
Phenotypic and Functional Heterogeneity of Monocyte Subsets in Chronic Heart Failure Patients



Abstract
Simple Summary
Abstract
1. Introduction
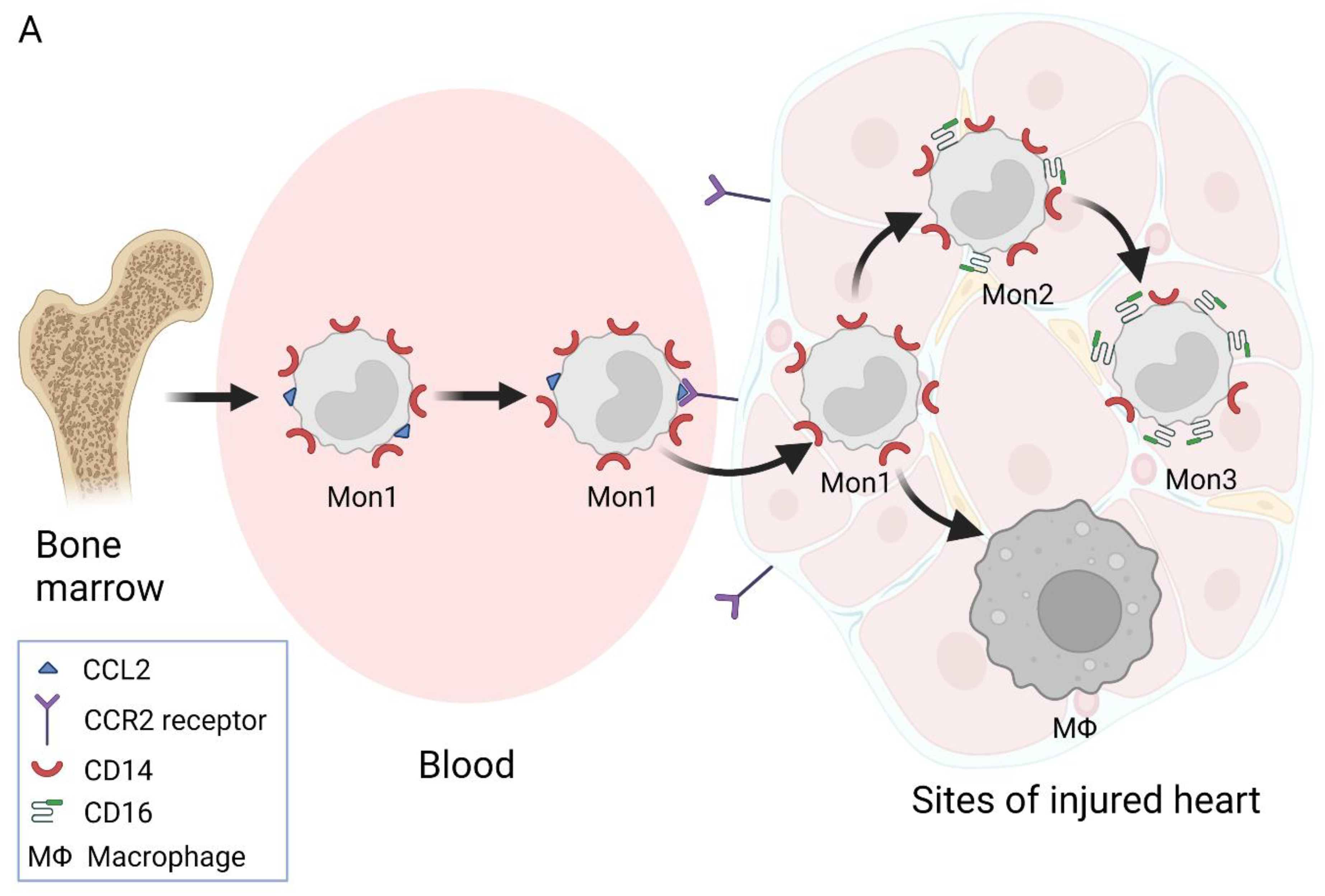
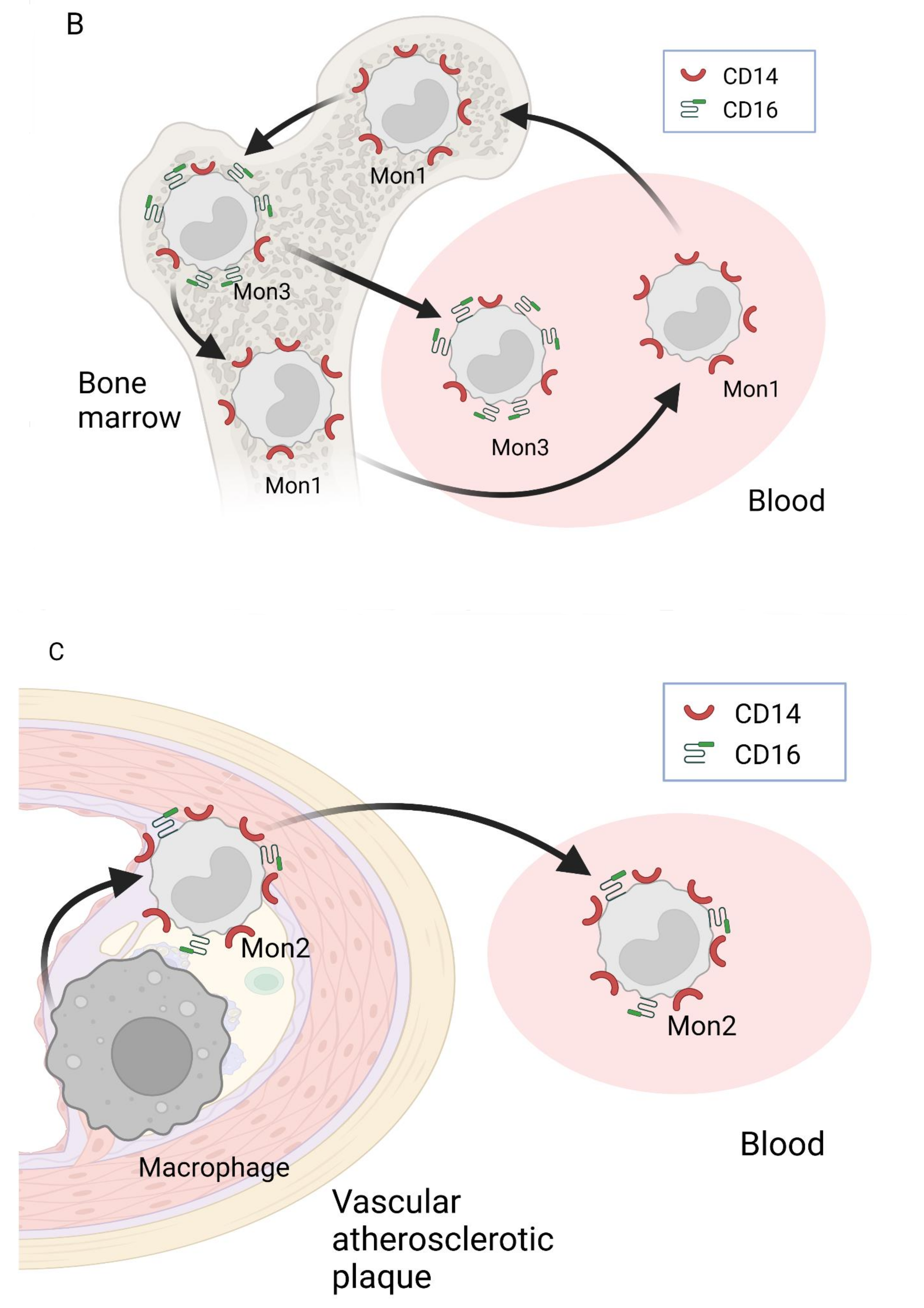
2. Nomenclature of Monocyte Subsets and Their Formation
3. Involvement of Different Monocyte Subsets in the Inflammatory Processes
4. Monocytes in CHF
4.1. The Distribution of Monocytes in CHF
4.2. Influence of Monocyte-Secreted Cytokines and Inflammatory Readings on HFrEF and HFpEF Development
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Glezeva, N.; Baugh, J.A. Role of inflammation in the pathogenesis of heart failure with preserved ejection fraction and its potential as a therapeutic target. Heart Fail. Rev. 2014, 19, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Bytyçi, I.; Bajraktari, G. Mortality in heart failure patients. Anatol. J. Cardiol. 2015, 15, 63–68. [Google Scholar] [CrossRef] [PubMed]
- James, S.; Barton, D.; O’Connell, E.; Voon, V.; Murtagh, G.; Watson, C.; Murphy, T.; Prendiville, B.; Brennan, D.; Henseym, M.; et al. Life expectancy for community-based patients with heart failure from time of diagnosis. Int. J. Cardiol. 2015, 178, 268–274. [Google Scholar] [CrossRef]
