain Barrier Permeability During Hyperglycemic Stroke and Prevents Edema Formation In Vivo. Stroke 2011, 42, 3252–3257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, K.; Bayarsaikhan, E.; Kim, D.; Son, M.; Hong, J.; Jeong, G.-B.; Paek, S.H.; Won, M.-H.; Lee, B. Activated microglial cells synthesize and secrete AGE-albumin. Anat. Cell Biol. 2012, 45, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Guerin-Dubourg, A.; Catan, A.; Bourdon, E.; Rondeau, P. Structural modifications of human albumin in diabetes. Diabetes Metab. 2012, 38, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Rehman, K.; Akash, M.S. Mechanisms of inflammatory responses and development of insulin resistance: How are they interlinked? J. Biomed. Sci. 2016, 23, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, K.F.; Sweat, V.; Yau, P.L.; Turchiano, M.M.; Convit, A. Impact of metabolic syndrome on cognition and brain: A selected review of the literature. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2060–2067. [Google Scholar] [CrossRef] [Green Version]
- Daiber, A.; Steven, S.; Vujacic-Mirski, K.; Kalinovic, S.; Oelze, M.; Di Lisa, F.; Münzel, T. Regulation of Vascular Function and Inflammation via Cross Talk of Reactive Oxygen and Nitrogen Species from Mitochondria or NADPH Oxidase-Implications for Diabetes Progression. Int. J. Mol. Sci. 2020, 21, 3405. [Google Scholar] [CrossRef]
- Cathcart, M.K. Regulation of superoxide anion production by NADPH oxidase in monocytes/macrophages: Contributions to atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Vendrov, A.E.; Hakim, Z.S.; Madamanchi, N.R.; Rojas, M.; Madamanchi, C.; Runge, M.S. Atherosclerosis Is Attenuated by Limiting Superoxide Generation in Both Macrophages and Vessel Wall Cells. Arter. Thromb. Vasc. Biol. 2007, 27, 2714–2721. [Google Scholar] [CrossRef] [Green Version]
- Rueckschloss, U.; Duerrschmidt, N.; Morawietz, H. NADPH Oxidase in Endothelial Cells: Impact on Atherosclerosis. Antioxidants Redox Signal. 2003, 5, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Tarafdar, A.; Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 2018, 19, 3824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Xue, Q.; Liu, Y.; Qi, H.; Ma, Q.; Xu, L.; Chen, W.; Chen, G.; Xu, X. A Novel Brain Neurovascular Unit Model with Neurons, Astrocytes and Microvascular Endothelial Cells of Rat. Int. J. Biol. Sci. 2013, 9, 174–189. [Google Scholar] [CrossRef]
- Bergaglio, T.; Luchicchi, A.; Schenk, G.J. Engine Failure in Axo-Myelinic Signaling: A Potential Key Player in the Pathogenesis of Multiple Sclerosis. Front. Cell. Neurosci. 2021, 15, 610295. [Google Scholar] [CrossRef]
- Nave, K.-A. Myelination and the trophic support of long axons. Nat. Rev. Neurosci. 2010, 11, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Hu, X.; Leak, R.K.; Shi, Y.; An, C.; Suenaga, J.; Chen, J.; Gao, Y. Demyelination as a rational therapeutic target for ischemic or traumatic brain injury. Exp. Neurol. 2015, 272, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Matute, C.; Ransom, B.R. Roles of white matter in central nervous system pathophysiologies. ASN Neuro. 2012, 4, e00079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, S.T.; McNeil, P.L. Membrane Repair: Mechanisms and Pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef] [Green Version]
- Gefen, T.; Kim, G.; Bolbolan, K.; Geoly, A.; Ohm, D.; Oboudiyat, C.; Shahidehpour, R.; Rademaker, A.; Weintraub, S.; Bigio, E.H.; et al. Activated Microglia in Cortical White Matter Across Cognitive Aging Trajectories. Front. Aging Neurosci. 2019, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nave, K.-A. Myelination and support of axonal integrity by glia. Nature 2010, 468, 244–252. [Google Scholar] [CrossRef]
