
5mC-Related lncRNAs as Potential Prognostic Biomarkers in Colon Adenocarcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
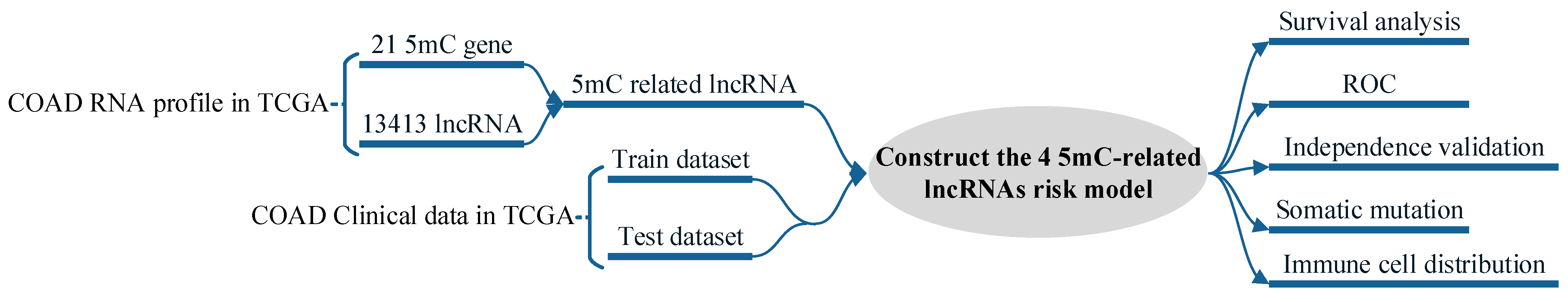
2. Materials and Methods
2.1. Data Obtaining and Correlation Analysis
2.2. Identification and Construction of Risk Signature
2.3. Evaluation and Verification of the 5mC-Related lncRNA Prognosis Signature
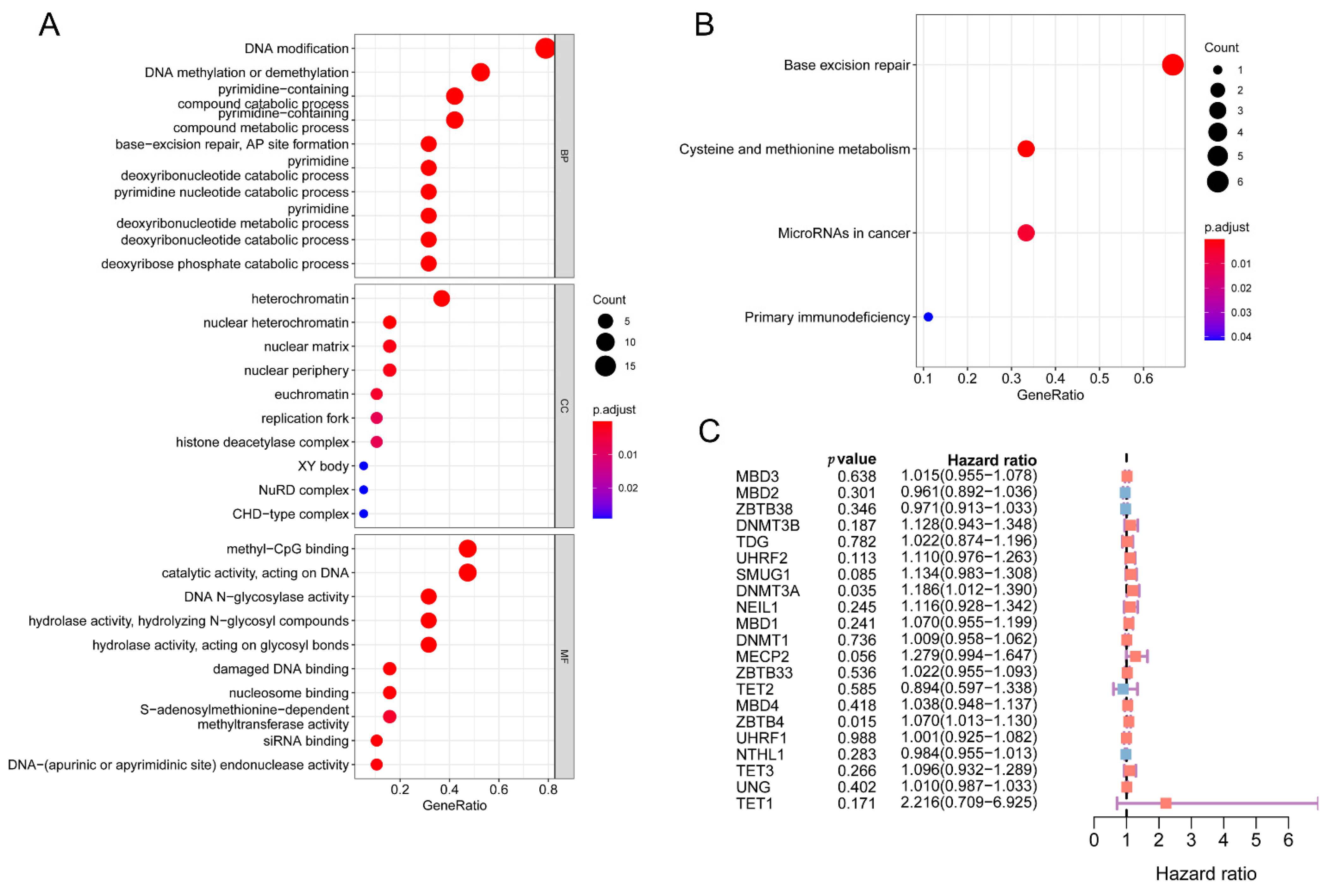
2.4. Function Enrichment Analysis and Immunization Score Analysis
2.5. Cell Culture and Reverse Transcription and Quantitative PCR Analysis (RT-qPCR)
2.6. Statistical Analyses
3. Results
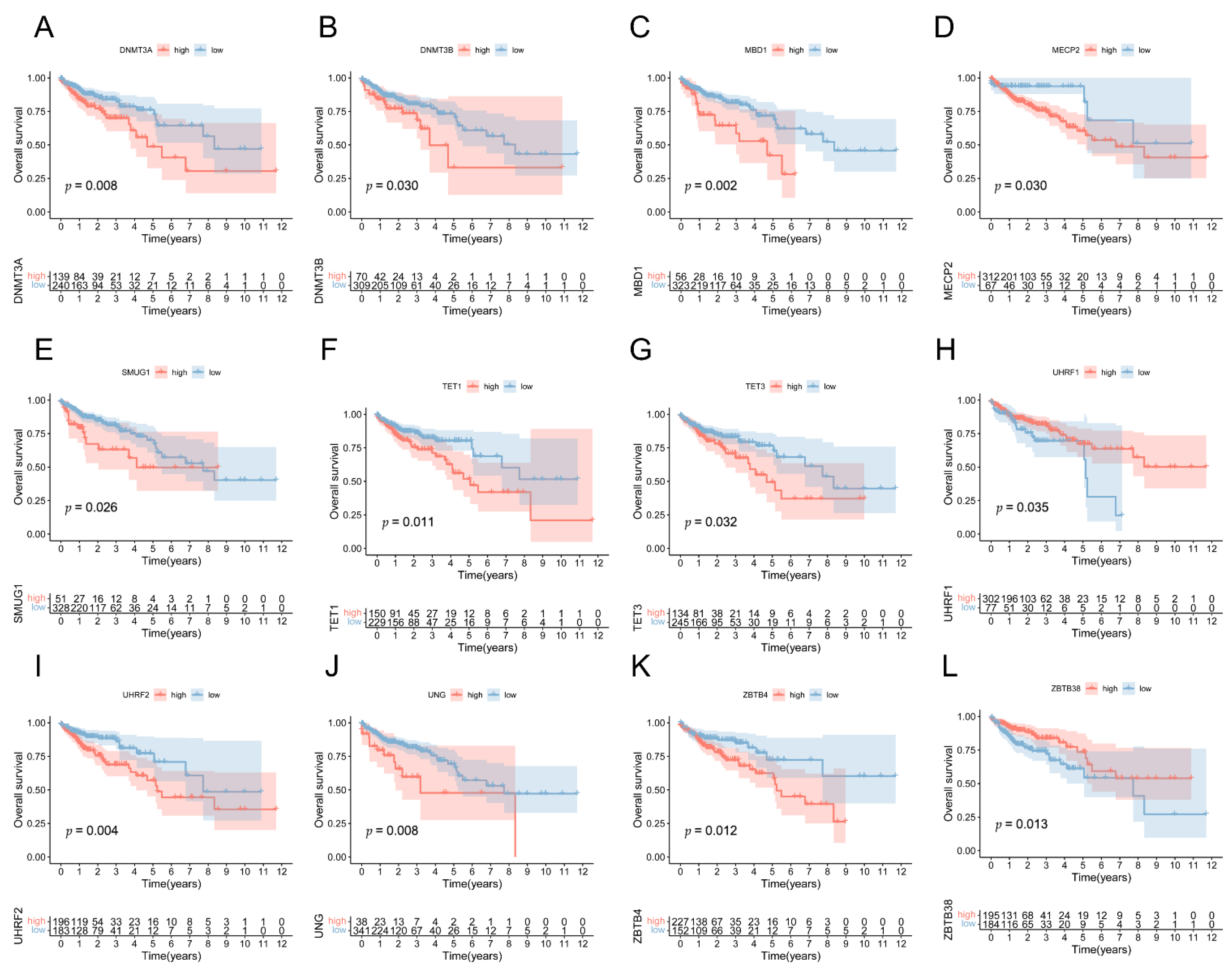
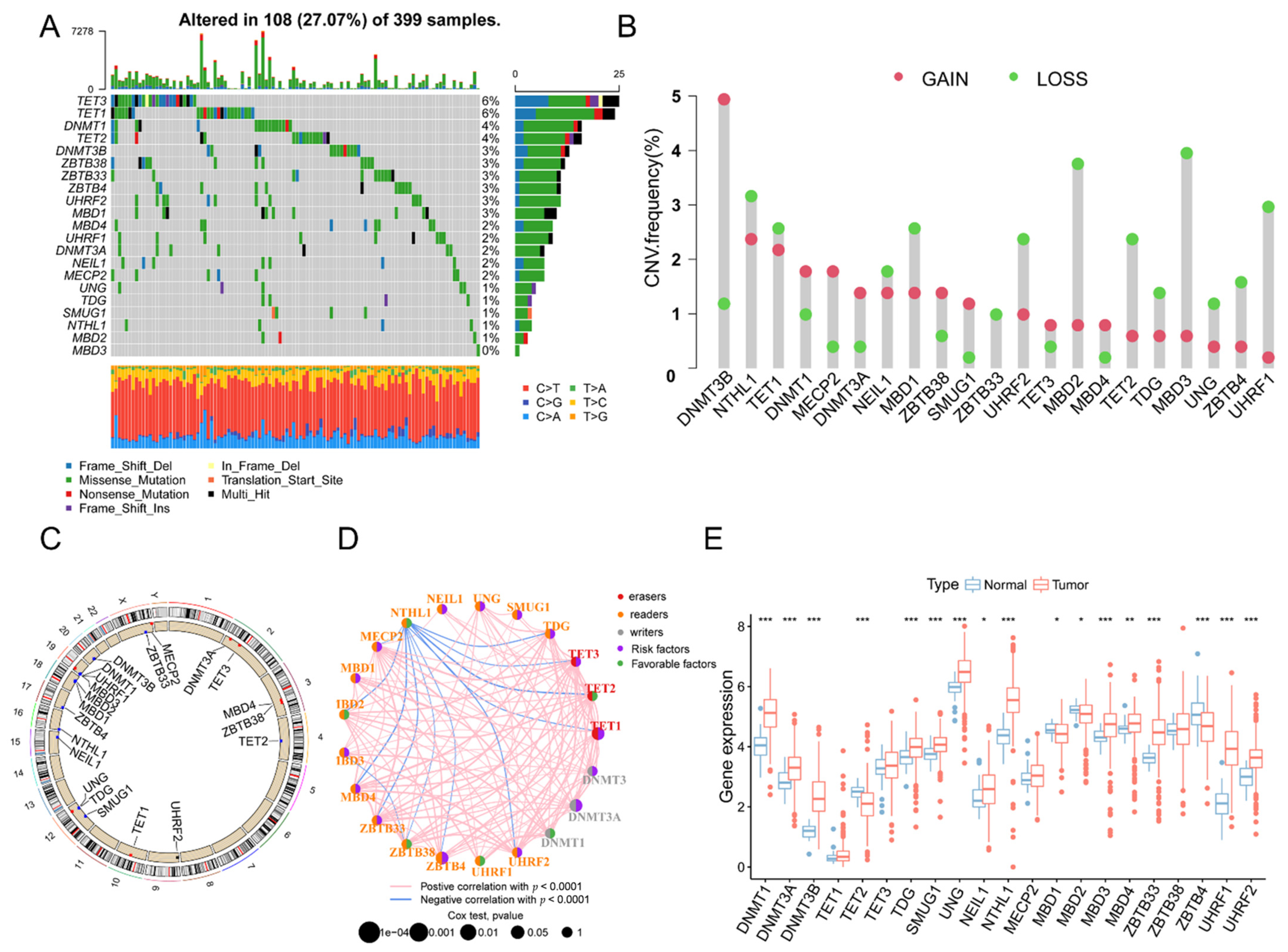
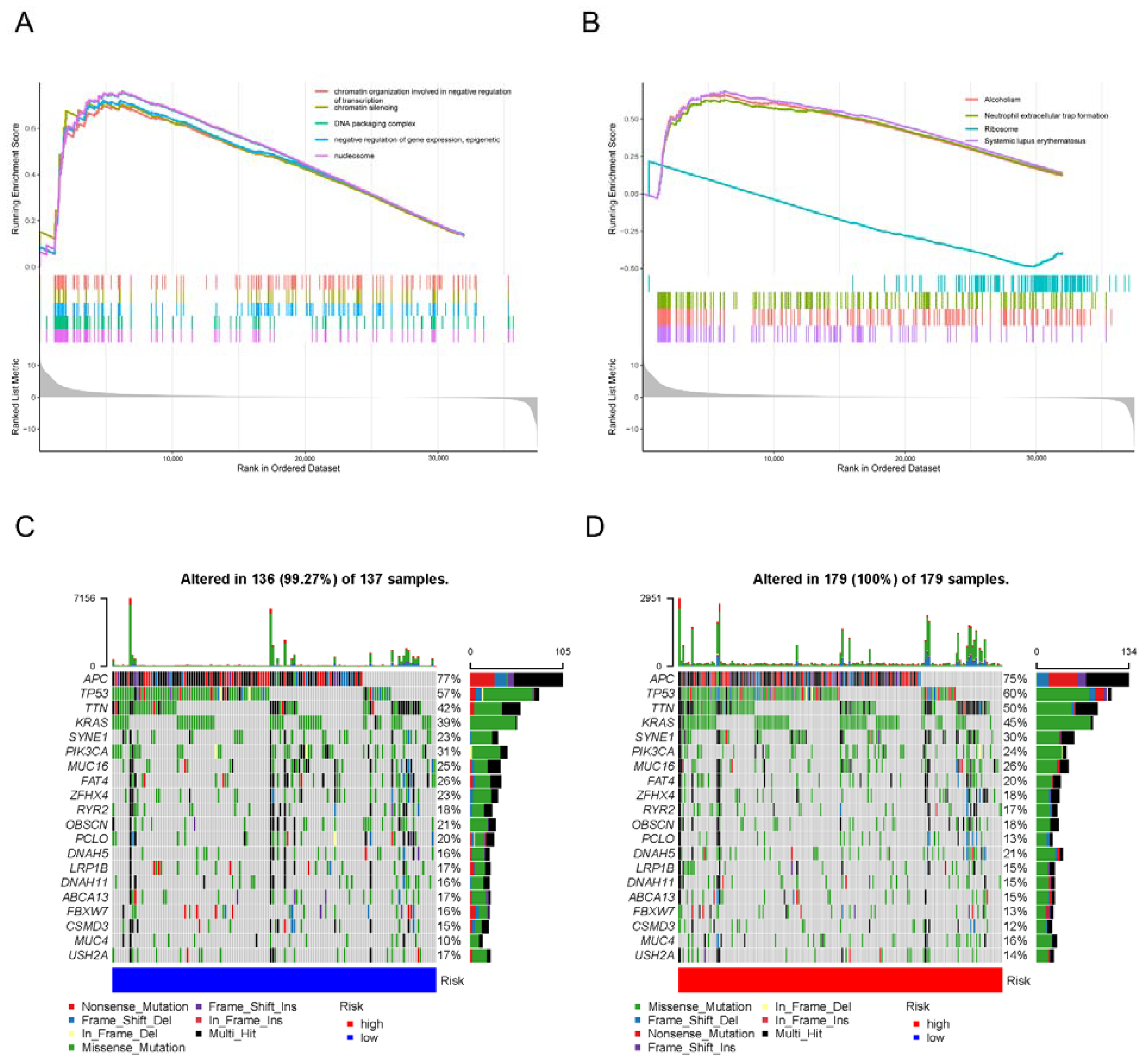
3.1. The Landscape of Genetic Variation of 5mC Regulatory Genes in COAD Patients
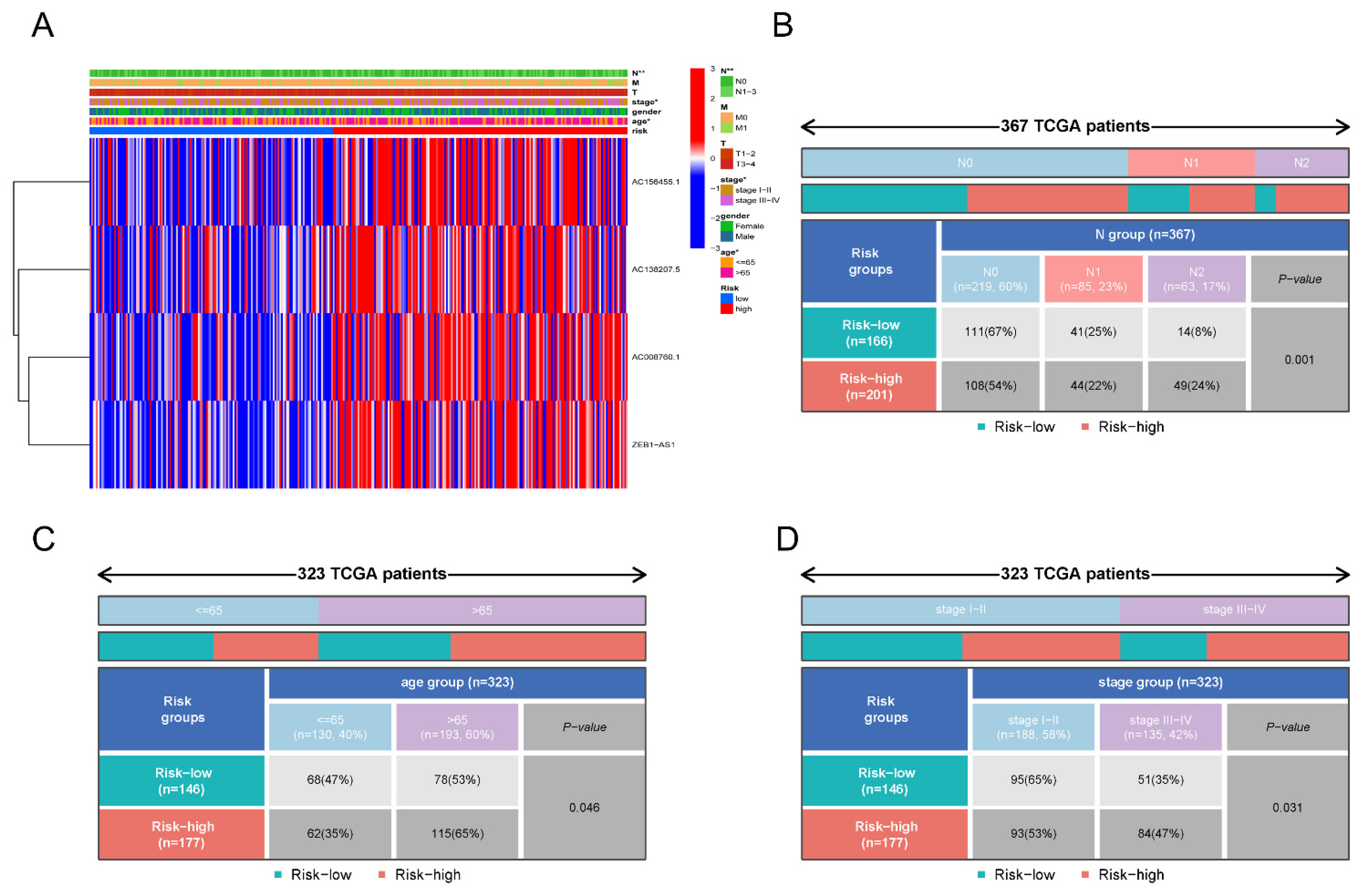
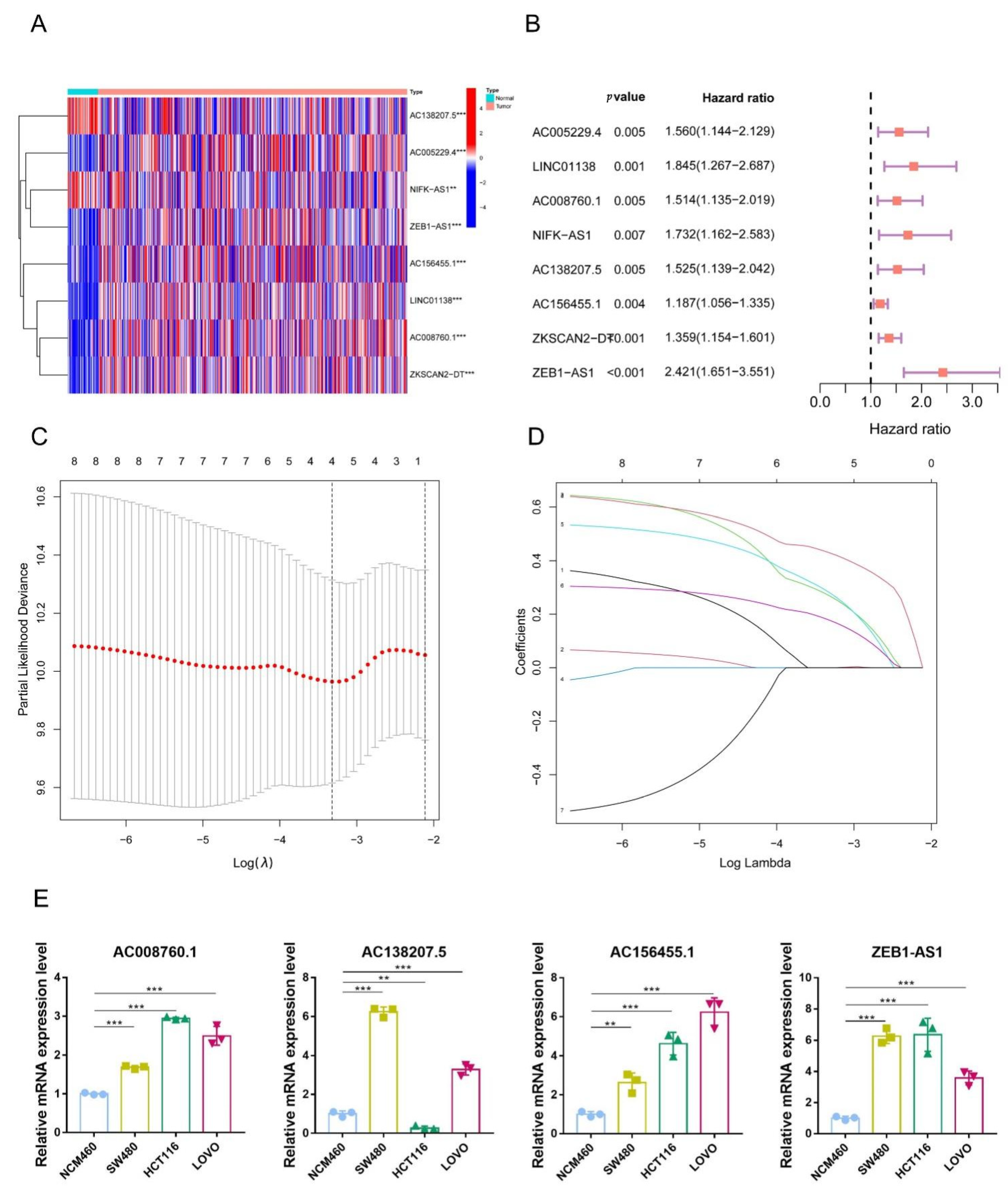
3.2. Identification of 5mC-Related lncRNAs in COAD Patients
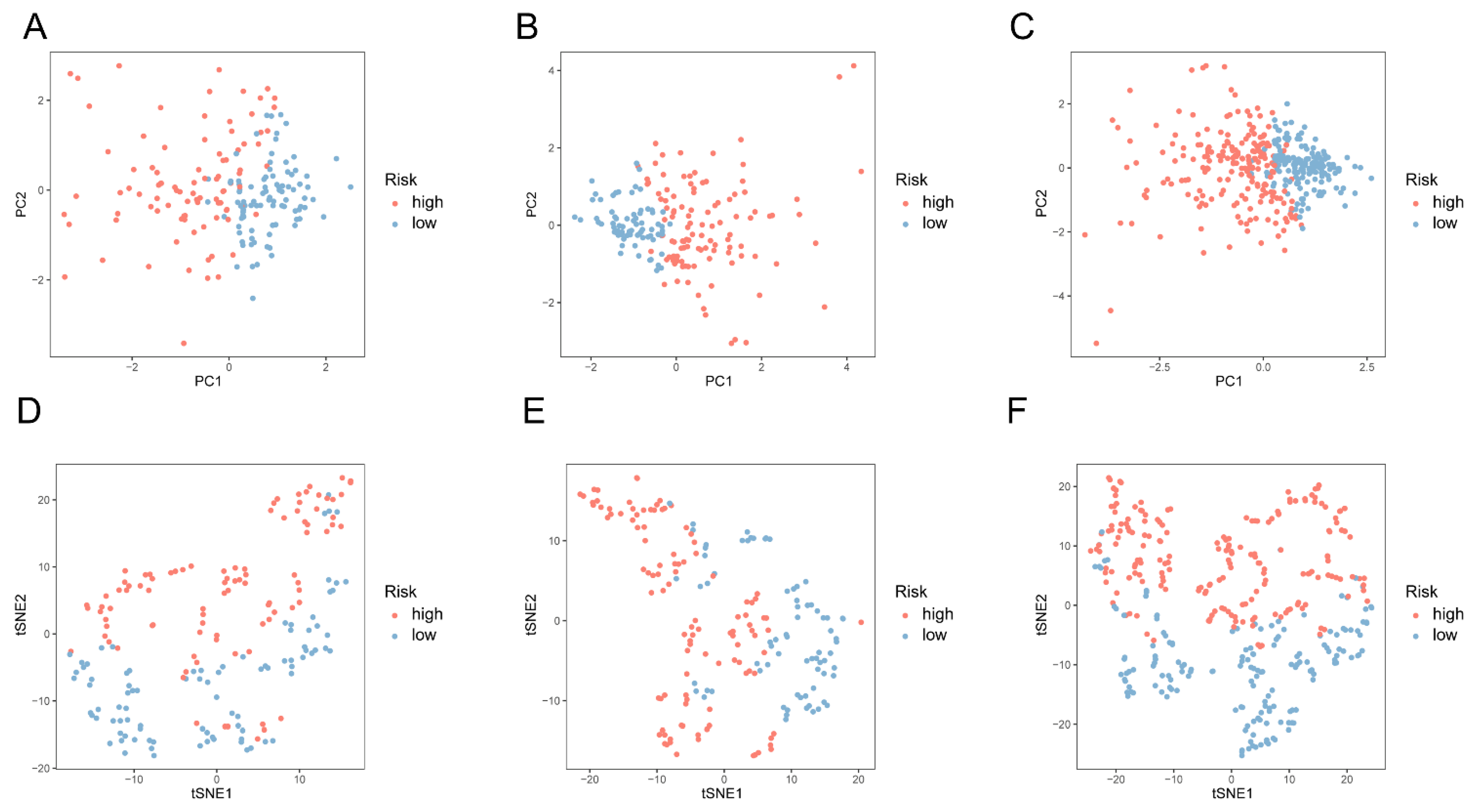
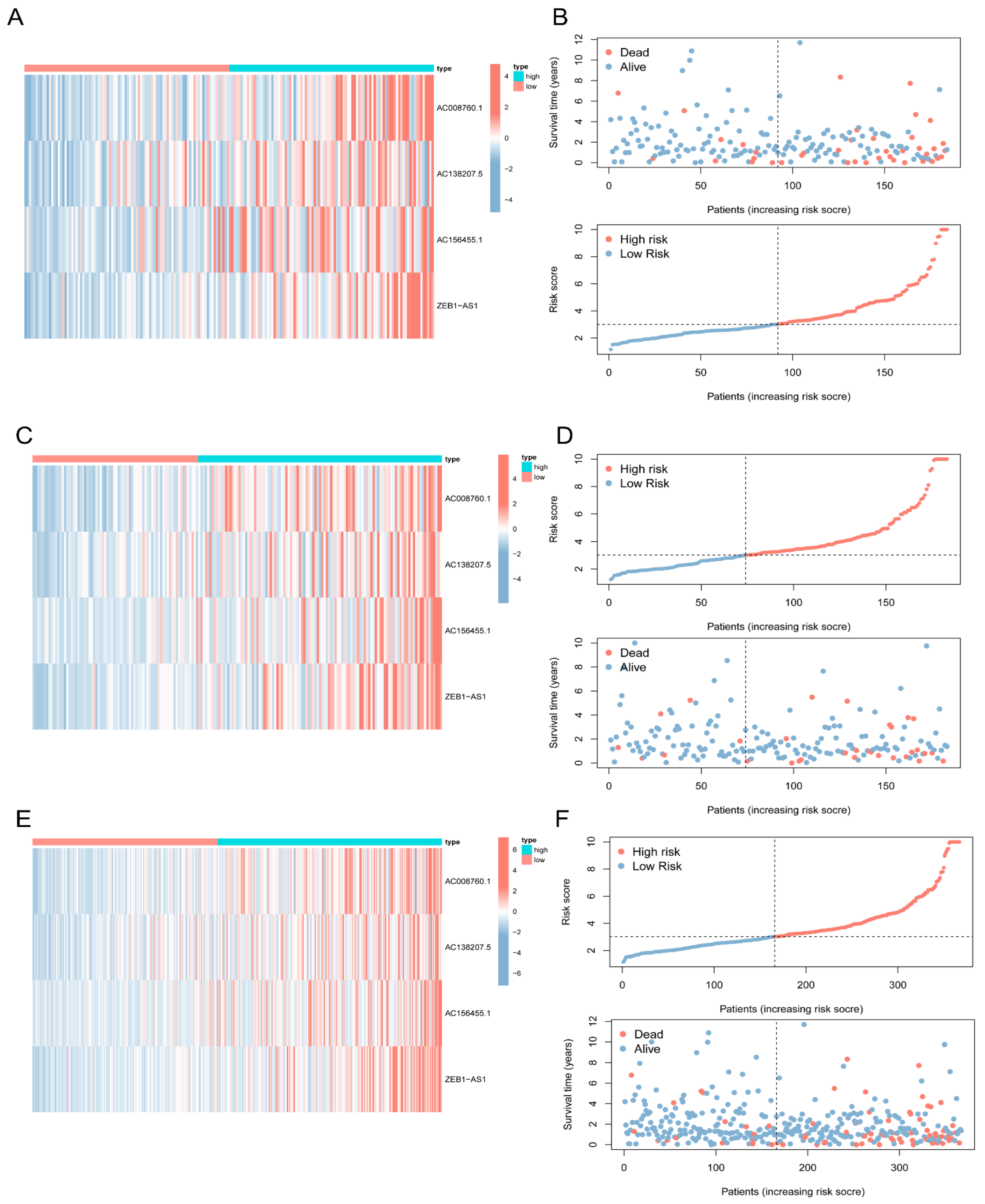
3.3. Construction of a Signature for 5mC-Related lncRNAs in COAD Patients
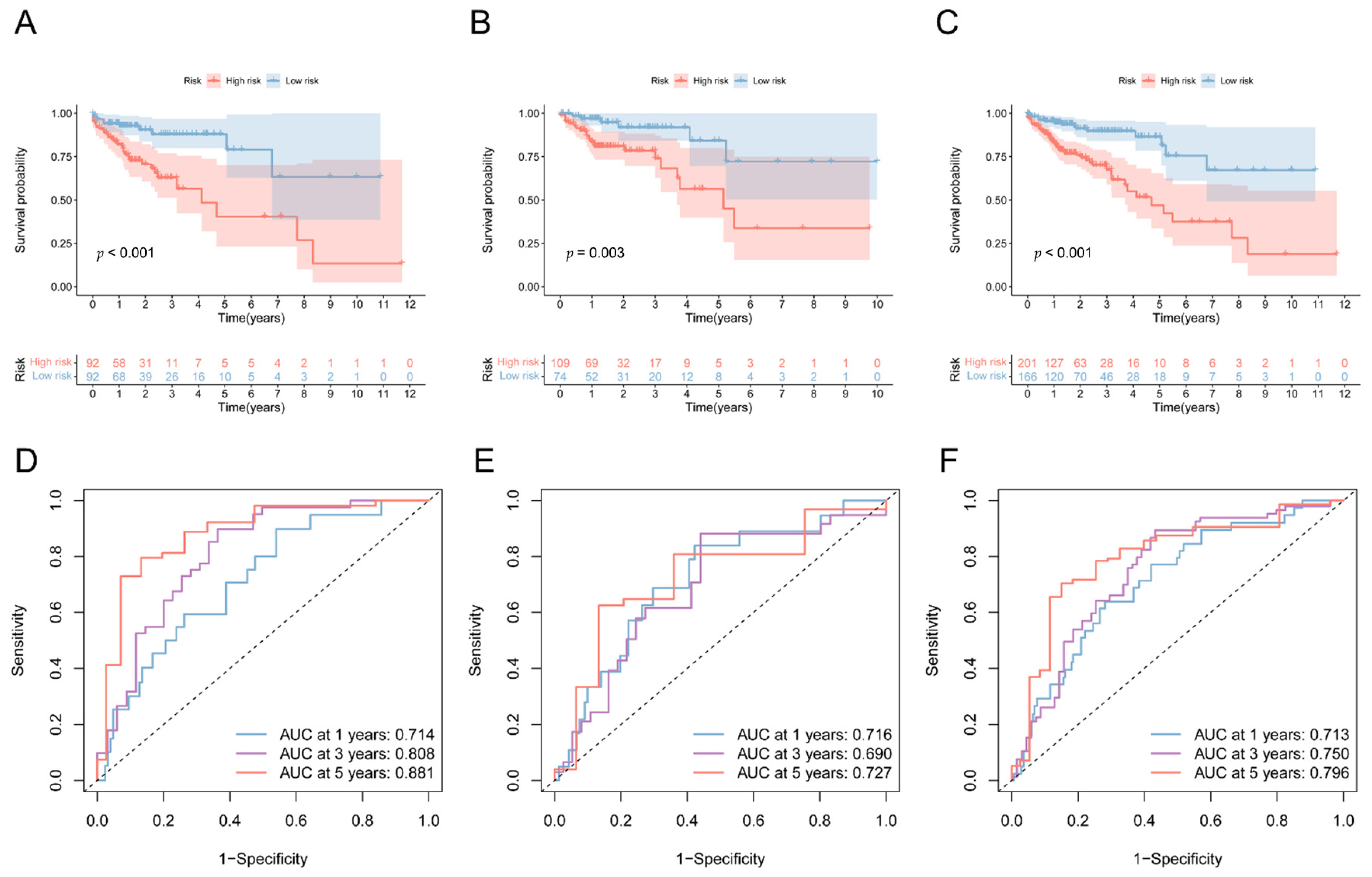
3.4. Validation of the 5mC-Related lncRNA Prognosis Signature
3.5. Prognostic Value of 5mC-Related lncRNA Prognosis Signature
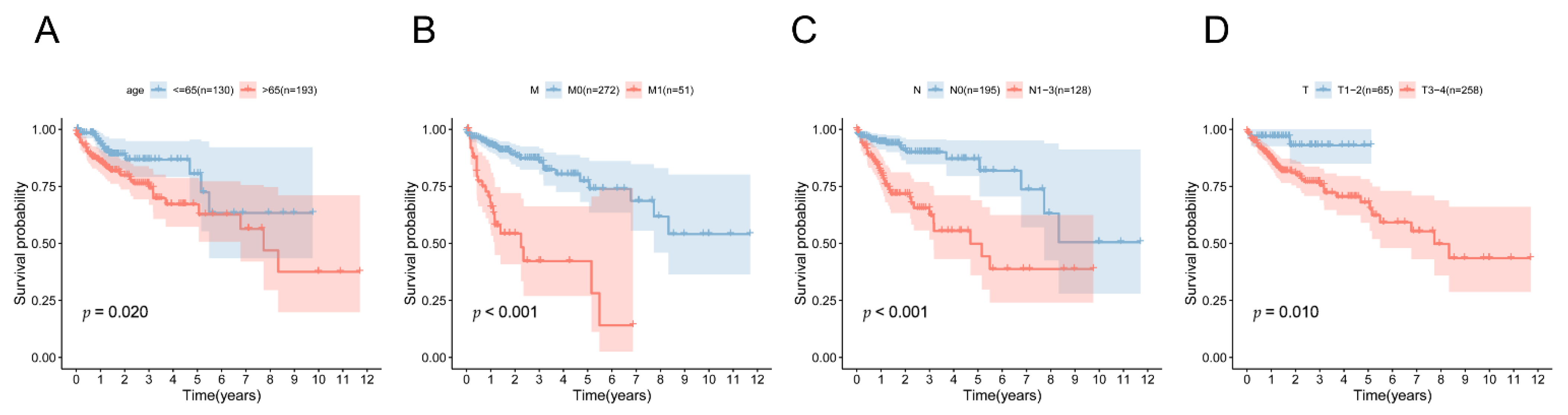
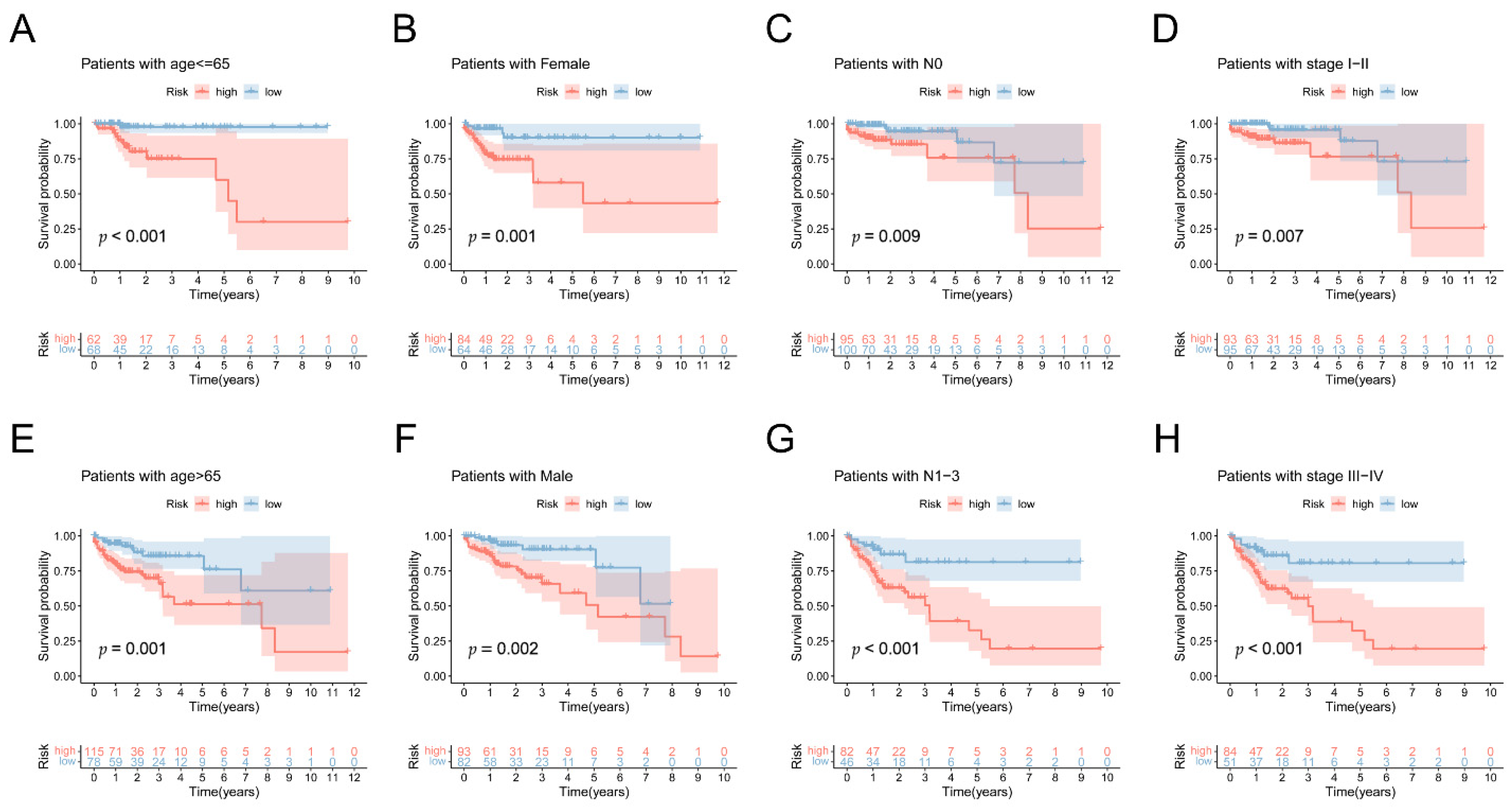
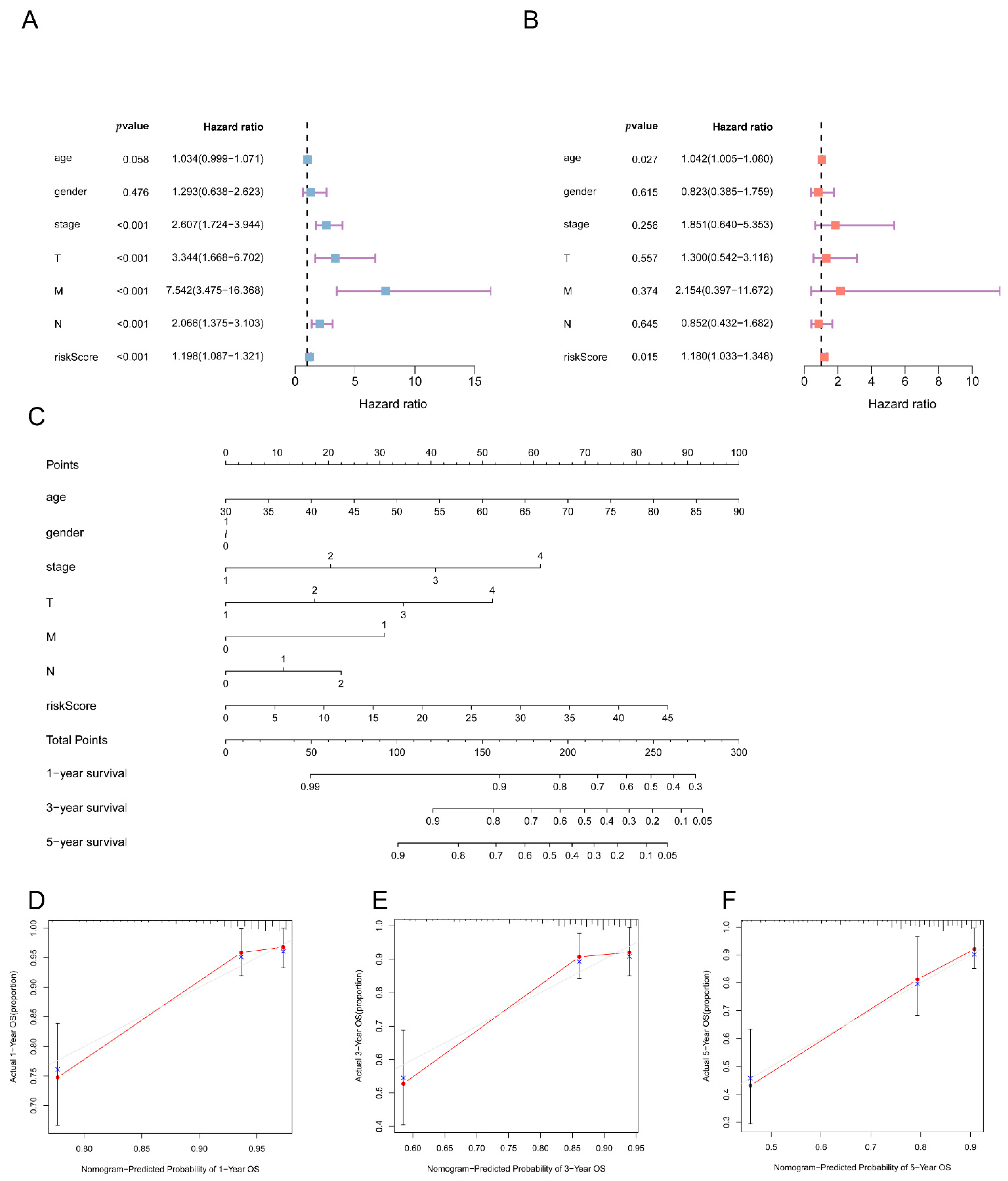
3.6. Independent Predictive Value of the 5mC-Related lncRNA Prognosis Signature
3.7. Molecular Characteristics of the 5mC-Related lncRNA Prognosis Signature
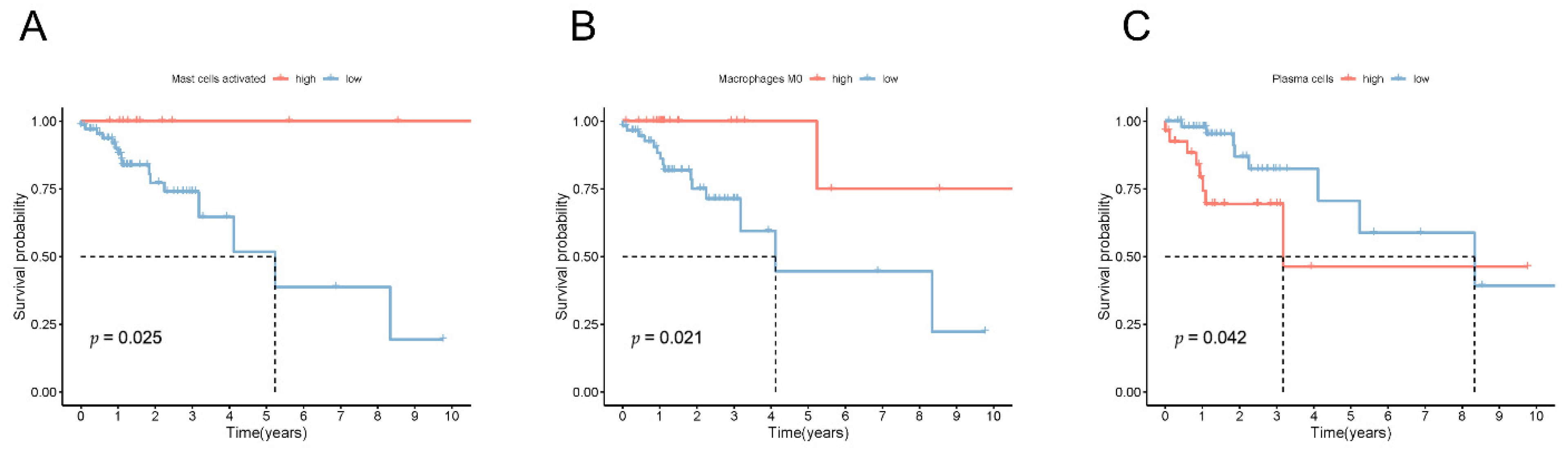
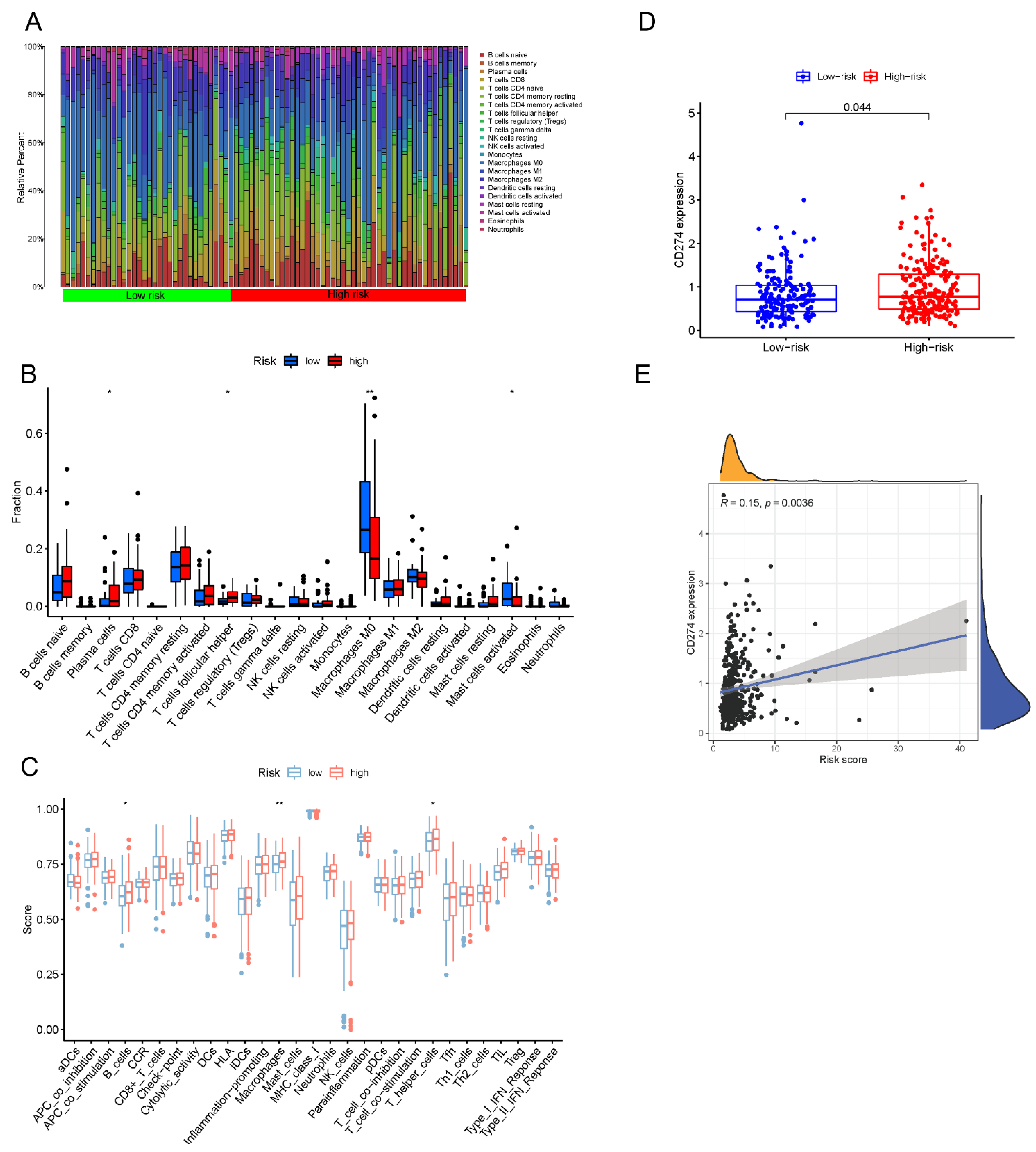
3.8. Estimation of the Tumor Immune Cell Types Utilizing the lncRNA Signature