- MacCarthy, P.A.; Shah, A.M. Impaired endothelium-dependent regulation of ventricular relaxation in pressure-overload cardiac hypertrophy. Circulation 2000, 101, 1854–1860. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Yndestad, A.; Damas, J.K.; Oie, E.; Ueland, T.; Gullestad, L.; Aukrust, P. Systemic inflammation in heart failure—the whys and wherefores. Heart Fail. Rev. 2006, 11, 83–92. [Google Scholar] [CrossRef]
- Heymans, S.; Hirsch, E.; Anker, S.D.; Aukrust, P.; Balligand, J.L.; Cohen-Tervaert, J.W.; Drexler, H.; Filippatos, G.; Felix, S.B.; Gullestad, L.; et al. Inflammation as a therapeutic target in heart failure? A scientific statement from the Translational Research Committee of the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2009, 11, 119–129. [Google Scholar] [CrossRef]
- Ridker, P.M.; Luscher, T.F. Anti-inflammatory therapies for cardiovascular disease. Eur. Heart J. 2014, 35, 1782–1791. [Google Scholar] [CrossRef]
- Fernandez-Velasco, M.; Gonzalez-Ramos, S.; Bosca, L. Involvement of monocytes/macrophages as key factors in the development and progression of cardiovascular diseases. Biochem J 2014, 458, 187–193. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L. Report on the Nomenclature of Monocytes and Dendritic Cells in Blood Sub-Committee 15 12 2017. Monocytomics Research, Herrsching, Germany. Available online: https://s3-eu-west-1.amazonaws.com/wp-iuis/app/uploads/2019/08/06110234/monocytes2017-9d369b75.pdf (accessed on 21 May 2021).