- Stone, D.B.; Tesche, C.D. Topological dynamics in spike-timing dependent plastic model neural networks. Front. Neural Circuits 2013, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Haight, T.J.; Landau, S.M.; Carmichael, O.; Schwarz, C.; DeCarli, C.; Jagust, W.J. Alzheimer’s Disease Neuroimaging Initiative. Dissociable effects of Alzheimer disease and white matter hyperintensities on brain metabolism. JAMA Neurol. 2013, 70, 1039–1045. [Google Scholar] [CrossRef]
- Lawrence, A.J.; Patel, B.; Morris, R.G.; MacKinnon, A.D.; Rich, P.M.; Barrick, T.R.; Markus, H.S. Mechanisms of cognitive impairment in cerebral small vessel disease: Multimodal MRI results from the St George’s cognition and neuroimaging in stroke (SCANS) study. PLoS ONE 2013, 8, e61014. [Google Scholar] [CrossRef]
- Sun, Y.-W.; Qin, L.-D.; Zhou, Y.; Xu, Q.; Qian, L.-J.; Tao, J.; Xu, J.-R. Abnormal functional connectivity in patients with vascular cognitive impairment, no dementia: A resting-state functional magnetic resonance imaging study. Behav. Brain Res. 2011, 223, 388–394. [Google Scholar] [CrossRef]
- Ferreira, D.; Perestelo-Pérez, L.; Westman, E.; Wahlund, L.O.; Sarría, A.; Serrano-Aguilar, P. Meta-Review of CSF Core Biomarkers in Alzheimer’s Disease: The State-of-the-Art after the New Revised Diagnostic Criteria. Front Aging Neurosci. 2014, 6, 47. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.S.H.; Hegde, V. Obesity and Diabetes Mediated Chronic Inflammation: A Potential Biomarker in Alzheimer’s Disease. J. Pers. Med. 2020, 10, 42. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [Google Scholar]
- Bature, F.; Guinn, B.; Pang, D.; Pappas, Y. Signs and symptoms preceding the diagnosis of Alzheimer’s disease: A systematic scoping review of literature from 1937 to 2016. BMJ Open 2017, 7, e015746. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Lin, X.; Lin, X.J.; Zheng, W.; Zheng, Z.K.; Lin, Z.M.; Chen, J.H. Therapeutic efficacy of neuromuscular electrical stimulation and electromyographic biofeedback on Alzheimer’s disease patients with dysphagia. Medicine 2017, 96, e8008. [Google Scholar] [CrossRef]
- Loera-Valencia, R.; Goikolea, J.; Parrado-Fernandez, C.; Merino-Serrais, P.; Maioli, S. Alterations in cholesterol metabolism as a risk factor for developing Alzheimer’s disease: Potential novel targets for treatment. J. Steroid Biochem. Mol. Biol. 2019, 190, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- US Burden of Disease Collaborators; Mokdad, A.H.; Ballestros, K.; Echko, M.; Glenn, S.; Olsen, H.E.; Mullany, E.; Lee, A.; Khan, A.R.; Ahmadi, A.; et al. The State of US Health, 1990–2016: Burden of Diseases, Injuries, and Risk Factors Among US States. JAMA 2018, 319, 1444–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanzi, R.E.; Bertram, L. Twenty Years of the Alzheimer’s Disease Amyloid Hypothesis: A Genetic Perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Blennow, K.; Hanse, E. Amyloid-beta and APP as biomarkers for Alzheimer’s disease. Exp. Gerontol. 2010, 45, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid-beta: Structure, biology, and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Tamagno, E.; Guglielmotto, M.; Vasciaveo, V.; Tabaton, M. Oxidative Stress and Beta-Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg? Antioxidants 2021, 10, 1479. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Abramov, A.Y. Mitochondrial Calcium Deregulation in the Mechanism of Beta-Amyloid and Tau Pathology. Cells 2020, 9, 2135. [Google Scholar] [CrossRef]
- Bhandary, B.; Marahatta, A.; Kim, H.-R.; Chae, H.-J. An Involvement of Oxidative Stress in Endoplasmic Reticulum Stress and Its Associated Diseases. Int. J. Mol. Sci. 2012, 14, 434–456. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, P.; Wu, J.; Yi, W.; Ping, L.; Xinxin, H. The Cascade of Oxidative Stress and Tau Protein Autophagic Dysfunction in Alzheimer’s Disease, Alzheimer’s Disease—Challenges for the Future; Zerr, I., Ed.; IntechOpen: London, UK, 2015; Available online: https://www.intechopen.com/chapters/48347 (accessed on 10 December 2021).