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Cederquist, L.; Chen, Y.J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Engstrom, P.F.; et al. NCCN Guidelines Insights: Colon Cancer, Version 2.2018. J. Natl. Compr. Canc. Netw. 2018, 16, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.; Pathak, S.; Subramanium, V.D.; Dharanivasan, G.; Murugesan, R.; Verma, R.S. Strategies for Targeted Drug Delivery in Treatment of Colon Cancer: Current Trends and Future Perspectives. Drug Discov. Today 2017, 22, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Li, Z.; Gao, C.Y.; Cho, C.H. Mechanisms of Drug Resistance in Colon Cancer and Its Therapeutic Strategies. World J. Gastroenterol. 2016, 22, 6876–6889. [Google Scholar] [CrossRef]
- Keum, N.; Giovannucci, E. Global Burden of Colorectal Cancer: Emerging Trends, Risk Factors and Prevention Strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef]
- Xia, F.; Yan, Y.; Shen, C. A Prognostic Pyroptosis-Related Lncrnas Risk Model Correlates with the Immune Microenvironment in Colon Adenocarcinoma. Front. Cell Dev. Biol. 2021, 9, 811734. [Google Scholar] [CrossRef]
- He, X.; Ding, J.; Cheng, X.; Xiong, M. Hypoxia-Related Gene-Based Signature Can Evaluate the Tumor Immune Microenvironment and Predict the Prognosis of Colon Adenocarcinoma Patients. Int. J. Gen. Med. 2021, 14, 9853–9862. [Google Scholar] [CrossRef]
- Schubeler, D. Function and Information Content of DNA Methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA Methylation: Islands, Start Sites, Gene Bodies and Beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Bird, A.P. Cpg-Rich Islands and the Function of DNA Methylation. Nature 1986, 321, 209–213. [Google Scholar] [CrossRef]
- Mohandas, T.; Sparkes, R.S.; Shapiro, L.J. Reactivation of an Inactive Human X Chromosome: Evidence for X Inactivation by DNA Methylation. Science 1981, 211, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Skvortsova, K.; Stirzaker, C.; Taberlay, P. The DNA Methylation Landscape in Cancer. Essays Biochem. 2019, 63, 797–811. [Google Scholar] [PubMed]