- Cesaroni, G.; Mureddu, G.F.; Agabiti, N.; PREDICTOR Study Group. Sex differences in factors associated with heart failure and diastolic left ventricular dysfunction: A cross-sectional population-based study. BMC Public Health 2021, 21, 415. [Google Scholar] [CrossRef] [PubMed]
- Connaughton, E.P.; Naicker, S.; Hanley, S.A.; Slevin, S.M.; Eykelenboom, J.K.; Lowndes, N.F.; O’Brien, T.; Ceredig, R.; Griffin, M.D.; Dennedy, M.C. Phenotypic and functional heterogeneity of human intermediate monocytes based on HLA-DR expression. Immunol. Cell Biol. 2018, 96, 742–758. [Google Scholar] [CrossRef] [PubMed]
- Peet, C.; Ivetic, A.; Bromage, D.I.; Shah, A.M. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc. Res. 2021, 116, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Cleland, J.G.; Puri, S. How do ACE inhibitors reduce mortality in patients with left ventricular dysfunction with and without heart failure: Remodelling, resetting, or sudden death? Br. Heart J. 1994, 72, S81–S86. [Google Scholar] [CrossRef]
- Rossol, M.; Kraus, S.; Pierer, M.; Baerwald, C.; Wagner, U. The CD14(bright) CD16+ monocyte subset is expanded in rheumatoid arthritis and promotes expansion of the TH17 cell population. Arthritis. Rheum. 2012, 64, 671–677.20. [Google Scholar] [CrossRef]
- Smedman, C.; Ernemar, T.; Gudmundsdotter, L.; Gille-Johnson, P.; Somell, A.; Nihlmark, K.; Gardlund, B.; Andersson, J.; Paulie, S. Fluorospot analysis of TLR-activated monocytes reveals several distinct cytokine-secreting subpopulations. Scand. J. Immunol. 2012, 75, 249–258. [Google Scholar] [CrossRef]
- Patel, H.; Davidson, D. Control of pro-inflammatory cytokine release from human monocytes with the use of an interleukin-10 monoclonal antibody. Methods Mol. Biol. 2014, 1172, 99–106. [Google Scholar] [CrossRef]
- Wong, K.L.; Tai, J.J.-Y.; Wong, W.C.; Han, H.; Sem, X.; Yeap, W.H.; Kourilsky, P.; Wong, S. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 2011, 118, e16–e31. [Google Scholar] [CrossRef]
- Mandl, M.; Schmitz, S.; Weber, C.; Hristov, M. Characterization of the CD14++CD16+ Monocyte Population in Human Bone Marrow. PLoS ONE 2014, 9, e112140. [Google Scholar] [CrossRef]
- Boyette, L.B.; Macedo, C.; Hadi, K.; Elinoff, B.D.; Walters, J.T.; Ramaswami, B.; Chalasani, G.; Taboas, J.M.; Lakkis, F.G.; Metes, D.M. Phenotype, function, and differentiation potential of human monocyte subsets. PLoS ONE 2017, 12, e0176460. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L. Blood monocytes and their subsets: Established features and open questions. Front. Immunol. 2015, 6, 423. [Google Scholar] [CrossRef] [PubMed]
- Stansfield, B.K.; Ingram, D.A. Clinical significance of monocyte heterogeneity. Clin Transl. Med. 2015, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Anbazhagan, K.; Duroux-Richard, I.; Jorgensen, C.; Apparailly, F. Transcriptomic network support distinct roles of classical and non-classical monocytes in human. Int. Rev. Immunol. 2014, 33, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Hijdra, D.; Vorselaars, A.D.; Grutters, J.C.; Claessen, A.M.; Rijkers, G.T. Phenotypic characterization of human intermediate monocytes. Front. Immunol. 2013, 4, 339. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.L.; Yeap, W.H.; Tai, J.J.; Ong, S.M.; Dang, T.M.; Wong, S.C. The three human monocyte subsets: Implications for health and disease. Immunol. Res. 2012, 53, 41–57. [Google Scholar] [CrossRef]
- Tallone, T.; Turconi, G.; Soldati, G.; Pedrazzini, G.; Moccetti, T.; Vassalli, G. Heterogeneity of human monocytes: An optimized four-color flow cytometry protocol for analysis of monocyte subsets. J. Cardiovasc. Trans. Res. 2011, 4, 211–219. [Google Scholar] [CrossRef]
- Zawada, A.M.; Rogacev, K.S.; Rotter, B.; Winter, P.; Marell, R.R.; Fliser, D.; Heine, G.H. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood 2011, 118, e50–e61. [Google Scholar] [CrossRef]
- Rogacev, K.S.; Seiler, S.; Zawada, A.M.; Reichart, B.; Herath, E.; Roth, D.; Ulrich, C.; Fliser, D.; Heine, G.H. CD14++CD16+ monocytes and cardiovascular outcome in patients with chronic kidney disease. Eur. Heart J. 2011, 32, 84–92. [Google Scholar] [CrossRef]
- Ulrich, C.; Heine, G.H.; Seibert, E.; Fliser, D.; Girndt, M. Circulating monocyte subpopulations with high expression of angiotensin-converting enzyme predict mortality in patients with end-stage renal disease. Nephrol. Dial. Transpl. 2010, 25, 2265–2272. [Google Scholar] [CrossRef][Green Version]
- Ingersoll, M.A.; Spanbroek, R.; Lottaz, C.; Gautier, E.L.; Frankenberger, M.; Hoffmann, R.; Lang, R.; Haniffa, M.; Collin, M.; Tacke, F.; et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood 2010, 115, e10–e19. [Google Scholar] [CrossRef]