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef] [PubMed]
- Kneynsberg, A.; Combs, B.; Christensen, K.; Morfini, G.; Kanaan, N.M. Axonal Degeneration in Tauopathies: Disease Relevance and Underlying Mechanisms. Front. Neurosci. 2017, 11, 572. [Google Scholar] [CrossRef]
- Alonso, A.D.; Cohen, L.S.; Corbo, C.; Morozova, V.; ElIdrissi, A.; Phillips, G.; Kleiman, F.E. Hyperphosphorylation of Tau Associated with Changes in Its Function Beyond Microtubule Stability. Front. Cell Neurosci. 2018, 12, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocahan, S.; Doğan, Z. Mechanisms of Alzheimer’s Disease Pathogenesis and Prevention: The Brain, Neural Pathology, N-Methyl-D-aspartate Receptors, Tau Protein, and Other Risk Factors. Clin. Psychopharmacol. Neurosci. 2017, 15, 1–8. [Google Scholar] [CrossRef]
- Wang, S.C.-Y.; Neil, D.L.; Home, P. 2020 vision—An overview of prospects for diabetes management and prevention in the next decade. Diabetes Res. Clin. Pract. 2018, 143, 101–112. [Google Scholar] [CrossRef]
- Weuve, J.; Hebert, L.E.; Scherr, P.A.; Evans, D.A. Prevalence of Alzheimer Disease in US States. Epidemiology 2015, 26, e4–e6. [Google Scholar] [CrossRef]
- Lee, H.J.; Seo, H.I.; Cha, H.Y.; Yang, Y.J.; Kwon, S.H.; Yang, S.J. Diabetes, and Alzheimer’s Disease: Mechanisms and Nutritional Aspects. Clin. Nutr. Res. 2018, 7, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Song, D.; Leng, S.X. Link between type 2 diabetes and Alzheimer’s disease: From epidemiology to mechanism and treatment. Clin. Interv. Aging 2015, 10, 549–560. [Google Scholar] [CrossRef] [Green Version]
- LeWitt, M.S.; Boyd, G.W. The Role of Insulin-Like Growth Factors and Insulin-Like Growth Factor–Binding Proteins in the Nervous System. Biochem. Insights 2019, 12, 1178626419842176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinohara, M.; Sato, N. Bidirectional interactions between diabetes and Alzheimer’s disease. Neurochem. Int. 2017, 108, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sankowski, R.; Mader, S.; Valdés-Ferrer, S.I. Systemic inflammation and the brain: Novel roles of genetic, molecular, and environmental cues as drivers of neurodegeneration. Front. Cell. Neurosci. 2015, 9, 28. [Google Scholar] [CrossRef] [Green Version]
- Chiroma, S.M.; Baharuldin, M.T.; Taib, C.N.; Amom, Z.; Jagadeesan, S.; Moklas, M.A.M. Inflammation in Alzheimer’s disease: A friend or foe? Biomed. Res. Ther. 2018, 5, 2552–2564. [Google Scholar] [CrossRef]
- Ma, Q.L.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Chen, P.P.; Hudspeth, B.; Chen, C.; Zhao, Y.; et al. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: Suppression by omega-3 fatty acids and curcumin. J. Neurosci. 2009, 29, 9078–9089. [Google Scholar] [CrossRef]
- Gabbouj, S.; Ryhänen, S.; Marttinen, M.; Wittrahm, R.; Takalo, M.; Kemppainen, S.; Martiskainen, H.; Tanila, H.; Haapasalo, A.; Hiltunen, M.; et al. Altered Insulin Signaling in Alzheimer’s Disease Brain—Special Emphasis on PI3K-Akt Pathway. Front. Neurosci. 2019, 13, 629. [Google Scholar] [CrossRef]
- Di Penta, A.; Moreno, B.; Reix, S.; Fernandez-Diez, B.; Villanueva, M.; Errea, O.; Escala, N.; Vandenbroeck, K.; Comella, J.; Villoslada, P. Oxidative stress and proinflammatory cytokines contribute to demyelination and axonal damage in a cerebellar culture model of neuroinflammation. PLoS ONE 2013, 8, e54722. [Google Scholar]
