


Interpretable and Predictive Deep Neural Network Modeling of the SARS-CoV-2 Spike Protein Sequence to Predict COVID-19 Disease Severity

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection and Pre-Processing
2.1.1. SARS-CoV-2 Spike Protein Sequences
2.1.2. GISAID Sequences Containing Patient (Clinical) Status Metadata
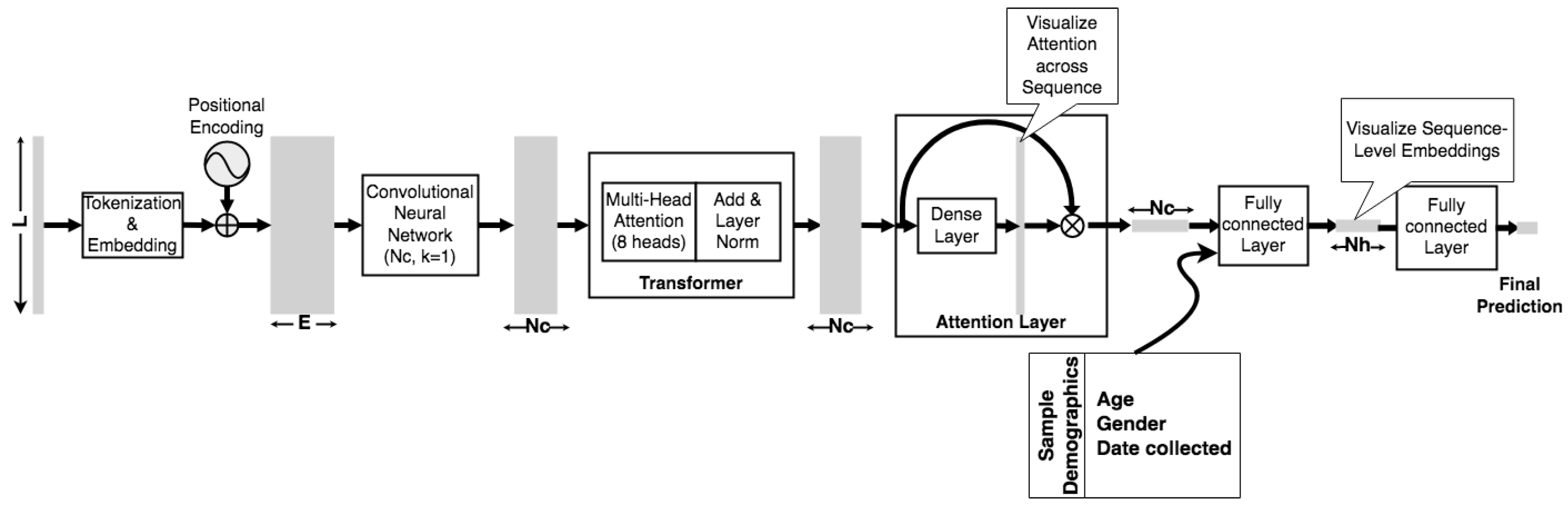
2.2. Model Architecture
2.3. Implementation
2.3.1. Model Parameters
2.3.2. Training and Validation
3. Results
3.1. Regression Based on Sample Collection Date
3.2. Predicting COVID-19 Patient Outcomes Using Machine Learning Models Based on Both Sequence and Demographic Information
3.3. Interpreting the Disease Severity Model by Visualizing Embeddings and Attention
3.4. Predicting Omicron Disease Severity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BERT | Bidirectional encoder representations from transformers |
| Bi-LSTM | Bidirectional long short term memory |
| CNN | Convolutional neural network |
| CTD | C-terminal domain |
| NLP | Natural language processing |
| NTD | N-terminal domain |
| RBD | Receptor binding domain |
| RNN | Recurrent neural network |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| t-SNE | t-distributed stochastic neighbor embedding |
| VOC | Variant Of Concern |
References
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, E.J.; Osterholm, M.; Gounder, C.R. A National Strategy for the “New Normal” of Life With COVID. JAMA 2022, 327, 211–212. [Google Scholar] [CrossRef] [PubMed]
- Sokhansanj, B.A.; Rosen, G.L. Mapping Data to Deep Understanding: Making the Most of the Deluge of SARS-CoV-2 Genome Sequences. mSystems 2022, 7, e00035-22. [Google Scholar] [CrossRef] [PubMed]
- Pillay, T.S. Gene of the Month: The 2019-nCoV/SARS-CoV-2 Novel Coronavirus Spike Protein. J. Clin. Pathol. 2020, 73, 366. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell Entry Mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Weinreich, D.M.; Sivapalasingam, S.; Norton, T.; Ali, S.; Gao, H.; Bhore, R.; Musser, B.J.; Soo, Y.; Rofail, D.; Im, J.; et al. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients with COVID-19. N. Engl. J. Med. 2021, 384, 238–251. [Google Scholar] [CrossRef]
- Thomas, S.J.; Moreira, E.D.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Polack, F.P.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine through 6 Months. N. Engl. J. Med. 2021, 385, 1761–1773. [Google Scholar] [CrossRef]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020, 383, 1920–1931. [Google Scholar] [CrossRef]
- Bosch, B.J.; van der Zee, R.; de Haan, C.A.M.; Rottier, P.J.M. The Coronavirus Spike Protein Is a Class I Virus Fusion Protein: Structural and Functional Characterization of the Fusion Core Complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef]
- Plante, J.A.; Mitchell, B.M.; Plante, K.S.; Debbink, K.; Weaver, S.C.; Menachery, V.D. The Variant Gambit: COVID-19’s next Move. Cell Host Microbe 2021, 29, 508–515. [Google Scholar] [CrossRef]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral Mutation Rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- Peacock, T.P.; Bauer, D.L.V.; Barclay Wendy, S. Putative Host Origins of RNA Insertions in SARS-CoV-2 Genomes. 2021. Available online: https://virological.org/t/putative-host-origins-of-rna-insertions-in-sars-cov-2-genomes/761 (accessed on 14 November 2022).