- Sunderkotter, C.; Nikolic, T.; Dillon, M.J.; Van Rooijen, N.; Stehling, M.; Drevets, D.A.; Leenen, P.J. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J. Immunol. 2004, 172, 4410–4417. [Google Scholar] [CrossRef] [PubMed]
- Ancuta, P.; Rao, R.; Moses, A.; Mehle, A.; Shaw, S.K.; Luscinskas, F.W.; Gabuzda, D. Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J. Exp. Med. 2003, 197, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.D.; Hamers, A.A.J.; Nakao, C.; Marcovecchio, P.; Taylor, A.M.; McSkimming, C.; Nguyen, A.T.; McNamara, C.A.; Hedrick, C.C. Human blood monocyte subsets. Arter. Thromb. Vasc. Biol. 2017, 37, 1548–1558. [Google Scholar] [CrossRef] [PubMed]
- Roussel, M.; Ferrell, P.B.; Greenplate, A.R.; Lhomme, F.; Le Gallou, S.; Diggins, K.E.; Johnson, D.B.; Irish, J.M. Mass cytometry deep phenotyping of human mononuclear phagocytes and myeloid-derived suppressor cells from human blood and bone marrow. J. Leukoc. Biol. 2017, 102, 437–447. [Google Scholar] [CrossRef]
- Cros, J.; Cagnard, N.; Woollard, K.; Patey, N.; Zhang, S.Y.; Senechal, B.; Puel, A.; Biswas, S.K.; Moshous, D.; Picard, C.; et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity 2010, 33, 375–386. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, L.; Yu, C.; Xiao-Feng, Y.; Hong, W. Monocyte and macrophage differentiation: Circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark. Res. 2014, 2, 1. [Google Scholar] [CrossRef]
- Gren, S.T.; Rasmussen, T.B.; Janciauskiene, S.; Håkansson, K.; Gerwien, J.G.; Grip, O. A single-cell gene-expression profile reveals inter-cellular heterogeneity within human monocyte subsets. PLoS ONE 2015, 10, e0144351. [Google Scholar] [CrossRef]
- Zawada, A.M.; Rogacev, K.S.; Schirmer, S.H.; Sester, M.; Böhm, M.; Fliser, D.; Heine, G.H. Monocyte heterogeneity in human cardiovascular disease. Immunobiology 2012, 217, 1273–1284. [Google Scholar] [CrossRef]
- Jakubzick, C.; Gautier, E.L.; Gibbings, S.L.; Sojka, D.K.; Schlitzer, A.; Johnson, T.E.; Ivanov, S.; Duan, Q.; Bala, S.; Condon, T.; et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity 2013, 39, 599–610. [Google Scholar] [CrossRef]
- Patel, A.A.; Ginhoux, F.; Yona, S. Monocytes, macropfages, dendritic cells and neutrophils: An update on lifespan in health and disease. Immunology 2021, 163, 250–261. [Google Scholar] [CrossRef]
- Amit, A.P.; Yan, Z.; James, N.F.; Lies, B.; Anthony, R.; Alexander, A.M.; Venetia, B.; Richard, A.F.; Derek, W.G.; Becca, A.; et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med. 2017, 214, 1913–1923. [Google Scholar] [CrossRef]
- Gainaru, G.; Papadopoulos, A.; Tsangaris, I.; Lada, M.; Giamarellos-Bourboulis, E.J.; Pistiki, A. Increases in inflammatory and CD14dim/CD16pos/CD45pos patrolling monocytes in sepsis: Correlation with final outcome. Crit. Care 2018, 22, 56. [Google Scholar] [CrossRef] [PubMed]
- Tapp, L.D.; Shantsila, E.; Wrigley, B.J.; Pamukcu, B.; Lip, G.Y. The CD 14++CD16+ monocyte subset and monocyte-platelet interactions in patients with ST-elevation myocardial infarction. J. Thromb. Hamost. 2012, 10, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Kapellos, T.S.; Bonaguro, L.; Gemünd, I.; Reusch, N.; Saglam, A.; Hinkley, E.R.; Schultze, J.L. Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front. Immunol. 2019, 10, 2035. [Google Scholar] [CrossRef]
- Poitou, C.; Dalmas, E.; Renovato, M.; Benhamo, V.; Hajduch, F.; Abdennour, M.; Kahn, J.F.; Veyrie, N.; Rizkalla, S.; Fridman, W.H.; et al. CD14dimCD16+ and CD14+CD16+ monocytes in obesity and during weight loss: Relationships with fat mass and subclinical atherosclerosis. Arter. Thromb. Vasc. Biol. 2011, 31, 2322–2330. [Google Scholar] [CrossRef]
- Kani, A.H.; Alavian, S.M.; Esmaillzadeh, A.; Adibi, P.; Haghighatdoost, F.; Azadbakht, L. Effects of a Low-calorie, low-carbohydrate soy containing diet on systemic inflammation among patients with nonalcoholic fatty liver disease: A parallel randomized clinical trial. Horm. Metab. Res. 2017, 49, 687–692. [Google Scholar] [CrossRef]
- Wei, M.; Brandhorst, S.; Shelehchi, M.; Mirzaei, H.; Cheng, C.W.; Budniak, J.; Cheng, C.W.; Villani, V.; Buono, R.; Wei, M.; et al. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci. Transl. Med. 2017, 9, eaai8700. [Google Scholar] [CrossRef]
- Shahid, F.; Lip, G.Y.H.; Shantsila, E. Role of Monocytes in Heart Failure and Atrial Fibrillation. J. Am. Heart. Assoc. 2018, 7, e007849. [Google Scholar] [CrossRef]
- Ożańska, A.; Szymczak, D.; Rybka, J. Pattern of human monocyte subpopulations in health and disease. Scand. J. Immunol. 2020, 92, e12883. [Google Scholar] [CrossRef]