- Yoo, M.-H.; Gu, X.; Xu, X.-M.; Kim, J.-Y.; Carlson, B.A.; Patterson, A.D.; Cai, H.; Gladyshev, V.N.; Hatfield, D.L. Delineating the role of glutathione peroxidase 4 in protecting cells against lipid hydroperoxide damage and in Alzheimer’s disease. Antioxid. Redox. Signal 2010, 12, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Riemer, J.; Kins, S. Axonal transport and mitochondrial dysfunction in Alzheimer’s disease. Neurodegener. Dis. 2013, 12, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Yin, X.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP, and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef]
- Rigotto, G.; Basso, E. Mitochondrial Dysfunctions: A Thread Sewing Together Alzheimer’s Disease, Diabetes, and Obesity. Oxid. Med. Cell Longev. 2019, 2019, 7210892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Felice, F.G.; Ferreira, S.T. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef] [Green Version]
- Kusminski, C.M.; Scherer, P.E. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol. Metab. 2012, 23, 435–443. [Google Scholar] [CrossRef] [Green Version]
- Boudina, S.; Graham, T.E. Mitochondrial function/dysfunction in white adipose tissue. Exp. Physiol. 2014, 99, 1168–1178. [Google Scholar] [CrossRef]
- Wang, C.H.; Wei, Y.H. Role of mitochondrial dysfunction and dysregulation of Ca2+ homeostasis in the pathophysiology of insulin resistance and type 2 diabetes. J. Biomed. Sci. 2017, 24, 70. [Google Scholar] [CrossRef]
- Sarasija, S.; Laboy, J.T.; Ashkavand, Z.; Bonner, J.; Tang, Y.; Norman, K.R. Presenilin mutations deregulate mitochondrial Ca2+ homeostasis and metabolic activity, causing neurodegeneration in Caenorhabditis elegans. Elife 2018, 7, e33052. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Deficient brain insulin signaling pathway in Alzheimer’s disease and diabetes. J. Pathol. 2011, 225, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Feldman, E.L. Insulin resistance in the nervous system. Trends Endocrinol. Metab. 2012, 23, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Duarte, A.I.; Moreira, P.I.; Oliveira, C.R. Insulin in Central Nervous System: More than Just a Peripheral Hormone. J. Aging Res. 2012, 2012, 384017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Mudher, A. Alzheimer’s Disease and Type 2 Diabetes: A Critical Assessment of the Shared Pathological Traits. Front Neurosci. 2018, 12, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdile, G.; Fuller, S.J.; Martins, R.N. The role of type 2 diabetes in neurodegeneration. Neurobiol. Dis. 2015, 84, 22–38. [Google Scholar] [CrossRef]
- Li, X.-H.; Du, L.-L.; Cheng, X.-S.; Jiang, X.; Zhang, Y.; Lv, B.-L.; Liu, R.; Wang, J.-Z.; Zhou, X.-W. Glycation exacerbates the neuronal toxicity of β-amyloid. Cell Death Dis. 2013, 4, e673. [Google Scholar] [CrossRef] [Green Version]
- Moreira, P.I.; Santos, M.S.; Moreno, A.M.; Seiça, R.; Oliveira, C.R. Increased vulnerability of brain mitochondria in diabetic (Goto-Kakizaki) rats with aging and amyloid-beta exposure. Diabetes 2003, 52, 1449–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef] [Green Version]
- Yin, F.; Boveris, A.; Cadenas, E. Mitochondrial Energy Metabolism and Redox Signaling in Brain Aging and Neurodegeneration. Antioxidants Redox Signal. 2014, 20, 353–371. [Google Scholar] [CrossRef] [Green Version]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Afridi, R.; Kim, J.-H.; Rahman, H.; Suk, K. Metabolic Regulation of Glial Phenotypes: Implications in Neuron–Glia Interactions and Neurological Disorders. Front. Cell. Neurosci. 2020, 14, 20. [Google Scholar] [CrossRef]