- Koyama, T.; Weeraratne, D.; Snowdon, J.L.; Parida, L. Emergence of Drift Variants That May Affect COVID-19 Vaccine Development and Antibody Treatment. Pathogens 2020, 9, 324. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Lu, X.; Chen, Q.; Xu, K.; Chen, Y.; Cheng, M.; Chen, K.; Cheng, L.; Weng, T.; Shi, D.; et al. Patient-Derived SARS-CoV-2 Mutations Impact Viral Replication Dynamics and Infectivity in Vitro and with Clinical Implications in Vivo. Cell Discov. 2020, 6, 76. [Google Scholar] [CrossRef]
- Vankadari, N. Overwhelming Mutations or SNPs of SARS-CoV-2: A Point of Caution. Gene 2020, 752, 144792. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The Biological and Clinical Significance of Emerging SARS-CoV-2 Variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike Mutation D614G Alters SARS-CoV-2 Fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Hanage, W.P.; Rasmussen, A.L. Making Sense of Mutation: What D614G Means for the COVID-19 Pandemic Remains Unclear. Cell 2020, 182, 794–795. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Davies, N.G.; Jarvis, C.I.; CMMID COVID-19 Working Group; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H. Increased Mortality in Community-Tested Cases of SARS-CoV-2 Lineage B.1.1.7. Nature 2021, 593, 270–274. [Google Scholar] [CrossRef]
- Challen, R.; Brooks-Pollock, E.; Read, J.M.; Dyson, L.; Tsaneva-Atanasova, K.; Danon, L. Risk of Mortality in Patients Infected with SARS-CoV-2 Variant of Concern 202012/1: Matched Cohort Study. BMJ (Clin. Res. Ed.) 2021, 372, n579. [Google Scholar] [CrossRef] [PubMed]
- Frampton, D.; Rampling, T.; Cross, A.; Bailey, H.; Heaney, J.; Byott, M.; Scott, R.; Sconza, R.; Price, J.; Margaritis, M.; et al. Genomic Characteristics and Clinical Effect of the Emergent SARS-CoV-2 B.1.1.7 Lineage in London, UK: A Whole-Genome Sequencing and Hospital-Based Cohort Study. Lancet Infect. Dis. 2021, 21, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Giles, B.; Meredith, P.; Robson, S.; Smith, G.; Chauhan, A.; PACIFIC-19 and COG-UK research groups. The SARS-CoV-2 B.1.1.7 Variant and Increased Clinical Severity-the Jury Is Out. Lancet Infect. Dis. 2021, 21, 1213–1214. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Rocklöv, J. The Reproductive Number of the Delta Variant of SARS-CoV-2 Is Far Higher Compared to the Ancestral SARS-CoV-2 Virus. J. Travel Med. 2021, 28, taab124. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta Spike P681R Mutation Enhances SARS-CoV-2 Fitness over Alpha Variant. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mlcochova, P.; Kemp, S.A.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.A.T.M.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S.; et al. SARS-CoV-2 B.1.617.2 Delta Variant Replication and Immune Evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef]
- Dite, G.S.; Murphy, N.M.; Allman, R. Development and Validation of a Clinical and Genetic Model for Predicting Risk of Severe COVID-19. Epidemiol. Infect. 2021, 149, e162. [Google Scholar] [CrossRef]
- Dite, G.S.; Murphy, N.M.; Allman, R. An Integrated Clinical and Genetic Model for Predicting Risk of Severe COVID-19: A Population-Based Case-Control Study. PLoS ONE 2021, 16, e0247205. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Wongtrakoongate, P.; Thawornwattana, Y.; Hongeng, S.; Thitithanyanont, A. SARS-CoV-2 Genetic Variations Associated with COVID-19 Severity. medRxiv 2020. [Google Scholar] [CrossRef]
- SeyedAlinaghi, S.; Mirzapour, P.; Dadras, O.; Pashaei, Z.; Karimi, A.; MohsseniPour, M.; Soleymanzadeh, M.; Barzegary, A.; Afsahi, A.M.; Vahedi, F.; et al. Characterization of SARS-CoV-2 Different Variants and Related Morbidity and Mortality: A Systematic Review. Eur. J. Med. Res. 2021, 26, 51. [Google Scholar] [CrossRef]
- Laskar, R.; Ali, S. Differential Mutation Profile of SARS-CoV-2 Proteins across Deceased and Asymptomatic Patients. Chem.-Biol. Interact. 2021, 347, 109598. [Google Scholar] [CrossRef] [PubMed]
- Clauwaert, J.; Menschaert, G.; Waegeman, W.; Dumonteil, E.; Fusco, D.; Drouin, A.; Herrera, C.; Esper, F.P.; Cheng, Y.W.; Adhikari, T.M.; et al. Geographical and Temporal Distribution of SARS-CoV-2 Globally: An Attempt to Correlate Case Fatality Rate with the Circulating Dominant SARS-CoV-2 Clades. medRxiv 2021, 193. [Google Scholar] [CrossRef]
- Hamed, S.M.; Elkhatib, W.F.; Khairalla, A.S.; Noreddin, A.M. Global Dynamics of SARS-CoV-2 Clades and Their Relation to COVID-19 Epidemiology. Sci. Rep. 2021, 11, 8435. [Google Scholar] [CrossRef] [PubMed]
- Voss, J.D.; Skarzynski, M.; McAuley, E.M.; Maier, E.J.; Gibbons, T.; Fries, A.C.; Chapleau, R.R. Variants in SARS-CoV-2 Associated with Mild or Severe Outcome. Evol. Med. Public Health 2021, 9, 267–275. [Google Scholar] [CrossRef]
- Agarwal, R.; Leblond, T.; McAuley, E.M.; Maier, E.J.; Skarzynski, M.; Voss, J.D.; Sozhamannan, S. Linking Genotype to Phenotype: Further Exploration of Mutations in SARS-CoV-2 Associated with Mild or Severe Outcomes—SARS-CoV-2 Coronavirus. 2022. Available online: https://virological.org/t/linking-genotype-to-phenotype-further-exploration-of-mutations-in-sars-cov-2-associated-with-mild-or-severe-outcomes/794 (accessed on 14 November 2022).