- Ficz, G.; Branco, M.R.; Seisenberger, S.; Santos, F.; Krueger, F.; Hore, T.A.; Marques, C.J.; Andrews, S.; Reik, W. Dynamic Regulation of 5-Hydroxymethylcytosine in Mouse Es Cells and During Differentiation. Nature 2011, 473, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Bogdanović, O.; Smits, A.H.; de la Calle Mustienes, E.; Tena, J.J.; Ford, E.; Williams, R.; Senanayake, U.; Schultz, M.D.; Hontelez, S.; van Kruijsbergen, I.; et al. Active DNA Demethylation at Enhancers During the Vertebrate Phylotypic Period. Nat. Genet. 2016, 48, 417–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlich, M.; Lacey, M. DNA Hypomethylation and Hemimethylation in Cancer. Adv. Exp. Med. Biol. 2013, 754, 31–56. [Google Scholar] [PubMed]
- Zhang, Y.; Yang, L.; Kucherlapati, M.; Hadjipanayis, A.; Pantazi, A.; Bristow, C.A.; Lee, E.A.; Mahadeshwar, H.S.; Tang, J.; Zhang, S.; et al. Global Impact of Somatic Structural Variation on the DNA Methylome of Human Cancers. Genome Biol. 2019, 20, 209. [Google Scholar] [CrossRef] [Green Version]
- Laird, P.W.; Jaenisch, R. DNA Methylation and Cancer. Hum. Mol. Genet. 1994, 3, 1487–1495. [Google Scholar] [CrossRef]
- Collignon, E.; Canale, A.; Al Wardi, C.; Bizet, M.; Calonne, E.; Dedeurwaerder, S.; Garaud, S.; Naveaux, C.; Barham, W.; Wilson, A.; et al. Immunity Drives Tet1 Regulation in Cancer through Nf-Kappab. Sci. Adv. 2018, 4, eaap7309. [Google Scholar] [CrossRef] [Green Version]
- Neri, F.; Dettori, D.; Incarnato, D.; Krepelova, A.; Rapelli, S.; Maldotti, M.; Parlato, C.; Paliogiannis, P.; Oliviero, S. Tet1 Is a Tumour Suppressor That Inhibits Colon Cancer Growth by Derepressing Inhibitors of the Wnt Pathway. Oncogene 2015, 34, 4168–4176. [Google Scholar] [CrossRef] [Green Version]
- Nair, L.; Chung, H.; Basu, U. Regulation of Long Non-Coding Rnas and Genome Dynamics by the Rna Surveillance Machinery. Nat. Rev. Mol. Cell Biol. 2020, 21, 123–136. [Google Scholar] [CrossRef]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding Rnas. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.X.; Koirala, P.; Mo, Y.Y. Lncrna-Mediated Regulation of Cell Signaling in Cancer. Oncogene 2017, 36, 5661–5667. [Google Scholar] [CrossRef] [PubMed]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long Noncoding Rna and Cancer: A New Paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.T.; Lin, J.F.; Li, T.; Li, J.J.; Xu, R.H.; Ju, H.Q. Lncrna-Mediated Posttranslational Modifications and Reprogramming of Energy Metabolism in Cancer. Cancer Commun. 2021, 41, 109–120. [Google Scholar] [CrossRef]
- Gao, W.; Chan, J.Y.; Wong, T.S. Long Non-Coding Rna Deregulation in Tongue Squamous Cell Carcinoma. Biomed. Res. Int. 2014, 2014, 405860. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.H.; Yang, F.; Wang, F.; Ma, J.Z.; Guo, Y.J.; Tao, Q.F.; Liu, F.; Pan, W.; Wang, T.T.; Zhou, C.C.; et al. A Long Noncoding Rna Activated by Tgf-Beta Promotes the Invasion-Metastasis Cascade in Hepatocellular Carcinoma. Cancer Cell 2014, 25, 666–681. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Bai, X.; Ni, J.; Zhang, H.; Duan, W.; Graham, P.; Li, Y. Exosomes and Breast Cancer Drug Resistance. Cell Death Dis. 2020, 11, 987. [Google Scholar] [CrossRef]
- Lim, Y.W.S.; Xiang, X.; Garg, M.; Le, M.T.; Wong, A.L.A.; Wang, L.; Goh, B.C. The Double-Edged Sword of H19 Lncrna: Insights into Cancer Therapy. Cancer Lett. 2021, 500, 253–262. [Google Scholar]
- Pan, J.; Fang, S.; Tian, H.; Zhou, C.; Zhao, X.; Tian, H.; He, J.; Shen, W.; Meng, X.; Jin, X.; et al. Lncrna Jpx/Mir-33a-5p/Twist1 Axis Regulates Tumorigenesis and Metastasis of Lung Cancer by Activating Wnt/Beta-Catenin Signaling. Mol. Cancer 2020, 19, 9. [Google Scholar] [CrossRef]
- Hu, H.; Shu, M.; He, L.; Yu, X.; Liu, X.; Lu, Y.; Chen, Y.; Miao, X.; Chen, X. Epigenomic Landscape of 5-Hydroxymethylcytosine Reveals Its Transcriptional Regulation of Lncrnas in Colorectal Cancer. Br. J. Cancer 2017, 116, 658–668. [Google Scholar] [CrossRef] [Green Version]