- Doll, D.N.; Rellick, S.L.; Barr, T.L.; Ren, X.; Simpkins, J.W. Rapid mitochondrial dysfunction mediates TNF-alpha-induced neurotoxicity. J. Neurochem. 2015, 132, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Wagner, P.D.; Breen, E.C. TNF-alpha-mediated reduction in PGC-1alpha may impair skeletal muscle function after cigarette smoke exposure. J. Cell Physiol. 2010, 222, 320–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, C.; Jiang, Y.; Wang, S.; Wu, X.; Wang, K. PPARγ coactivator-1α (PGC-1α) protects neuroblastoma cells against amyloid-beta (Aβ) induced cell death and neuroinflammation via NF-κB pathway. BMC Neurosci. 2017, 18, 69. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, C.; Di Domenico, F.; Perluigi, M.; Butterfield, D.A. Targeting Mitochondria in Alzheimer Disease: Rationale and Perspectives. CNS Drugs 2019, 33, 957–969. [Google Scholar] [CrossRef]
- Moya-Alvarado, G.; Gershoni-Emek, N.; Perlson, E.; Bronfman, F.C. Neurodegeneration and Alzheimer’s disease (AD). What Can Proteomics Tell Us About the Alzheimer’s Brain? Mol. Cell Proteom. 2016, 15, 409–425. [Google Scholar] [CrossRef] [Green Version]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid-beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef]
- Rajmohan, R.; Reddy, P.H. Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer’s disease Neurons. J. Alzheimer’s Dis. 2017, 57, 975–999. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-K.; Nam, K.I.; Song, J. The Glymphatic System in Diabetes-Induced Dementia. Front. Neurol. 2018, 9, 867. [Google Scholar] [CrossRef]
- Sripetchwandee, J.; Chattipakorn, N.; Chattipakorn, S.C. Links Between Obesity-Induced Brain Insulin Resistance, Brain Mitochondrial Dysfunction, and Dementia. Front. Endocrinol. 2018, 9, 496. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P.; Wood, I.S. Signaling role of adipose tissue: Adipokines and inflammation in obesity. Biochem. Soc. Trans. 2005, 33, 1078–1081. [Google Scholar] [CrossRef] [Green Version]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, V.; Werner, E.D.; Giraud, J.; Lee, Y.H.; Shoelson, S.E.; White, M.F. Phosphorylation of Ser307 in Insulin Receptor Substrate-1 Blocks Interactions with the Insulin Receptor and Inhibits Insulin Action. J. Biol. Chem. 2002, 277, 1531–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Zhang, X.; Zuberi, A.; Hwang, D.; Quon, M.; Lefevre, M.; Ye, J. Inhibition of Insulin Sensitivity by Free Fatty Acids Requires Activation of Multiple Serine Kinases in 3T3-L1 Adipocytes. Mol. Endocrinol. 2004, 18, 2024–2034. [Google Scholar] [CrossRef] [PubMed]
- Werner, E.D.; Lee, J.; Hansen, L.; Yuan, M.; Shoelson, S.E. Insulin Resistance Due to Phosphorylation of Insulin Receptor Substrate-1 at Serine 302. J. Biol. Chem. 2004, 279, 35298–35305. [Google Scholar] [CrossRef] [Green Version]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Le Marchand-Brustel, Y.; Gual, P.; Grémeaux, T.; Gonzalez, T.; Barrès, R.; Tanti, J.F. Fatty acid-induced insulin resistance: Role of insulin receptor substrate 1 serine phosphorylation in the retroregulation of insulin signaling. Biochem. Soc. Trans. 2003, 31, 1152–1156. [Google Scholar] [CrossRef] [PubMed]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Arita, Y.; Kiharaa, S.; Ouchia, N.; Takahashia, M.; Maedaa, K.; Ichiromiyagawaa, J.; Hottaa, K.; Shimomuraa, I.; Nakamuraa, T.; Miyaokaa, K.; et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun. 1999, 257, 79–83. [Google Scholar] [CrossRef]