- Nagy, Á.; Ligeti, B.; Szebeni, J.; Pongor, S.; Gyorffy, B. COVIDOUTCOME—Estimating COVID Severity Based on Mutation Signatures in the SARS-CoV-2 Genome. Database 2021, 2021, baab020. [Google Scholar] [CrossRef]
- Sawmya, S.; Saha, A.; Tasnim, S.; Toufikuzzaman, M.; Anjum, N.; Rafid, A.H.M.; Rahman, M.S.; Rahman, M.S. Analyzing hCov Genome Sequences: Predicting Virulence and Mutation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Obermeyer, F.; Schaffner, S.F.; Jankowiak, M.; Barkas, N.; Pyle, J.D.; Park, D.J.; MacInnis, B.L.; Luban, J.; Sabeti, P.C.; Lemieux, J.E. Analysis of 2.1 Million SARS-CoV-2 Genomes Identifies Mutations Associated with Transmissibility. medRxiv 2021. [Google Scholar] [CrossRef]
- Sun, Q.; Shu, C.; Shi, W.; Luo, Y.; Fan, G.; Nie, J.; Bi, Y.; Wang, Q.; Qi, J.; Lu, J.; et al. VarEPS: An Evaluation and Prewarning System of Known and Virtual Variations of SARS-CoV-2 Genomes. Nucleic Acids Res. 2022, 50, D888–D897. [Google Scholar] [CrossRef]
- Mullick, B.; Magar, R.; Jhunjhunwala, A.; Barati Farimani, A. Understanding Mutation Hotspots for the SARS-CoV-2 Spike Protein Using Shannon Entropy and K-means Clustering. Comput. Biol. Med. 2021, 138, 104915. [Google Scholar] [CrossRef]
- Beguir, K.; Skwark, M.J.; Fu, Y.; Pierrot, T.; Carranza, N.L.; Laterre, A.; Kadri, I.; Lui, B.G.; Sänger, B.; Liu, Y.; et al. Early Computational Detection of Potential High Risk SARS-CoV-2 Variants. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sokhansanj, B.A.; Rosen, G.L. Predicting COVID-19 Disease Severity from SARS-CoV-2 Spike Protein Sequence by Mixed Effects Machine Learning. Comput. Biol. Med. 2022, 149, 105969. [Google Scholar] [CrossRef] [PubMed]
- Bahdanau, D.; Cho, K.; Bengio, Y. Neural Machine Translation by Jointly Learning to Align and Translate. arXiv 2014, arXiv:1409.0473. [Google Scholar]
- Xu, K.; Ba, J.; Kiros, R.; Cho, K.; Courville, A.C.; Salakhutdinov, R.; Zemel, R.S.; Bengio, Y. Show, Attend and Tell: Neural Image Caption Generation with Visual Attention. arXiv 2015, arXiv:1502.03044. [Google Scholar]
- Rush, A.M.; Chopra, S.; Weston, J. A Neural Attention Model for Abstractive Sentence Summarization. arXiv 2015, arXiv:1509.00685. [Google Scholar]
- Yang, Z.; Yang, D.; Dyer, C.; He, X.; Smola, A.; Hovy, E. Hierarchical Attention Networks for Document Classification. In Proceedings of the Proceedings of the 2016 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, San Diego, CA, USA, 12–17 June 2016; pp. 1480–1489. [Google Scholar]
- Zhou, P.; Shi, W.; Tian, J.; Qi, Z.; Li, B.; Hao, H.; Xu, B. Attention-Based Bidirectional Long Short-Term Memory Networks for Relation Classification. In Proceedings of the Proceedings of the 54th Annual Meeting of the Association for Computational Linguistics (Volume 2: Short Papers), Berlin, Germany, 7–12 August 2016; pp. 207–212. [Google Scholar]
- Deming, L.; Targ, S.; Sauder, N.; Almeida, D.; Ye, C.J. Genetic Architect: Discovering Genomic Structure with Learned Neural Architectures. arXiv 2016, arXiv:1605.07156. [Google Scholar]
- Lanchantin, J.; Singh, R.; Lin, Z.; Qi, Y. Deep Motif: Visualizing Genomic Sequence Classifications. arXiv 2016, arXiv:1605.01133. [Google Scholar]
- Zhao, Z.; Woloszynek, S.; Agbavor, F.; Mell, J.C.; Sokhansanj, B.A.; Rosen, G.L. Learning, Visualizing and Exploring 16S rRNA Structure Using an Attention-Based Deep Neural Network. PLoS Comput. Biol. 2021, 17, e1009345. [Google Scholar] [CrossRef]
- Zhao, Z.; Rosen, G. Visualizing and Annotating Protein Sequences Using A Deep Neural Network. In Proceedings of the 2020 54th Asilomar Conference on Signals, Systems, and Computers, Pacific Grove, CA, USA, 1–4 November 2020; pp. 506–510. [Google Scholar] [CrossRef]
- Clauwaert, J.; Menschaert, G.; Waegeman, W. Explainability in Transformer Models for Functional Genomics. Briefings Bioinform. 2021, 22, bbab060. [Google Scholar] [CrossRef]
