
Effective Quality Breeding Directions—Comparison and Conservative Analysis of Hepatic Super-Enhancers between Chinese and Western Pig Breeds

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Available Public Data
2.2. ChIP-Seq Data Processing
2.3. Identification of cis-Regulatory Elements
2.4. Differential Analysis of Promoters and TEs
2.5. Identification of Conserved and Specific SEs
2.6. Motif Enrichment of Specific SEs
2.7. Comparison of Orthologous SEs and TEs
2.8. Other Bioinformatic Analysis
3. Result
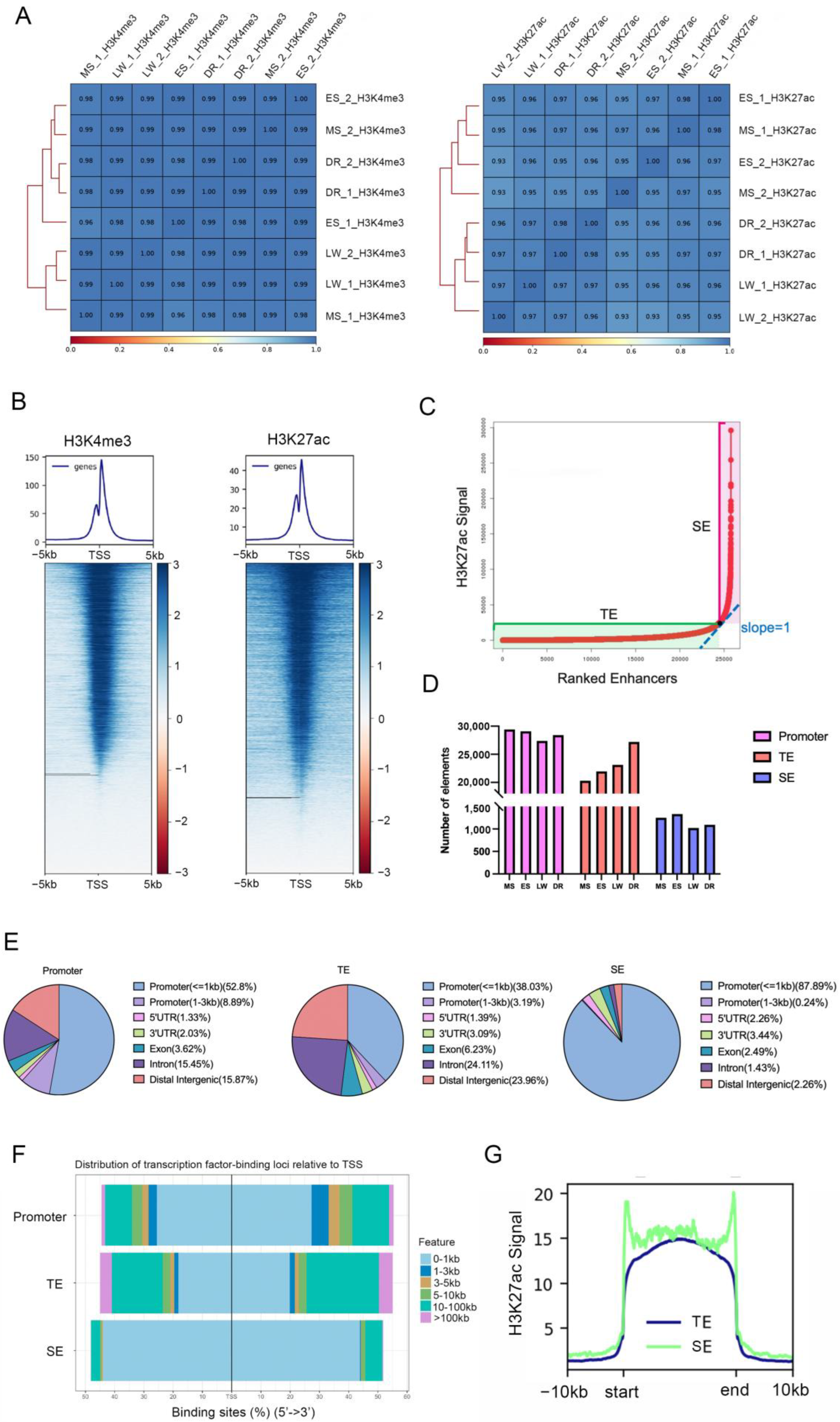
3.1. Profiling of Promoter, TE, and SE in Liver
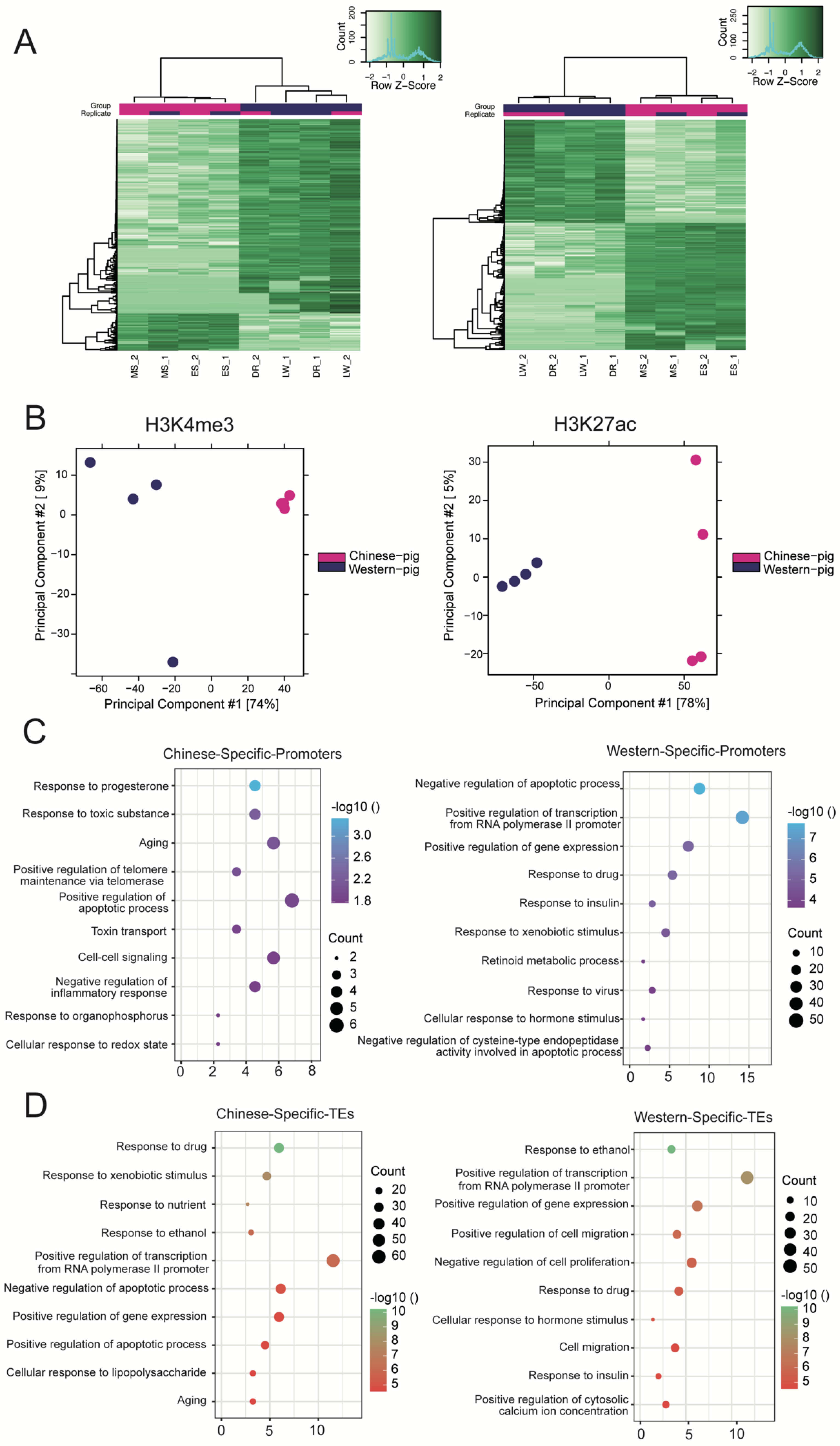
3.2. Comparison of Promoter and TE Variants across Different Porcine Breeds
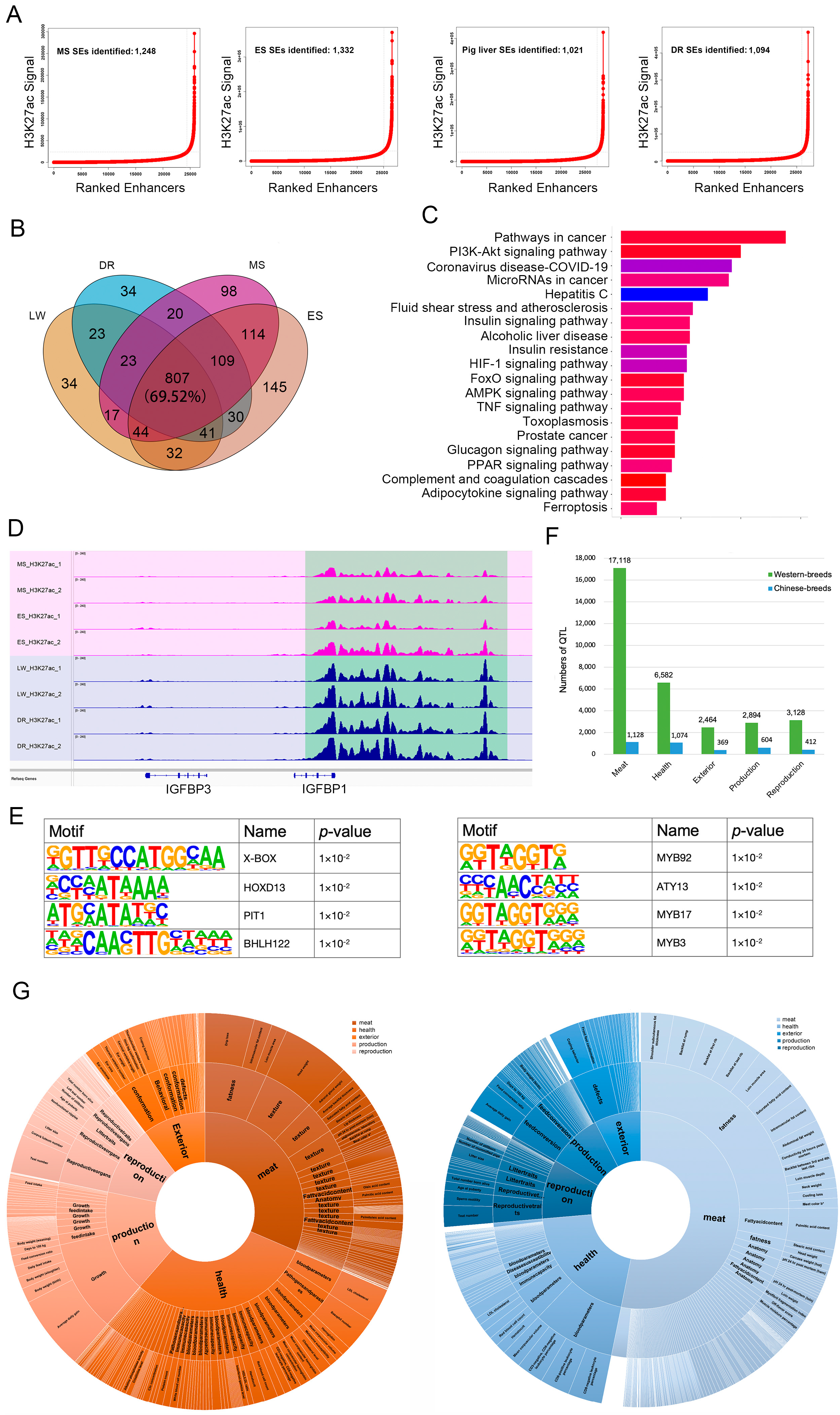
3.3. Conservation and Variants of SEs in Different Breeds
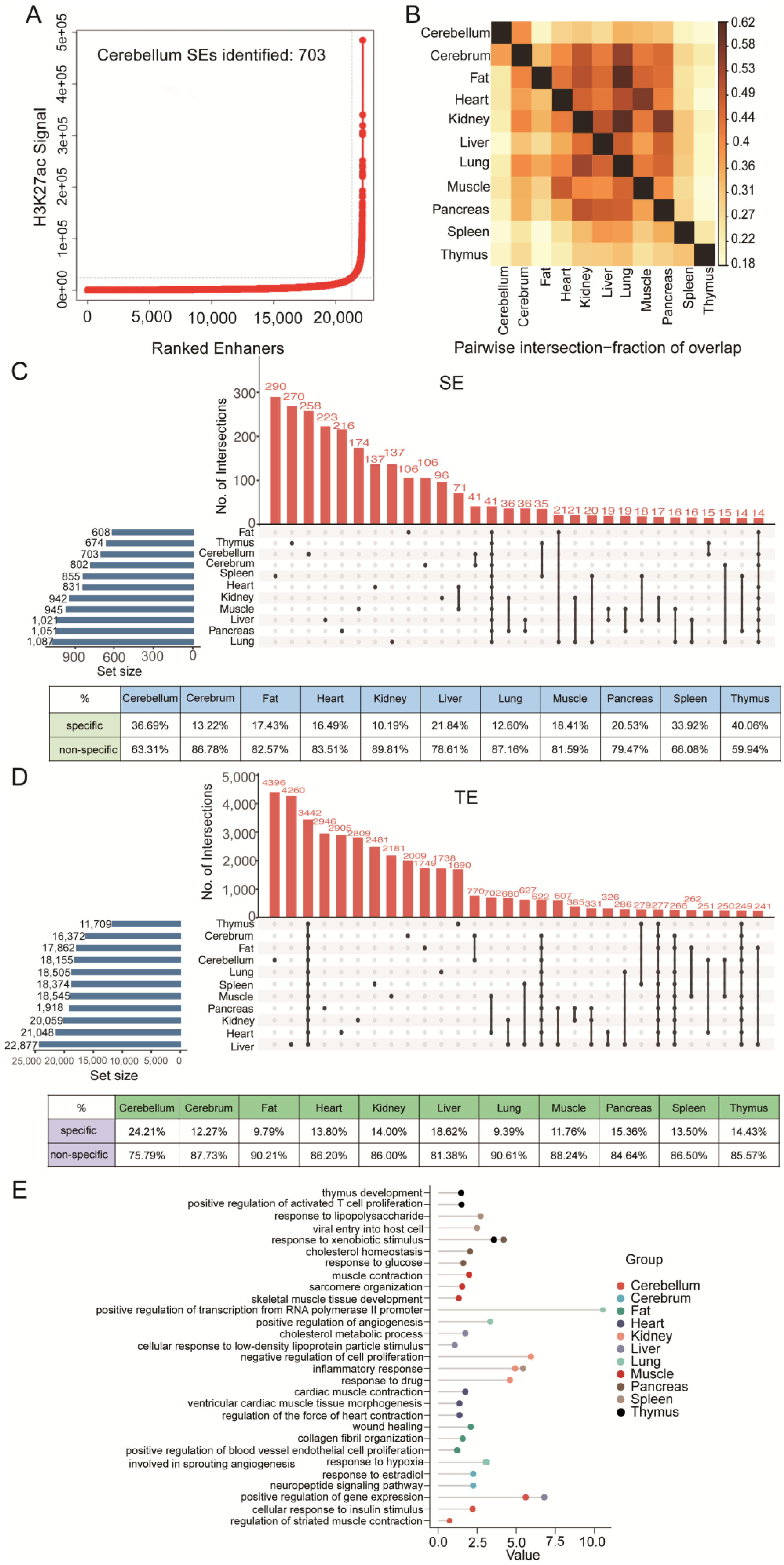
3.4. Tissue Specificity of SEs
3.5. Species Specificity of SEs
4. Discussion
4.1. SEs Are Tissue-Specific ‘Network Hubs’ of cis-Elements
4.2. The Promising Application of SE in Genomic Selection Breeding
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Field, A.; Adelman, K. Evaluating Enhancer Function and Transcription. Annu. Rev. Biochem. 