- Russo, L.; Lumeng, C.N. Properties and functions of adipose tissue macrophages in obesity. Immunology 2018, 155, 407–417. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Adlung, L.; Thaiss, C.A.; Weiner, A.; Li, B.; Descamps, H.; Lundgren, P.; Bleriot, C.; Liu, Z.; Deczkowska, A.; et al. Lipid-associated macrophages control metabolic homeostasis in a trem2-dependent manner. Cell 2019, 178, 686–698.e14. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.W.; Villani, V.; Buono, R.; Wei, M.; Kumar, S.; Yilmaz, O.H.; Cohen, P.; Sneddon, J.B.; Perin, L.; Longo, V.D. Fasting-mimicking diet promotes Ngn3-Driven β-cell regeneration to reverse diabetes. Cell 2017, 168, 775–788.e12. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.; Tung, N.; Casanova-Acebes, M.; Chang, C.; Cantoni, C.; Zhang, D.; Wirtz, T.H.; Naik, S.; Rose, S.A.; Brocker, C.N.; et al. Dietary Intake Regulates the Circulating Inflammatory Monocyte Pool. Cell 2019, 22, 1102–1114.e17. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef]
- Strauss-Ayali, D.; Conrad, S.M.; Mosser, D.M. Monocyte subpopulations and their differentiation patterns during infection. J. Leukoc. Biol. 2007, 82, 244–252. [Google Scholar] [CrossRef]
- Kelley, J.L.; Ozment, T.R.; Li, C.; Schweitzer, J.B.; Williams, D.L. Scavenger receptor-A (CD204): A two-edged sword in health and disease. Crit. Rev. Immunol. 2014, 34, 241–261. [Google Scholar] [CrossRef]
- Jing, J.; Yang, I.V.; Hui, L.; Patel, J.A.; Evans, C.M.; Prikeris, R.; Kobzik, L.; O’Connor, B.P.; Schwartz, D.A. Role of macrophage receptor with collagenous structure in innate immune tolerance. J. Immunol. 2013, 15, 6360–6367. [Google Scholar] [CrossRef]
- Menezes, S.; Melandri, D.; Anselmi, G.; Perchet, T.; Loschko, J.; Dubro, J.; Patel, R.; Gautier, E.L.; Hugues, S.; Longhi, M.P.; et al. The heterogeneity of Ly6C(hi) monocytes controls their differentiation into iNOS(+) macrophages or monocyte-derived dendritic cells. Immunity 2016, 45, 1205–1218. [Google Scholar] [CrossRef]
- Serbina, N.V.; Cherny, M.; Shi, C.; Bleau, S.A.; Collins, N.H.; Young, J.W.; Eric, G.P. Distinct responses of human monocyte subsets to aspergillus fumigatus conidia. J. Immunol. 2009, 183, 2678–2687. [Google Scholar] [CrossRef]
- Weber, C.; Belge, K.U.; von Hundelshausen, P.; Draude, G.; Steppich, B.; Mack, M.; Frankenberger, M.; Weber, K.S.; Ziegler-Heitbrock, H.W. Differential chemokine receptor expression and function in human monocyte subpopulations. J. Leukoc. Biol. 2000, 67, 699–704. [Google Scholar] [CrossRef]
- Gerszten, R.E.; Tager, A.M. The monocyte in atherosclerosis—should I stay or should I go now? N. Engl. J. Med. 2012, 366, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Tam, H.; Adler, L.; Ilstad-Minnihan, A.; Macaubas, C.; Mellins, E.D. The MHC class II antigen presentation pathway in human monocytes differs by subset and is regulated by cytokines. PLoS ONE 2017, 12, e0183594. [Google Scholar] [CrossRef] [PubMed]
- Belge, K.U.; Dayyani, F.; Horelt, A.; Siedlar, M.; Frankenberger, M.; Frankenberger, B.; Espevik, T.; Ziegler-Heitbrock, L. The proinflammatory CD14+CD16+DR++ monocytes are a major source of TNF. J. Immunol. 2002, 168, 3536–3542. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K. The spatial and developmental relationships in the macrophage family. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1517–1522. [Google Scholar] [CrossRef]
- Castagna, A.; Polati, R.; Bossi, A.M.; Girelli, D. Monocyte/macro phage proteomics: Recent findings and biomedical applications. Expert Rev. Proteom. 2012, 9, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Oertelt-Prigione, S. The influence of sex and gender on the immune response. Autoimmun. Rev. 2012, 11, A479–A485. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Dieterlen, M.T.; John, K.; Reichenspurner, H.; Mohr, F.W.; Barten, M.J. Dendritic Cells and Their Role in Cardiovascular Diseases: A View on Human Studies. J. Immunol. Res. 2016, 2016, 5946807. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Sica, A.; Erreni, M.; Allavena, P.; Porta, C. Macrophage polarization in pathology. Cell Mol. Life Sci. 2015, 72, 4111–4126. [Google Scholar] [CrossRef]
- Sreejit, G.; Fleetwood, A.J.; Nagareddy, P.R. Origins and diversity of macrophages in health and disease. Clin. Transl. Immunol. 2020, 9, e1222. [Google Scholar] [CrossRef] [PubMed]
- Elchinova, E.; Teubel, S.I.; Fernandes, M.A.; Lupon, J.; Galvez-Monton, C.; de Antonio, M.; Moliner, P.; Domingo, M.; Zamora, E.; Nunez, J.; et al. Circulating Monocyte Subsets and Heart Failure Prognosis. PLoS ONE 2018, 13, e0204074. [Google Scholar] [CrossRef] [PubMed]
- Charach, G.; Rogovski, O.; Karniel, E.; Charach, L.; Groskopf, I.; Novikov, I. Monocytes may be favorable biomarker and predictor of ling-term outcome in patients with chronic heart failure. Medicine 2019, 98, e17108. [Google Scholar] [CrossRef] [PubMed]