- Karim, S.S.A.; Karim, Q.A. Omicron SARS-CoV-2 Variant: A New Chapter in the COVID-19 Pandemic. Lancet 2021, 398, 2126–2128. [Google Scholar] [CrossRef]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef]
- Zhao, M.; Lee, W.P.; Garrison, E.P.; Marth, G.T. SSW Library: An SIMD Smith-Waterman C/C++ Library for Use in Genomic Applications. PLoS ONE 2013, 8, e82138. [Google Scholar] [CrossRef] [PubMed]
- The Scikit-Bio Development Team. Scikit-Bio: A Bioinformatics Library for Data Scientists, Students, and Developers. 2020. Available online: https://www.youtube.com/watch?v=ZpgkRQooGqo (accessed on 14 November 2022).
- Wang, C.; Liu, Z.; Chen, Z.; Huang, X.; Xu, M.; He, T.; Zhang, Z. The Establishment of Reference Sequence for SARS-CoV-2 and Variation Analysis. J. Med. Virol. 2020, 92, 667–674. [Google Scholar] [CrossRef]
- National Institutes of Health. Clinical Spectrum of SARS-CoV-2 Infection; National Institutes of Health: Bethesda, MD, USA, 2021.
- Luong, M.T.; Pham, H.; Manning, C.D. Effective Approaches to Attention-based Neural Machine Translation. arXiv 2015, arXiv:1508.04025. [Google Scholar]
- Kobayashi, G.; Kuribayashi, T.; Yokoi, S.; Inui, K. Attention Is Not Only a Weight: Analyzing Transformers with Vector Norms. arXiv 2020, arXiv:2004.10102. [Google Scholar]
- Woloszynek, S.; Zhao, Z.; Chen, J.; Rosen, G.L. 16S rRNA Sequence Embeddings: Meaningful Numeric Feature Representations of Nucleotide Sequences That Are Convenient for Downstream Analyses. PLoS Comput. Biol. 2019, 15, e1006721. [Google Scholar] [CrossRef] [PubMed]
- van der Maaten, L.; Hinton, G. Visualizing Data Using T-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; Association for Computing Machinery: New York, NY, USA, 2016; pp. 785–794. [Google Scholar] [CrossRef]
- Angeletti, S.; Giovanetti, M.; Fogolari, M.; Cella, E.; De Florio, L.; Lintas, C.; Veralli, R.; Francesconi, M.; Caccuri, F.; De Cesaris, M.; et al. SARS-CoV-2 AY.4.2 Variant Circulating in Italy: Genomic Preliminary Insight. J. Med Virol. 2021, 94, 1689–1692. [Google Scholar] [CrossRef]
- Aoki, A.; Adachi, H.; Mori, Y.; Ito, M.; Sato, K.; Okuda, K.; Sakakibara, T.; Okamoto, Y.; Jinno, H. A Rapid Screening Assay for L452R and T478K Spike Mutations in SARS-CoV-2 Delta Variant Using High-Resolution Melting Analysis. J. Toxicol. Sci. 2021, 46, 471–476. [Google Scholar] [CrossRef]
- van Dorp, C.H.; Goldberg, E.E.; Hengartner, N.; Ke, R.; Romero-Severson, E.O. Estimating the Strength of Selection for New SARS-CoV-2 Variants. Nat. Commun. 2021, 12, 7239. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 Variant through Europe in the Summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef]
- Singh, A.; Steinkellner, G.; Köchl, K.; Gruber, K.; Gruber, C.C. Serine 477 Plays a Crucial Role in the Interaction of the SARS-CoV-2 Spike Protein with the Human Receptor ACE2. Sci. Rep. 2021, 11, 4320. [Google Scholar] [CrossRef] [PubMed]
- Jangra, S.; Ye, C.; Rathnasinghe, R.; Stadlbauer, D.; Personalized Virology Initiative study group; Krammer, F.; Simon, V.; Martinez-Sobrido, L.; García-Sastre, A.; Schotsaert, M. SARS-CoV-2 Spike E484K Mutation Reduces Antibody Neutralisation. Lancet Microbe 2021, 2, e283–e284. [Google Scholar] [CrossRef] [PubMed]
- Saha, I.; Ghosh, N.; Sharma, N.; Nandi, S. Hotspot Mutations in SARS-CoV-2. Front. Genet. 2021, 12, 753440. [Google Scholar] [CrossRef]
- Adiga, R.; Nayak, V. Emergence of Novel SARS-CoV-2 Variants in India: Second Wave. J. Infect. Dev. Ctries. 2021, 15, 1578–1583. [Google Scholar] [CrossRef] [PubMed]
- Lubinski, B.; Fernandes, M.H.V.; Frazier, L.; Tang, T.; Daniel, S.; Diel, D.G.; Jaimes, J.A.; Whittaker, G.R. Functional Evaluation of the P681H Mutation on the Proteolytic Activation the SARS-CoV-2 Variant B.1.1.7 (Alpha) Spike. iScience 2021, 25, 103589. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Malhotra, A.G.; Biswas, D.; Shankar, P.; Lokhande, L.; Yadav, A.K.; Raghuvanshi, A.; Kale, D.; Nema, S.; Saigal, S.; et al. Relative Consolidation of the Kappa Variant Pre-Dates the Massive Second Wave of COVID-19 in India. Genes 2021, 12, 1803. [Google Scholar] [CrossRef]