- Mo, Z.; Cao, Z.; Luo, S.; Chen, Y.; Zhang, S. Novel Molecular Subtypes Associated with 5mc Methylation and Their Role in Hepatocellular Carcinoma Immunotherapy. Front. Mol. Biosci. 2020, 7, 562441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cao, H.; Ye, L.; Wen, X.; Wang, S.; Zheng, W.; Zhang, Y.; Huang, D.; Gao, Y.; Liu, H.; et al. Cancer-Associated Methylated Lncrnas in Patients with Bladder Cancer. Am. J. Transl. Res. 2019, 11, 3790–3800. [Google Scholar] [PubMed]
- Lio, C.J.; Yuita, H.; Rao, A. Dysregulation of the Tet Family of Epigenetic Regulators in Lymphoid and Myeloid Malignancies. Blood 2019, 134, 1487–1497. [Google Scholar] [CrossRef] [PubMed]
- Hlady, R.A.; Sathyanarayan, A.; Thompson, J.J.; Zhou, D.; Wu, Q.; Pham, K.; Lee, J.H.; Liu, C.; Robertson, K.D. Integrating the Epigenome to Identify Drivers of Hepatocellular Carcinoma. Hepatology 2019, 69, 639–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martisova, A.; Holcakova, J.; Izadi, N.; Sebuyoya, R.; Hrstka, R.; Bartosik, M. DNA Methylation in Solid Tumors: Functions and Methods of Detection. Int. J. Mol. Sci. 2021, 22, 4247. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Shen, J.Y.; Chen, D.P.; Wu, C.F.; Guo, R.; Zhang, P.P.; Lv, J.W.; Li, W.F.; Wang, Z.X.; Chen, Y.P. Identification of Cross-Talk between M (6)a and 5mc Regulators Associated with Onco-Immunogenic Features and Prognosis across 33 Cancer Types. J. Hematol. Oncol. 2020, 13, 22. [Google Scholar] [CrossRef] [Green Version]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. Pd-1 and Pd-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef]
- Constantinidou, A.; Alifieris, C.; Trafalis, D.T. Targeting Programmed Cell Death -1 (Pd-1) and Ligand (Pd-L1): A New Era in Cancer Active Immunotherapy. Pharmacol. Ther. 2019, 194, 84–106. [Google Scholar] [CrossRef]