2020, 89, 213–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerstein, M.B.; Kundaje, A.; Hariharan, M.; Landt, S.G.; Yan, K.K.; Cheng, C.; Mu, X.J.; Khurana, E.; Rozowsky, J.; Alexander, R.; et al. Architecture of the human regulatory network derived from ENCODE data. Nature 2012, 489, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Zhang, Y. Enhancer and super-enhancer: Positive regulators in gene transcription. Anim. Model Exp. Med. 2018, 1, 169–179. [Google Scholar] [CrossRef]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef]
- Villar, D.; Berthelot, C.; Aldridge, S.; Rayner, T.F.; Lukk, M.; Pignatelli, M.; Park, T.J.; Deaville, R.; Erichsen, J.T.; Jasinska, A.J.; et al. Enhancer evolution across 20 mammalian species. Cell 2015, 160, 554–566. [Google Scholar] [CrossRef] [Green Version]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [Green Version]
- Pott, S.; Lieb, J.D. What are super-enhancers? Nat. Genet. 2015, 47, 8–12. [Google Scholar] [CrossRef]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thandapani, P. Super-enhancers in cancer. Pharmacol. Ther. 2019, 199, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Vodicka, P.; Smetana, K., Jr.; Dvorankova, B.; Emerick, T.; Xu, Y.Z.; Ourednik, J.; Ourednik, V.; Motlik, J. The miniature pig as an animal model in biomedical research. Ann. N. Y. Acad. Sci. 2005, 1049, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Meurens, F.; Summerfield, A.; Nauwynck, H.; Saif, L.; Gerdts, V. The pig: A model for human infectious diseases. Trends Microbiol. 2012, 20, 50–57. [Google Scholar] [CrossRef]