- Gao, Y.D.; Ding, M.; Dong, X.; Zhang, J.J.; Kursat Azkur, A.; Azkur, D.; Gan, H.; Sun, Y.L.; Fu, W.; Li, W.; et al. Risk Factors for Severe and Critically Ill COVID-19 Patients: A Review. Allergy 2021, 76, 428–455. [Google Scholar] [CrossRef]
- Grasselli, G.; Greco, M.; Zanella, A.; Albano, G.; Antonelli, M.; Bellani, G.; Bonanomi, E.; Cabrini, L.; Carlesso, E.; Castelli, G.; et al. Risk Factors Associated With Mortality Among Patients With COVID-19 in Intensive Care Units in Lombardy, Italy. JAMA Intern. Med. 2020, 180, 1345–1355. [Google Scholar] [CrossRef]
- Holt, H.; Talaei, M.; Greenig, M.; Zenner, D.; Symons, J.; Relton, C.; Young, K.S.; Davies, M.R.; Thompson, K.N.; Ashman, J.; et al. Risk Factors for Developing COVID-19: A Population-Based Longitudinal Study (COVIDENCE UK). Thorax 2021, 77, 900–912. [Google Scholar] [CrossRef]
- Fisman, D.N.; Tuite, A.R. Evaluation of the Relative Virulence of Novel SARS-CoV-2 Variants: A Retrospective Cohort Study in Ontario, Canada. CMAJ Can. Med Assoc. J. 2021, 193, E1619–E1625. [Google Scholar] [CrossRef]
- Paredes, M.I.; Lunn, S.M.; Famulare, M.; Frisbie, L.A.; Painter, I.; Burstein, R.; Roychoudhury, P.; Xie, H.; Mohamed Bakhash, S.A.; Perez, R.; et al. Associations between SARS-CoV-2 Variants and Risk of COVID-19 Hospitalization among Confirmed Cases in Washington State: A Retrospective Cohort Study. medRxiv 2021. [Google Scholar] [CrossRef]
- Twohig, K.A.; Nyberg, T.; Zaidi, A.; Thelwall, S.; Sinnathamby, M.A.; Aliabadi, S.; Seaman, S.R.; Harris, R.J.; Hope, R.; Lopez-Bernal, J.; et al. Hospital Admission and Emergency Care Attendance Risk for SARS-CoV-2 Delta (B.1.617.2) Compared with Alpha (B.1.1.7) Variants of Concern: A Cohort Study. Lancet Infect. Dis. 2021, 22, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Bager, P.; Wohlfahrt, J.; Fonager, J.; Rasmussen, M.; Albertsen, M.; Michaelsen, T.Y.; Møller, C.H.; Ethelberg, S.; Legarth, R.; Button, M.S.F.; et al. Risk of Hospitalisation Associated with Infection with SARS-CoV-2 Lineage B.1.1.7 in Denmark: An Observational Cohort Study. Lancet Infect. Dis. 2021, 21, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Bager, P.; Wohlfahrt, J.; Bhatt, S.; Stegger, M.; Legarth, R.; Møller, C.H.; Skov, R.L.; Valentiner-Branth, P.; Voldstedlund, M.; Fischer, T.K.; et al. Risk of Hospitalisation Associated with Infection with SARS-CoV-2 Omicron Variant versus Delta Variant in Denmark: An Observational Cohort Study. Lancet Infect. Dis. 2022. [Google Scholar] [CrossRef]
- Despres, H.W.; Mills, M.G.; Shirley, D.J.; Schmidt, M.M.; Huang, M.L.; Jerome, K.R.; Greninger, A.L.; Bruce, E.A. Quantitative Measurement of Infectious Virus in SARS-CoV-2 Alpha, Delta and Epsilon Variants Reveals Higher Infectivity (Viral Titer:RNA Ratio) in Clinical Samples Containing the Delta and Epsilon Variants. medRxiv 2021. [Google Scholar] [CrossRef]
- Gunadi; Hakim, M.S.; Wibawa, H.; Marcellus; Trisnawati, I.; Supriyati, E.; Afiahayati; Khair, R.E.; Iskandar, K.; Siswanto; et al. Association between Prognostic Factors and the Outcomes of Patients Infected with SARS-CoV-2 Harboring Multiple Spike Protein Mutations. Sci. Rep. 2021, 11, 21352. [Google Scholar] [CrossRef]
- Hsu, S.H.; Chang, S.H.; Gross, C.P.; Wang, S.Y. Relative Risks of COVID-19 Fatality between the First and Second Waves of the Pandemic in Ontario, Canada. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 2021, 109, 189–191. [Google Scholar] [CrossRef]
- Wang, J.; Gribskov, M. IRESpy: An XGBoost Model for Prediction of Internal Ribosome Entry Sites. BMC Bioinform. 2019, 20, 409. [Google Scholar] [CrossRef]