- Ishii, M.; Iadecola, C. Adipocyte-derived factors in age-related dementia and their contribution to vascular and Alzheimer pathology. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hu, W.; Jiang, S.; Wang, B.; Li, Y.; Fan, C.; Di, S.; Ma, Z.; Lau, W.B.; Qu, Y. The emerging role of adiponectin in cerebrovascular and neurodegenerative diseases. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 1887–1894. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, M.; Kawano, T.; Saito, T.; Yuzawa, M.; Saito, T.; Ikoma, A.; Tamemoto, H.; Kawakami, M.; Ishikawa, S.-E. Hypoadiponectinemia in Patients with Cerebral Infarction: Comparison with Other Atherosclerotic Disorders. Am. J. Med Sci. 2007, 333, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.P.; Tsai, J.C.; Chung, F.M.; Yang, S.S.; Hsing, L.L.; Shin, S.J.; Lee, Y.J. Hypoadiponectinemia is associated with ischemic cerebrovascular disease. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 821–826. [Google Scholar] [CrossRef] [Green Version]
- Gorelick, P.B.; Counts, S.E.; Nyenhuis, D. Vascular cognitive impairment and dementia. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 860–868. [Google Scholar] [CrossRef]
- Iliff, J.J.; Goldman, S.; Nedergaard, M. Implications of the discovery of brain lymphatic pathways. Lancet Neurol. 2015, 14, 977–979. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Ineichen, B.V.; Detmar, M.; Proulx, S.T. Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nat. Commun. 2017, 8, 1434. [Google Scholar] [CrossRef] [Green Version]
- Achariyar, T.M.; Li, B.; Peng, W.; Verghese, P.B.; Shi, Y.; McConnell, E.; Benraiss, A.; Kasper, T.; Song, W.; Takano, T.; et al. Glymphatic distribution of CSF-derived apoE into brain is isoform specific and suppressed during sleep deprivation. Mol. Neurodegener. 2016, 11, 74. [Google Scholar] [CrossRef] [Green Version]
- Thrane, V.R.; Thrane, A.S.; Plog, B.A.; Thiyagarajan, M.; Iliff, J.J.; Deane, R.; Nagelhus, E.A.; Nedergaard, M. Paravascular microcirculation facilitates rapid lipid transport and astrocyte signaling in the brain. Sci. Rep. 2013, 3, 2582. [Google Scholar] [CrossRef]
- Berridge, C.W.; Waterhouse, B.D. The locus coeruleus–noradrenergic system: Modulation of behavioral state and state-dependent cognitive processes. Brain Res. Rev. 2003, 42, 33–84. [Google Scholar] [CrossRef]
- Jessen, N.A.; Munk, A.S.F.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef] [Green Version]
- Grandner, M.A.; Patel, N.P.; Jean-Louis, G.; Jackson, N.; Gehrman, P.R.; Perlis, M.L.; Gooneratne, N.S. Sleep-Related Behaviors and Beliefs Associated With Race/Ethnicity in Women. J. Natl. Med Assoc. 2013, 105, 4–15. [Google Scholar] [CrossRef] [Green Version]
- Parashar, A.; Mehta, V.; Malairaman, U. Type 2 Diabetes Mellitus Is Associated with Social Recognition Memory Deficit and Altered Dopaminergic Neurotransmission in the Amygdala. Ann. Neurosci. 2018, 24, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Al Khatib, H.K.; Harding, S.V.; Darzi, J.; Pot, G.K. The effects of partial sleep deprivation on energy balance: A systematic review and meta-analysis. Eur. J. Clin. Nutr. 2017, 71, 614–624. [Google Scholar] [CrossRef]
- Verheggen, I.; Van Boxtel, M.; Verhey, F.; Jansen, J.; Backes, W. Interaction between blood-brain barrier and glymphatic system in solute clearance. Neurosci. Biobehav. Rev. 2018, 90, 26–33. [Google Scholar] [CrossRef]