- Robbins, C.S.; Chudnovskiy, A.; Rauch, P.J.; Figueiredo, J.L.; Iwamoto, Y.; Gorbatov, R.; Etzrodt, M.; Weber, G.F.; Ueno, T.; Van Rooijen, N.; et al. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation 2012, 125, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Sam, F.; Nahrendorf, M. Monocyte and macrophage contributions to cardiac remodeling. J. Mol. Cell Cardiol. 2016, 93, 149–155. [Google Scholar] [CrossRef]
- Van Craenenbroeck, A.H.; Van Ackeren, K.; Hoymans, V.Y.; Johan Roeykens, J.; Gert, A.; Verpooten, G.A.; Vrints, C.J.; Couttenye, M.M.; Van Craenenbroeck, E.M. Acute exercise-induced response of monocyte subtypes in chronic heart and renal failure. Mediat. Inflamm. 2014, 216534. [Google Scholar] [CrossRef]
- Barisione, C.; Garibaldi, S.; Ghigliotti, G.; Fabbi, P.; Altieri, P.; Casale, M.C.; Spallarossa, P.; Bertero, G.; Balbi, M.; Corsiglia, L.; et al. CD14CD16 monocyte subset levels in heart failure patients. Dis. Markers 2010, 28, 115–124. [Google Scholar] [CrossRef]
- Amir, O.; Spivak, I.; Lavi, I.; Rahat, M.A. Changes in the monocytic subsets CD14(dim)CD16(+) and CD14(++)CD16(-) in chronic systolic heart failure patients. Mediat. Inflamm. 2012, 2012, 616384. [Google Scholar] [CrossRef]
- Mongirdienė, A.; Laukaitienė, J.; Skipskis, V.; Kuršvietienė, L.; Liobikas, J. Platelet activity and its correlation with inflammation and cell count readings in chronic heart failure patients with reduced ejection fraction. Medicina 2021, 57, 176. [Google Scholar] [CrossRef]
- Mongirdienė, A.; Laukaitienė, J.; Skipskis, V.; Kuršvietienė, L.; Liobikas, J. The Difference of Cholesterol, Platelet and Cortisol Levels in Patients Diagnosed with Chronic Heart Failure with Reduced Ejection Fraction Groups According to Neutrophil Count. Medicina 2021, 57, 557. [Google Scholar] [CrossRef]
- Wrigley, B.J.; Shantsila, E.; Tapp, L.D.; Lip, G.Y. CD14++CD16+ monocytes in patients with acute ischaemic heart failure. Eur. J. Clin. Invest. 2013, 43, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Yan, L.F.; Luo, Y.W.; Liu, X.L.; Liu, J.X.; Guo, Z.Z.; Xu, Z.W.; Li, Y.M.; Ji, W.J.; Zhou, X. Trajectories of Circulating Monocyte Subsets After ST-Elevation Myocardial Infarction During Hospitalization: Latent Class Growth Modeling for High-Risk Patient Identification. J. Cardiovasc. Transl. Res. 2018, 11, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Zhang, Z.; Fu, C.; Ma, G. Intermediate monocytes lead to enhanced myocardial remodelling in STEMI patients with diabetes. Int. Heart J. 2015, 56, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Tromp, J.; Khan, M.A.; Klip, I.T.; Meyer, S.; de Boer, R.A.; Jaarsma, T.; Hillege, H.; van Veldhuisen, D.J.; van der Meer, P.; Voors, A.A. Biomarker Profiles in Heart Failure Patients With Preserved and Reduced Ejection Fraction. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Tromp, J.; Westenbrink, B.D.; Ouwerkerk, W.; van Veldhuisen, D.J.; Samani, N.J.; Ponikowski, P.; Metra, M.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; et al. dentifying Pathophysiological Mechanisms in Heart Failure With Reduced Versus Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2018, 72, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Sanders-van Wijk, S.; van Empel, V.; Davarzani, N.; Maeder, M.T.; Handschin, R.; Pfisterer, M.E.; Brunner-La Rocca, H.P. Circulating biomarkers of distinct pathophysiological pathways in heart failure with preserved vs. reduced left ventricular ejection fraction. Eur. J. Heart Fail. 2015, 17, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; Van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef]
- Bosco, M.C.; Puppo, M.; Blengio, F.; Fraone, T.; Cappello, P.; Giovarelli, M.; Varesio, L. Monocytes and dendritic cells in a hypoxic environment: Spotlights on chemotaxis and migration. Immunobiology 2008, 213, 733–749. [Google Scholar] [CrossRef]
- Bradham, W.S.; Moe, G.; Wendt, K.A.; Scott, A.A.; Konig, A.; Romanova, M.; Naik, G.; Spinale, F.G. TNF-α and myocardial matrix metalloproteinases in heart failure: Relationship to LV remodeling. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1288–H1295. [Google Scholar] [CrossRef][Green Version]
- Van Loon, R.B.; Veen, G.; Kamp, O.; Baur, L.H.; Van Rossum, A.C. Left ventricular remodeling after acute myocardial infarction: The influence of viability and revascularization—an echocardiographic substudy of the VIAMI-trial. Trials 2014, 15, 329. [Google Scholar] [CrossRef][Green Version]
- Kaikita, K.; Hayasaki, T.; Okuma, T.; Kuziel, W.A.; Ogawa, H.; Takeya, M. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am. J. Pathol. 2004, 165, 439–447. [Google Scholar] [CrossRef]