- ValizadehAslani, T.; Zhao, Z.; Sokhansanj, B.A.; Rosen, G.L. Amino Acid K-Mer Feature Extraction for Quantitative Antimicrobial Resistance (AMR) Prediction by Machine Learning and Model Interpretation for Biological Insights. Biology 2020, 9, 365. [Google Scholar] [CrossRef]
- Borisov, V.; Leemann, T.; Seßler, K.; Haug, J.; Pawelczyk, M.; Kasneci, G. Deep Neural Networks and Tabular Data: A Survey. arXiv 2022, arXiv:2110.01889. [Google Scholar]
- Otto, S.P.; Day, T.; Arino, J.; Colijn, C.; Dushoff, J.; Li, M.; Mechai, S.; Van Domselaar, G.; Wu, J.; Earn, D.J.; et al. The Origins and Potential Future of SARS-CoV-2 Variants of Concern in the Evolving COVID-19 Pandemic. Curr. Biol. 2021, 31, R918–R929. [Google Scholar] [CrossRef]
- Duong, D. Alpha, Beta, Delta, Gamma: What’s Important to Know about SARS-CoV-2 Variants of Concern? CMAJ Can. Med Assoc. J. 2021, 193, E1059–E1060. [Google Scholar] [CrossRef] [PubMed]
- Ramraj, S.; Uzir, N.; Sunil, R.; Banerjee, S. Experimenting XGBoost Algorithm for Prediction and Classification of Different Datasets. Int. J. Control. Theory Appl. 2016, 9, 651–662. [Google Scholar]
- Elith, J.; Leathwick, J.R.; Hastie, T. A Working Guide to Boosted Regression Trees. J. Anim. Ecol. 2008, 77, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Triche, T.J.; Bard, J.D.; Biegel, J.A.; Judkins, A.R.; Gai, X. Spike Protein NTD Mutation G142D in SARS-CoV-2 Delta VOC Lineages Is Associated with Frequent Back Mutations, Increased Viral Loads, and Immune Evasion. medRxiv 2021. [Google Scholar] [CrossRef]
- Joshi, N.; Tyagi, A.; Nigam, S. Molecular Level Dissection of Critical Spike Mutations in SARS-CoV-2 Variants of Concern (VOCs): A Simplified Review. ChemistrySelect 2021, 6, 7981–7998. [Google Scholar] [CrossRef]
- Rocheleau, L.; Laroche, G.; Fu, K.; Stewart, C.M.; Mohamud, A.O.; Côté, M.; Giguère, P.M.; Langlois, M.A.; Pelchat, M. Identification of a High-Frequency Intrahost SARS-CoV-2 Spike Variant with Enhanced Cytopathic and Fusogenic Effects. mBio 2021. [Google Scholar] [CrossRef]
- Yang, X.J. δ Subvariants of SARS-COV-2 in Israel, Qatar and Bahrain: Optimal Vaccination as an Effective Strategy to Block Viral Evolution and Control the Pandemic. medRxiv 2021. [Google Scholar] [CrossRef]
- Meng, B.; Ferreira, I.; Abdullahi, A.; Kemp, S.A.; Goonawardane, N.; Papa, G.; Fatihi, S.; Charles, O.; Collier, D.; Collaboration, C.N.B.C.; et al. SARS-CoV-2 Omicron Spike Mediated Immune Escape, Infectivity and Cell-Cell Fusion. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhao, H.; Lu, L.; Peng, Z.; Chen, L.L.; Meng, X.; Zhang, C.; Ip, J.D.; Chan, W.M.; Chu, A.W.H.; Chan, K.H.; et al. SARS-CoV-2 Omicron Variant Shows Less Efficient Replication and Fusion Activity When Compared with Delta Variant in TMPRSS2-expressed Cells. Emerg. Microbes Infect. 2022, 11, 277–283. [Google Scholar] [CrossRef]
- Abdelnabi, R.; Foo, C.S.Y.; Zhang, X.; Lemmens, V.; Maes, P.; Slechten, B.; Raymenants, J.; Andre, E.; Weynand, B.; Dallmeier, K.; et al. The Omicron (B.1.1.529) SARS-CoV-2 Variant of Concern Does Not Readily Infect Syrian Hamsters. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ryan, K.A.; Watson, R.J.; Bewley, K.R.; Burton, C.A.; Carnell, O.; Cavell, B.E.; Challis, A.R.; Coombes, N.S.; Emery, K.; Fell, R.; et al. Convalescence from Prototype SARS-CoV-2 Protects Syrian Hamsters from Disease Caused by the Omicron Variant. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wolter, N.; Jassat, W.; Walaza, S.; Welch, R.; Moultrie, H.; Groome, M.; Amoako, D.G.; Everatt, J.; Bhiman, J.N.; Scheepers, C.; et al. Early Assessment of the Clinical Severity of the SARS-CoV-2 Omicron Variant in South Africa. medRxiv 2021. [Google Scholar] [CrossRef]
- Ferguson, N.; Ghani, A.; Hinsley, W.; Volz, E. Report 50—Hospitalisation Risk for Omicron Cases in England. 2021. Available online: http://www.imperial.ac.uk/medicine/departments/school-public-health/infectious-disease-epidemiology/mrc-global-infectious-disease-analysis/covid-19/report-50-severity-omicron/ (accessed on 14 November 2022).