- Winer, A.; Ghatalia, P.; Bubes, N.; Anari, F.; Varshavsky, A.; Kasireddy, V.; Liu, Y.; El-Deiry, W.S. Dual Checkpoint Inhibition with Ipilimumab Plus Nivolumab after Progression on Sequential Pd-1/Pdl-1 Inhibitors Pembrolizumab and Atezolizumab in a Patient with Lynch Syndrome, Metastatic Colon, and Localized Urothelial Cancer. Oncologist 2019, 24, 1416–1419. [Google Scholar] [CrossRef] [Green Version]
- Angell, H.K.; Bruni, D.; Barrett, J.C.; Herbst, R.; Galon, J. The Immunoscore: Colon Cancer and Beyond. Clin. Cancer Res. 2020, 26, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Koliaraki, V.; Pallangyo, C.K.; Greten, F.R.; Kollias, G. Mesenchymal Cells in Colon Cancer. Gastroenterology 2017, 152, 964–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M. Colon Cancer: A Clinician’s Perspective in 2019. Gastroenterol. Res. 2020, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Zhang, J.; Zeng, D.; Sun, H.; Rong, X.; Shi, M.; Bin, J.; Liao, Y.; Liao, W. Immune Cell Infiltration as a Biomarker for the Diagnosis and Prognosis of Stage I-Iii Colon Cancer. Cancer Immunol. Immunother. 2019, 68, 433–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tie, J.; Cohen, J.D.; Wang, Y.; Christie, M.; Simons, K.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; McKendrick, J.; et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage Iii Colon Cancer. JAMA Oncol. 2019, 5, 1710–1717. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Zhang, H.; Wu, W.; Cheng, S.; Dai, X.; Zhu, X.; Fu, Q.; Tong, Z.; Liu, L.; Zheng, Y.; et al. Analysis of the Molecular Nature Associated with Microsatellite Status in Colon Cancer Identifies Clinical Implications for Immunotherapy. J. Immunother. Cancer 2020, 8, e001437. [Google Scholar] [CrossRef]
- Zhou, M.; Zhang, Z.; Zhao, H.; Bao, S.; Cheng, L.; Sun, J. An Immune-Related Six-Lncrna Signature to Improve Prognosis Prediction of Glioblastoma Multiforme. Mol. Neurobiol. 2018, 55, 3684–3697. [Google Scholar] [CrossRef]
- Shen, Y.; Peng, X.; Shen, C. Identification and Validation of Immune-Related Lncrna Prognostic Signature for Breast Cancer. Genomics 2020, 112, 2640–2646. [Google Scholar] [CrossRef]