- Morris, A.W.; Carare, R.O.; Schreiber, S.; Hawkes, C.A. The Cerebrovascular Basement Membrane: Role in the Clearance of β-amyloid and Cerebral Amyloid Angiopathy. Front Aging Neurosci. 2014, 6, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sironi, L.; Guerrini, U.; Tremoli, E.; Miller, I.; Gelosa, P.; Lascialfari, A.; Zucca, I.; Eberini, I.; Gemeiner, M.; Paoletti, R.; et al. Analysis of pathological events at the onset of brain damage in stroke-prone rats: A proteomics and magnetic resonance imaging approach. J. Neurosci. Res. 2004, 78, 115–122. [Google Scholar] [CrossRef]
- Van de Haar, H.J.; Burgmans, S.; Jansen, J.F.; van Osch, M.J.P.; van Buchem, M.A.; Muller, M.; Hofman, P.A.M.; Verhey, F.R.J.; Backes, W.H. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef]
- Brandon, J.A.; Farmer, B.C.; Williams, H.C.; Johnson, L.A. APOE and Alzheimer’s Disease: Neuroimaging of Metabolic and Cerebrovascular Dysfunction. Front. Aging Neurosci. 2018, 10, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaraman, A.; Pike, C.J. Alzheimer’s disease and type 2 diabetes: Multiple mechanisms contribute to interactions. Curr. Diab. Rep. 2014, 14, 476. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms, and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vagelatos, N.T.; Eslick, G.D. Type 2 diabetes as a risk factor for Alzheimer’s disease: The confounders, interactions, and neuropathology associated with this relationship. Epidemiol. Rev. 2013, 35, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.C.; Zimmer, E.R.; Rosa-Neto, P.; Yoon, S.O. Consequences of Metabolic Disruption in Alzheimer’s Disease Pathology. Neurotherapeutics 2019, 16, 600–610. [Google Scholar] [CrossRef]
- Delikkaya, B.; Moriel, N.; Tong, M.; Gallucci, G.; Suzanne, M. Altered expression of insulin-degrading enzyme and regulator of calcineurin in the rat intracerebral streptozotocin model and human apolipoprotein E-ε4-associated Alzheimer’s disease. Alzheimer’s Dement. 2019, 11, 392–404. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.-C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Morris, J.K.; Uy, R.A.Z.; Vidoni, E.D.; Wilkins, H.M.; Archer, A.E.; Thyfault, J.P.; Miles, J.M.; Burns, J.M. Effect of APOE ε4 Genotype on Metabolic Biomarkers in Aging and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 1129–1135. [Google Scholar] [CrossRef] [Green Version]
- Geijselaers, S.L.; Aalten, P.; Ramakers, I.H.; De Deyn, P.P.; Heijboer, A.C.; Koek, H.L.; OldeRikkert, M.G.; Papma, J.M.; Reesink, F.E.; Smits, L.L.; et al. Association of Cerebrospinal Fluid (CSF) Insulin with Cognitive Performance and CSF Biomarkers of Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 61, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimer’s Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claxton, A.; Baker, L.D.; Wilkinson, C.W.; Trittschuh, E.H.; Chapman, D.; Watson, G.S.; Cholerton, B.; Plymate, S.R.; Arbuckle, M.; Craft, S. Sex and ApoE genotype differences in treatment response to two doses of intranasal insulin in adults with mild cognitive impairment or Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 35, 789–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Zhang, X.; Zhao, L. Human ApoE Isoforms Differentially Modulate Brain Glucose and Ketone Body Metabolism: Implications for Alzheimer’s Disease Risk Reduction and Early Intervention. J. Neurosci. 2018, 38, 6665–6681. [Google Scholar] [CrossRef]