- Wang, L.; Berger, N.A.; Davis, P.B.; Kaelber, D.C.; Volkow, N.D.; Xu, R. Comparison of Outcomes from COVID Infection in Pediatric and Adult Patients before and after the Emergence of Omicron. medRxiv 2022. [Google Scholar] [CrossRef]
- Glocker, M.O.; Opuni, K.F.M.; Thiesen, H.J. Compared with SARS-CoV2 Wild Type’s Spike Protein, the SARS-CoV2 Omicron’s Receptor Binding Motif Has Adopted a More SARS-CoV1 and/or Bat/Civet-like Structure. bioRxiv 2021. [Google Scholar] [CrossRef]
- Jones, D.T. Setting the Standards for Machine Learning in Biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 659–660. [Google Scholar] [CrossRef]
- Allegretti, S.; Bulai, I.M.; Marino, R.; Menandro, M.A.; Parisi, K. Vaccination Effect Conjoint to Fraction of Avoided Contacts for a Sars-Cov-2 Mathematical Model. Math. Model. Numer. Simul. Appl. 2021, 1, 56–66. [Google Scholar] [CrossRef]
- Wang, Z.; Zheutlin, A.; Kao, Y.H.; Ayers, K.; Gross, S.; Kovatch, P.; Nirenberg, S.; Charney, A.; Nadkarni, G.; De Freitas, J.K.; et al. Hospitalised COVID-19 Patients of the Mount Sinai Health System: A Retrospective Observational Study Using the Electronic Medical Records. BMJ Open 2020, 10, e040441. [Google Scholar] [CrossRef]
- Monod, M.; Blenkinsop, A.; Xi, X.; Hebert, D.; Bershan, S.; Tietze, S.; Baguelin, M.; Bradley, V.C.; Chen, Y.; Coupland, H.; et al. Age Groups That Sustain Resurging COVID-19 Epidemics in the United States. Science 2021, 371. [Google Scholar] [CrossRef]
- Islam, M.R.; Hoque, M.N.; Rahman, M.S.; Alam, A.S.M.R.U.; Akther, M.; Puspo, J.A.; Akter, S.; Sultana, M.; Crandall, K.A.; Hossain, M.A. Genome-Wide Analysis of SARS-CoV-2 Virus Strains Circulating Worldwide Implicates Heterogeneity. Sci. Rep. 2020, 10, 14004. [Google Scholar] [CrossRef]
- Chen, Z.; Chong, K.C.; Wong, M.C.S.; Boon, S.S.; Huang, J.; Wang, M.H.; Ng, R.W.Y.; Lai, C.K.C.; Chan, P.K.S. A Global Analysis of Replacement of Genetic Variants of SARS-CoV-2 in Association with Containment Capacity and Changes in Disease Severity. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2021, 27, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Sokhansanj, B.A.; Malhotra, C.; Zheng, K.; Rosen, G.L. Genetic Grouping of SARS-CoV-2 Coronavirus Sequences Using Informative Subtype Markers for Pandemic Spread Visualization. PLoS Comput. Biol. 2020, 16, e1008269. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.A.; Yavuz, M.; Qureshi, S.; Zu, J.; Townley, S. Modeling and Analysis of COVID-19 Epidemics with Treatment in Fractional Derivatives Using Real Data from Pakistan. Eur. Phys. J. Plus 2020, 135, 795. [Google Scholar] [CrossRef]
- Schriml, L.M.; Chuvochina, M.; Davies, N.; Eloe-Fadrosh, E.A.; Finn, R.D.; Hugenholtz, P.; Hunter, C.I.; Hurwitz, B.L.; Kyrpides, N.C.; Meyer, F.; et al. COVID-19 Pandemic Reveals the Peril of Ignoring Metadata Standards. Sci. Data 2020, 7, 188. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Status (Based on Table S1) | Classification |
|---|---|---|
| Alive | Alive | Unknown |
| Asymptomatic | Asymptomatic, Asymptomatic/Hospitalized, Asymptomatic/Mild, Asymptomatic/Mild, Paucisymptomatic, Paucisymptomatic/Reinfection | Mild |
| Dead | Dead, Dead/Vaccinated | Severe |
| Hospitalized | Hospitalized, Hospitalized/Stable, Hospitalized/Symptomatic | Severe |
| Mild | Ambulatory, Ambulatory/Under treatment, Home, Hospitalized/Mild, House Assistance, Mild, Mild/Reinfection, Mild/Moderate, Not hospitalized, Not severe, Outpatient, Vaccinated/Mild | Mild |