- Larson, G.; Dobney, K.; Albarella, U.; Fang, M.; Matisoo-Smith, E.; Robins, J.; Lowden, S.; Finlayson, H.; Brand, T.; Willerslev, E.; et al. Worldwide phylogeography of wild boar reveals multiple centers of pig domestication. Science 2005, 307, 1618–1621. [Google Scholar] [CrossRef] [Green Version]
- Ramayo-Caldas, Y.; Mach, N.; Esteve-Codina, A.; Corominas, J.; Castello, A.; Ballester, M.; Estelle, J.; Ibanez-Escriche, N.; Fernandez, A.I.; Perez-Enciso, M.; et al. Liver transcriptome profile in pigs with extreme phenotypes of intramuscular fatty acid composition. BMC Genom. 2012, 13, 547. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Xiong, X.; Yang, J.; Zhou, L.; Yang, B.; Ai, H.; Ma, H.; Xie, X.; Huang, Y.; Fang, S.; et al. Genome-wide association analyses for meat quality traits in Chinese Erhualian pigs and a Western Duroc x (Landrace x Yorkshire) commercial population. Genet. Sel. Evol. 2015, 47, 44. [Google Scholar] [CrossRef] [Green Version]
- Rubin, C.J.; Megens, H.J.; Barrio, A.M.; Maqbool, K.; Sayyab, S.; Schwochow, D.; Wang, C.; Carlborg, O.; Jern, P.; Jorgensen, C.B.; et al. Strong signatures of selection in the domestic pig genome. Proc. Natl. Acad. Sci. USA 2012, 109, 19529–19536. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Yu, Y.; Feng, W.; Du, H.; Yu, J.; Kang, H.; Zheng, X.; Wang, Z.; Liu, G.E.; Ernst, C.W.; et al. Evidence of evolutionary history and selective sweeps in the genome of Meishan pig reveals its genetic and phenotypic characterization. Gigascience 2018, 7, giy058. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Hou, Y.; Liu, F.; Liu, A.; Jing, L.; Zhao, C.; Luan, Y.; Miao, Y.; Zhao, S.; Li, X. Transcriptome Analysis Reveals that Vitamin A Metabolism in the Liver Affects Feed Efficiency in Pigs. G3 (Bethesda) 2016, 6, 3615–3624. [Google Scholar] [CrossRef]
- Kubes, P.; Jenne, C. Immune Responses in the Liver. Annu. Rev. Immunol. 2018, 36, 247–277. [Google Scholar] [CrossRef] [PubMed]
- Horodyska, J.; Hamill, R.M.; Reyer, H.; Trakooljul, N.; Lawlor, P.G.; McCormack, U.M.; Wimmers, K. RNA-Seq of Liver From Pigs Divergent in Feed Efficiency Highlights Shifts in Macronutrient Metabolism, Hepatic Growth and Immune Response. Front. Genet. 2019, 10, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittkopp, P.J.; Kalay, G. Cis-regulatory elements: Molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 2011, 13, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Yao, Y.; Yin, H.; Cai, Z.; Wang, Y.; Bai, L.; Kern, C.; Halstead, M.; Chanthavixay, G.; Trakooljul, N.; et al. Pig genome functional annotation enhances the biological interpretation of complex traits and human disease. Nat. Commun. 2021, 12, 5848. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, Y.; Liu, H.; Gao, Y.; Liu, Y.; Chen, L.; Liu, L.; Irwin, D.M.; Hou, C.; Zhou, Z.; et al. Genome-wide identification of functional enhancers and their potential roles in pig breeding. J. Anim. Sci. Biotechnol. 2022, 13, 75. [Google Scholar] [CrossRef] [PubMed]
- Kai, Y.; Li, B.E.; Zhu, M.; Li, G.Y.; Chen, F.; Han, Y.; Cha, H.J.; Orkin, S.H.; Cai, W.; Huang, J.; et al. Mapping the evolving landscape of super-enhancers during cell differentiation. Genome Biol. 2021, 22, 269. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Ramirez, F.; Dundar, F.; Diehl, S.; Gruning, B.A.; Manke, T. deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, R.; Brown, G. DiffBind: Differential binding analysis of ChIP-Seq peak data. R Package Version 2011, 100, 1–38. [Google Scholar]
- Hu, Z.L.; Park, C.A.; Wu, X.L.; Reecy, J.M. Animal QTLdb: An improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 2013, 41, D871–D879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fronicke, L.; Chowdhary, B.P.; Scherthan, H.; Gustavsson, I. A