- Simmonds, S.J.; Cuijpers, I.; Heymans, S.; Jones, E.A.V. Cellular and molecular differences between HFpEF and HFrEF: A step ahed in an improved pathological understanding. Cells 2020, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Sager, H.B.; Roh, J.D.; Valero-Munoz, M.; Houstis, N.E.; Iwamoto, Y.; Sun, Y.; Wilson, R.M.; Woitkewicz, G.; Tricot, B.; et al. Cardiac macrophages promote diastolic dysfunction. J. Exp. Med. 2018, 215, 423–440. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Adamopoulos, S.; Anker, S.D.; Auricchio, A.; Böhm, M.; Dickstein, K.; Falk, V.; Filippatos, G.; Fonseca, C.; Gomez-Sanchez, M.A.; et al. ESC Committee for Practice Guidelines. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2012, 33, 1787–1847. [Google Scholar] [CrossRef]
- Mewhort, H.E.; Lipon, B.D.; Svystonyuk, D.A.; Teng, G.; Guzzardi, D.G.; Silva, C.; Yong, V.W.; Fedak, P.W. Monocytes increase human cardiac myofibroblast-mediated extracellular matrix remodeling through TGF β1. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H716–H724. [Google Scholar] [CrossRef]
- Glezeva, N.; Voon, V.; Watson, C.; Horgan, S.; McDonald, K.; Ledwidge, M.; Baugh, J. Exaggerated inflammation and monocytosis associate with diastolic dysfunction in heart failure with preserved ejection fraction: Evidence of M2 macrophage activation in disease pathogenesis. J. Card. Fail. 2015, 21, 167–177. [Google Scholar] [CrossRef]
- Collier, P.; Watson, C.J.; Voon, V.; Phelan, D.; Jan, A.; Mak, G.; Martos, R.; Baugh, J.A.; Ledwidge, M.T.; McDonald, K.M. Can emerging biomarkers of myocardial remodelling identify asymptomatic hypertensive patients at risk for diastolic dysfunction and diastolic heart failure? Eur. J. Heart Fail. 2011, 13, 1087–1095. [Google Scholar] [CrossRef]
- Abernethy, A.; Raza, S.; Sun, J.L.; Anstrom, K.J.; Tracy, R.; Steiner, J.; VanBuren, P.; LeWinter, M.M. Pro-Inflammatory Biomarkers in Stable Versus Acutely Decompensated Heart Failure With Preserved Ejection Fraction. J. Am. Heart Assoc. 2018, 7, e007385. [Google Scholar] [CrossRef]
- Westermann, D.; Lindner, D.; Kasner, M.; Zietsch, C.; Savvatis, K.; Escher, F.; von Schlippenbach, J.; Skurk, C.; Steendijk, P.; Riad, A.; et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ. Heart Fail. 2011, 4, 44–52. [Google Scholar] [CrossRef]
- Zhang, S.; Weinberg, S.; DeBerge, M.; Gainullina, A.; Schipma, M.; Kinchen, J.M.; Ben-Sahra, I.; Gius, D.R.; Yvan-Charvet, L.; Chandel, N.S.; et al. Efferocytosis Fuels Requirements of Fatty Acid Oxidation and the Electron Transport Chain to Polarize Macrophages for Tissue Repair. Cell Metab. 2019, 29, 443–456. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Rajasingh, J.; Lambers, E.; Qin, G.; Losordo, D.W.; Kishore, R. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ. Res. 2009, 104, e9–e18. [Google Scholar] [CrossRef] [PubMed]
- Kalogeropoulos, A.; Georgiopoulou, V.; Psaty, B.M.; Rodondi, N.; Smith, A.L.; Harrison, D.G.; Liu, Y.; Hoffmann, U.; Bauer, D.C.; Newman, A.B.; et al. Inflammatory markers and incident heart failure risk in older adults: The Health ABC (Health, Aging, and Body Composition) study. J. Am. Coll. Cardiol. 2010, 55, 2129–2137. [Google Scholar] [CrossRef] [PubMed]
- Fujiu, K.; Wang, J.; Nagai, R. Cardioprotective function of cardiac macrophages. Cardiovasc. Res. 2014, 10, 232–239. [Google Scholar] [CrossRef] [PubMed]
- van der Laan, A.M.; Ter Horst, E.N.; Delewi, R.; Begieneman, M.P.; Krijnen, P.A.; Hirsch, A.; Lavaei, M.; Nahrendorf, M.; Horrevoets, A.J.; Niessen, H.W.; et al. Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of the spleen as monocyte reservoir. Eur. Heart J. 2014, 35, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Deswal, A.; Petersen, N.J.; Feldman, A.M.; Young, J.B.; White, B.G.; Mann, D.L. Cytokines and cytokine receptors in advanced heart failure: An analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 2001, 103, 2055–2059.11. [Google Scholar] [CrossRef] [PubMed]
- Testa, M.; Yeh, M.; Lee, P.; Fanelli, R.; Loperfido, F.; Berman, J.W.; LeJemtel, T.H. Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J. Am. Coll. Cardiol. 1996, 28, 964–971. [Google Scholar] [CrossRef]
- Bernhagen, J.; Krohn, R.; Lue, H.; Gregory, J.L.; Zernecke, A.; Koenen, R.R.; Dewor, M.; Georgiev, I.; Schober, A.; Leng, L.; et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 2007, 13, 587–596. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Classical (Mon1) Subset CD14++/CD16- | Intermediate (Mon2) Subset CD14++/CD16+ | Non-classical (Mon3) Subset CD14+/CD16++ | Reference | |
|---|---|---|---|---|
| Highly expressed surface markers | CCR1, CCR2, CD1d, CD9, CD11b, CD33, CD36, CD62L, CD64, CD99, CLEC4D, CLEC5A, CXCR1-4 | CCR5, CD11b, CD32, CD40, CD47, CD54, CD64, CD80, CD86, CD163, GFRα2, HLA-ABC, HLA-DR, TNFR1 | CD45, CD97, CD116, CD123, CD294, CD11c, CX3CR1, P2RX1, Siglec10, SIRPα, SLAN, TNFR2 | [19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35] |
| High levels of cytokines | IL13Rα1, G-CSF, CCL2, MCP-1 | IL-6, IL-8, IL-10, TNF- α | TNFα, IL-1β, IL-6, IL-8 | [21,27,36,37] |