- Li, X.; Jin, F.; Li, Y. A Novel Autophagy-Related Lncrna Prognostic Risk Model for Breast Cancer. J. Cell Mol. Med. 2021, 25, 4–14. [Google Scholar] [CrossRef]
- Wu, C.Y.; Zhang, B.; Kim, H.; Anderson, S.K.; Miller, J.S.; Cichocki, F. Ascorbic Acid Promotes Kir Demethylation During Early Nk Cell Differentiation. J. Immunol. 2020, 205, 1513–1523. [Google Scholar] [CrossRef]
- Bowman, R.L.; Levine, R.L. Tet2 in Normal and Malignant Hematopoiesis. Cold Spring Harb. Perspect. Med. 2017, 7, a026518. [Google Scholar] [CrossRef] [Green Version]
- Tulstrup, M.; Soerensen, M.; Hansen, J.W.; Gillberg, L.; Needhamsen, M.; Kaastrup, K.; Helin, K.; Christensen, K.; Weischenfeldt, J.; Gronbaek, K. Tet2 Mutations Are Associated with Hypermethylation at Key Regulatory Enhancers in Normal and Malignant Hematopoiesis. Nat. Commun. 2021, 12, 6061. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Ding, Y.; Yuan, H.; Shao, J.; Yan, Y.; Guo, R.; Luan, W.; Xu, M. Long Non-Coding Rna Zeb1-As1 Promotes Colon Adenocarcinoma Malignant Progression Via Mir-455-3p/Pak2 Axis. Cell Prolif. 2020, 53, e12723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Hao, Q.; Prasanth, K.V. Nuclear Long Noncoding Rnas: Key Regulators of Gene Expression. Trends Genet. 2018, 34, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Chong, W.; Shang, L.; Liu, J.; Fang, Z.; Du, F.; Wu, H.; Liu, Y.; Wang, Z.; Chen, Y.; Jia, S.; et al. M (6)a Regulator-Based Methylation Modification Patterns Characterized by Distinct Tumor Microenvironment Immune Profiles in Colon Cancer. Theranostics 2021, 11, 2201–2217. [Google Scholar] [CrossRef]
- Jia, Q.; Wang, J.; He, N.; He, J.; Zhu, B. Titin Mutation Associated with Responsiveness to Checkpoint Blockades in Solid Tumors. JCI Insight 2019, 4, e127901. [Google Scholar] [CrossRef]