- Zhao, N.; Liu, C.-C.; Van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Johnson, L.; Torres, E.R.; Boutros, S.; Patel, E.; Akinyeke, T.; Alkayed, N.J.; Raber, J. Apolipoprotein E4 mediates insulin resistance-associated cerebrovascular dysfunction and the post-prandial response. J. Cereb. Blood Flow Metab. 2019, 39, 770–781. [Google Scholar] [CrossRef] [Green Version]
- Ravona-Springer, R.; Schnaider-Beeri, M. The association of diabetes and dementia and possible implications for non-diabetic populations. Expert Rev. Neurother. 2011, 11, 1609–1617. [Google Scholar] [CrossRef] [Green Version]
- de la Monte, S.M. Early intranasal insulin therapy halts progression of neurodegeneration: Progress in Alzheimer’s disease therapeutics. Aging Health 2012, 8, 61–64. [Google Scholar] [CrossRef] [Green Version]
- Bethel, M.A.; A Patel, R.; Merrill, P.; Lokhnygina, Y.; Buse, J.B.; Mentz, R.J.; Pagidipati, N.J.; Chan, J.C.; Gustavson, S.M.; Iqbal, N.; et al. Cardiovascular outcomes with glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes: A meta-analysis. Lancet Diabetes Endocrinol. 2018, 6, 105–113. [Google Scholar] [CrossRef]
- Ballard, C.; Nørgaard, C.H.; Friedrich, S.; Mørch, L.S.; Gerds, T.; Møller, D.V.; Knudsen, L.B.; Kvist, K.; Zinman, B.; Holm, E.; et al. Liraglutide and semaglutide: Pooled post hoc analysis to evaluate risk of dementia in patients with type 2 diabetes. Alzheimer’s Dement. 2020, 16, e042909. [Google Scholar] [CrossRef]
- Diabetes drug on trial for Alzheimer’s. Nat. Biotechnol. 2021, 39, 127. [CrossRef]
- Femminella, G.D.; Frangou, E.; Love, S.B.; Busza, G.; Holmes, C.; Ritchie, C.; Lawrence, R.; McFarlane, B.; Tadros, G.; Ridha, B.H.; et al. Evaluating the effects of the novel GLP-1 analog liraglutide in Alzheimer’s disease: Study protocol for a randomized controlled trial (ELAD study). Trials 2019, 20, 191. [Google Scholar] [CrossRef]
- Hansen, H.H.; Fabricius, K.; Barkholt, P.; Niehoff, M.L.; Morley, J.E.; Jelsing, J.; Pyke, C.; Knudsen, L.B.; Farr, S.A.; Vrang, N. The GLP-1 Receptor Agonist Liraglutide Improves Memory Function and Increases Hippocampal CA1 Neuronal Numbers in a Senescence-Accelerated Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 46, 877–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biessels, G.J.; Despa, F. Cognitive decline and dementia in diabetes mellitus: Mechanisms and clinical implications. Nat. Rev. Endocrinol. 2018, 14, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Cholerton, B.; Baker, L.D.; Montine, T.J.; Craft, S. Type 2 Diabetes, Cognition, and Dementia in Older Adults: Toward a Precision Health Approach. Diabetes Spectr. 2016, 29, 210–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, R.; Chowdhury, K.; Kumar, S.; Irfan, M.; Reddy, G.S.; Akter, F.; Jahan, D.; Haque, M. Diabetes Mellitus: A Path to Amnesia, Personality, and Behavior Change. Biology 2022, 11, 382. https://doi.org/10.3390/biology11030382
Ahmad R, Chowdhury K, Kumar S, Irfan M, Reddy GS, Akter F, Jahan D, Haque M. Diabetes Mellitus: A Path to Amnesia, Personality, and Behavior Change. Biology. 2022; 11(3):382. https://doi.org/10.3390/biology11030382
Chicago/Turabian StyleAhmad, Rahnuma, Kona Chowdhury, Santosh Kumar, Mohammed Irfan, Govindool Sharaschandra Reddy, Farhana Akter, Dilshad Jahan, and Mainul Haque. 2022. "Diabetes Mellitus: A Path to Amnesia, Personality, and Behavior Change" Biology 11, no. 3: 382. https://doi.org/10.3390/biology11030382
APA StyleAhmad, R., Chowdhury, K., Kumar, S., Irfan, M., Reddy, G. S., Akter, F., Jahan, D., & Haque, M. (2022). Diabetes Mellitus: A Path to Amnesia, Personality, and Behavior Change. Biology, 11(3), 382. https://doi.org/10.3390/biology11030382