| Activated function | Phagocytosis; adhesion to the endothelium; migration; anti-microbial responses; inflammation | Antigen presentation; participation in proliferation and inflammatory responses; regulation of apoptosis; trans-endothelial migration; high ROS production | Complement and FcR-mediated phagocytosis; trans-endothelial migration; adhesion; anti-viral responses; patrolling the endothelium | [20,38] |
| Part of total monocyte count in the blood (%) | 80.1 ± 7 | 3.7 ± 2 | 6.2 ± 2.8 | [20,26,37] |
| Implicit place of formation/persistency | Bone marrow/tissues | Peripheral blood flow or tissues/blood | Peripheral blood flow or tissues | [39,40] |
| Lifespan | 1 day | 3–4 days | 4–7 days | [41] |
| Investigated Person | CHF (IDC (65% of Investigated Population) and ISH) | Healthy | Ambulatory Treated CHF I-IV NYHA Functional Class | CHF I-III NYHA Functional Class, 57% ISH, 43% IDC | Healthy | Stabile CVD where LVEF > 43% | Healthy | Systolic CHF II-IV NYHA Functional Class | Healthy | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Alive | Deceased | ||||||||||
| Reference | [77] | [73] | [78] | [27] | [79] | ||||||
| n | 20 | 15 | 293 | 107 | 30 | 26 | 14 | 13 | 59 | 29 | |
| Gender F/M | 7/13 | 6/9 | 80/213 | 29/78 | M | M | 5/9 | 8/5 | 14/45 | 14/15 | |
| Age | 51,2 (9,3) | 43,5 (5,0) | 66,7 (11,9) | 76,9 (9,7) | 70,9 (2,1) | 69,5 (2,2) | 60 (9) | 59 11) | 58,1 (13,9) | 59,7 (6,4) | |
| BMI | 26,6 (3,8) | 24,2 (2,3) | |||||||||
| Exclusion criteria /Inclusion criteria | Active inflammatory or malignant disease and treatment with immunosuppressive agents /CHF patients | Active inflammatory disease /HF irrespective of etiology (at least 1 HF hospitalization or reduced LVEF) | Inflammatory, cancer, autoimmune diseases, malnutrition /CHF lasting longer than 1 year, clinical stability and the same treatment in the last 3 weeks, LVEF≤45% | ACS or coronary revascularization within the last 6 months, current inflammation within the last 6 months, autoimmune or malignant diseases, dialysis-requiring renal failure /stable CAD (1–3 vessel disease) | Acute heart failure or acute coronary syndrome, or haemodialysis, or known systemic inflammatory disease /LVEF<40%, no recent cardiac decompensation | ||||||
| Leukocyte count (106/mL) | 8.24 (1.82) | 7.17 (1.60) | 8.34 (0.62) | 6.45 (0.26) | 7.0 (4.2–9.4) | 6.7 (4.3–15.6) | |||||
| Monocytes | % of leukocytes | 7.72 (1.88) | 6.28 (1.24) | 5.1 (3.6–10.8) | 3.7 (3.2–8.0) | ||||||
| Count (cells/µL) | 628 (159) | 450 (128) | 629 (61) | 509 (34) | 354 (131–452) | 308 (187–440) | |||||
| Monocyte subsets (% of monocytes) | % Mon1 | 87.34 (3.54) | 88.09 (4.73) | 50.4 (16.5) | 48.9 (19.08) | 73.5 (1.8) | 84.3 (1.9) | ||||
| % Mon2 | 4.74 (2.46) | 4.51 (2.05) | 41.2 (16.5) | 44.0 (18.8) | 12.3 (8.7–14.8) | 5.9 (4.7–6.9) | |||||
| % Mon3 | 7.92 (2.19) | 7.39 (3.17) | 8.42 (4.0) | 7.1 (4.0) | |||||||
| Monocyte subsets (cells/L) | Mon1 | 550.3 (143.9) | 395.2 (107) | 327 (222–435) | 363 (227–451) | 303 (113–437) | 266 (161–412) | ||||
| Mon2 | 29.3 (17.1) | 20.7 (13.5) | 253 (170–374) | 303 (186–470) | |||||||
| Mon3 | 49.3 (17.3) | 34.1 (20.9) | 48 (35–71) | 44 (27–73) | |||||||
| HFpEF | HFrEF | |
|---|---|---|
| Predominant monocyte subset in the myocardium | CD14++, CD16+ (Mon2) | CD14++, CD16- (Mon1) |
| Differences in pathogenesis | Low-grade systemic inflammation; monocytes produce chemokines (MCP-1, TNF-α, TGF-β, IL-6). | Cardiac inflammation; fibrosis is associated with monocyte surface TLRs and the migration of Mon1 monocytes into the myocardium due to increased levels of IL-1β and CCR2 expression. |
| Macrophage subset | M2 | M1 |
| Myocardial changes | LV stiffness is caused by reduced Ca2+ signaling; conversion of titin into a less flexible form; perivascular and interstitial fibrosis; fibrotic changes in extracellular matrix and cardiomyocyte hypertrophy; impaired relaxation of the heart muscle | Collagen scar formation; cardiomyocyte apoptosis; impaired myocardial contraction |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mongirdienė, A.; Liobikas, J. Phenotypic and Functional Heterogeneity of Monocyte Subsets in Chronic Heart Failure Patients. Biology 2022, 11, 195. https://doi.org/10.3390/biology11020195
Mongirdienė A, Liobikas J. Phenotypic and Functional Heterogeneity of Monocyte Subsets in Chronic Heart Failure Patients. Biology. 2022; 11(2):195. https://doi.org/10.3390/biology11020195
Chicago/Turabian StyleMongirdienė, Aušra, and Julius Liobikas. 2022. "Phenotypic and Functional Heterogeneity of Monocyte Subsets in Chronic Heart Failure Patients" Biology 11, no. 2: 195. https://doi.org/10.3390/biology11020195
APA StyleMongirdienė, A., & Liobikas, J. (2022). Phenotypic and Functional Heterogeneity of Monocyte Subsets in Chronic Heart Failure Patients. Biology, 11(2), 195. https://doi.org/10.3390/biology11020195