| Moderate | Hospitalized/Moderate, Hospitalized/Not Serious, Moderate, Moderate/Reinfection, Moderate/Severe, Not critically hospitalized, Pneumonia | Mild |
| Released | Released | Severe |
| Screening | Screening | Mild |
| Symptomatic | Acute infection, Bronchitis, Chronic infection, Flu-like, Stable, Symptomatic, Symptomatic/Reinfection | Uknown |
| Severe | Casualty, Critical, Hospitalized/Serious, Hospitalized/Serious, ICU, Serious, Severe, Severe/Hospitalized | Severe |
| Unknown | Active, Not vaccinated, Recovered, Reinfection, Unknown, Unknown/Vaccinated, Vaccinated | Unknown |
| Lineage | Mean Age | % Severe | % Male | Mean Date | Frequency |
|---|---|---|---|---|---|
| AY.4 | 39.4 | 26% | 53% | 631 | 817 |
| B.1 | 49.8 | 73% | 42% | 314 | 994 |
| B.1.1 | 50.3 | 70% | 49% | 314 | 696 |
| B.1.1.519 | 49.9 | 68% | 47% | 485 | 1583 |
| B.1.1.7 | 48.3 | 60% | 50% | 509 | 3298 |
| B.1.351 | 45.7 | 52% | 50% | 481 | 849 |
| B.1.617.2 | 40.0 | 42% | 48% | 604 | 3901 |
| P.1 | 48.3 | 64% | 48% | 548 | 877 |
| Precision | Recall | F1 Score | Accuracy | |
|---|---|---|---|---|
| XGBoost | ||||
| No Sequence | 0.56/0.72 | 0.65/0.64 | 0.60/0.68 | 0.64 |
| Sequence Only | 0.57/0.74 | 0.66/0.65 | 0.61/0.69 | 0.65 |
| Sequence + Age, Gender, Date | 0.64/0.77 | 0.67/0.74 | 0.66/0.76 | 0.71 |
| Transformer | ||||
| No Sequence | 0.53/0.75 | 0.72/0.56 | 0.61/0.64 | 0.63 |
| Sequence Only | 0.58/0.69 | 0.52/0.74 | 0.55/0.72 | 0.65 |
| Sequence + Age, Gender, Date | 0.59/0.76 | 0.70/0.66 | 0.64/0.71 | 0.69 |
| Attention | Amino Acid | Mean Attention | XGBoost | XGBoost |
|---|---|---|---|---|
| Rank | Position | Score | Rank | Score |
| 1 | 1258 | 5.42 | 73 | 163 |
| 2 | 732 | 5.1 | 10 | 2090 |
| 3 | 142 | 4.96 | 1 | 6421 |
| 4 | 501 | 3.79 | 8 | 2272 |
| 5 | 69 | 2.14 | 5 | 2813 |
| 6 | 70 | 2.13 | 31 | 573 |
| 7 | 716 | 2.08 | 25 | 756 |
| 8 | 570 | 2.06 | 27 | 709 |
| 9 | 19 | 1.99 | 26 | 720 |
| 10 | 950 | 1.92 | 17 | 1352 |
| 11 | 144 | 1.34 | 6 | 2306 |
| 12 | 95 | 1.25 | 2 | 4698 |
| 13 | 222 | 1.07 | 4 | 3474 |
| 14 | 417 | 1.06 | 24 | 757 |
| 15 | 655 | 1.01 | 62 | 225 |
| 16 | 614 | 0.94 | 7 | 2288 |
| 17 | 1104 | 0.91 | 9 | 2240 |
| 18 | 241 | 0.87 | 134 | 78 |
| 19 | 243 | 0.86 | 335 | 7 |
| 20 | 242 | 0.86 | 250 | 23 |
| 21 | 477 | 0.86 | 14 | 1488 |
| 22 | 681 | 0.84 | 3 | 3559 |
| 23 | 1074 | 0.83 | 22 | 962 |
| 24 | 97 | 0.81 | 18 | 1168 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sokhansanj, B.A.; Zhao, Z.; Rosen, G.L. Interpretable and Predictive Deep Neural Network Modeling of the SARS-CoV-2 Spike Protein Sequence to Predict COVID-19 Disease Severity. Biology 2022, 11, 1786. https://doi.org/10.3390/biology11121786
Sokhansanj BA, Zhao Z, Rosen GL. Interpretable and Predictive Deep Neural Network Modeling of the SARS-CoV-2 Spike Protein Sequence to Predict COVID-19 Disease Severity. Biology. 2022; 11(12):1786. https://doi.org/10.3390/biology11121786
Chicago/Turabian StyleSokhansanj, Bahrad A., Zhengqiao Zhao, and Gail L. Rosen. 2022. "Interpretable and Predictive Deep Neural Network Modeling of the SARS-CoV-2 Spike Protein Sequence to Predict COVID-19 Disease Severity" Biology 11, no. 12: 1786. https://doi.org/10.3390/biology11121786
APA StyleSokhansanj, B. A., Zhao, Z., & Rosen, G. L. (2022). Interpretable and Predictive Deep Neural Network Modeling of the SARS-CoV-2 Spike Protein Sequence to Predict COVID-19 Disease Severity. Biology, 11(12), 1786. https://doi.org/10.3390/biology11121786