- Gao, J.; Xi, L.; Yu, R.; Xu, H.; Wu, M.; Huang, H. Differential Mutation Detection Capability through Capture-Based Targeted Sequencing in Plasma Samples in Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 596789. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and Prioritization of Somatic Mutations in Diffuse Large B-Cell Lymphoma (Dlbcl) by Whole-Exome Sequencing. Proc. Natl. Acad. Sci USA 2012, 109, 3879–3884. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; Lin, A.; Li, K.; Lin, W.; Wang, Q.; Wei, T.; Zhu, W.; Luo, P.; Zhang, J. A Novel Mutation Panel for Predicting Etoposide Resistance in Small-Cell Lung Cancer. Drug Des. Devel. Ther. 2019, 13, 2021–2041. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Zitvogel, L.; Sautes-Fridman, C.; Kroemer, G. The Immune Contexture in Cancer Prognosis and Treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The Prognostic Landscape of Genes and Infiltrating Immune Cells across Human Cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.; Smyth, M.J. Resistance to Pd1/Pdl1 Checkpoint Inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Taube, J.M.; Pardoll, D.M. Neoadjuvant Checkpoint Blockade for Cancer Immunotherapy. Science 2020, 367, eaax0182. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Huang, H.; Wang, Y.; Liu, H.; Huang, Y. 5mC-Related lncRNAs as Potential Prognostic Biomarkers in Colon Adenocarcinoma. Biology 2022, 11, 231. https://doi.org/10.3390/biology11020231
Huang Y, Huang H, Wang Y, Liu H, Huang Y. 5mC-Related lncRNAs as Potential Prognostic Biomarkers in Colon Adenocarcinoma. Biology. 2022; 11(2):231. https://doi.org/10.3390/biology11020231
Chicago/Turabian StyleHuang, Yinghui, Huiqian Huang, Yong Wang, Hui Liu, and Yingdan Huang. 2022. "5mC-Related lncRNAs as Potential Prognostic Biomarkers in Colon Adenocarcinoma" Biology 11, no. 2: 231. https://doi.org/10.3390/biology11020231
APA StyleHuang, Y., Huang, H., Wang, Y., Liu, H., & Huang, Y. (2022). 5mC-Related lncRNAs as Potential Prognostic Biomarkers in Colon Adenocarcinoma. Biology, 11(2), 231. https://doi.org/10.3390/biology11020231