comparative map of the porcine and human genomes demonstrates ZOO-FISH and gene mapping-based chromosomal homologies. Mamm. Genome 1996, 7, 285–290. [Google Scholar] [CrossRef]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef] [PubMed]
- Ansari-Mahyari, S.; Berg, P.; Lund, M.S. Fine mapping quantitative trait loci under selective phenotyping strategies based on linkage and linkage disequilibrium criteria. J. Anim. Breed. Genet. 2015, 126, 443–454. [Google Scholar] [CrossRef]
- Bibiana, M.C.; Marco, B.M.; Arelí, B.; Ledesma-Colunga, M.G.; Xarubet, R.H.; Pamela, R.O.; De, L.; Yazmín, M.; Gonzalo, M.; Carmen, C. Prolactin Regulates Liver Growth During Postnatal Development in Mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R902–R908. [Google Scholar]
- Anzo, M.; Cobb, L.J.; Hwang, D.L.; Mehta, H.; Said, J.W.; Yakar, S.; Leroith, D.; Cohen, P. Targeted Deletion of Hepatic Igf1 in TRAMP Mice Leads to Dramatic Alterations in the Circulating Insulin-Like Growth Factor Axis but Does Not Reduce Tumor Progression. Cancer Res. 2008, 68, 3342–3349. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yao, W.; Pan, Z.; Liu, H. Long-distance chromatin interaction of IGF1 during embryonic and postnatal development in the liver of Sus scrofa. Funct. Integr. Genom. 2021, 21, 59–72. [Google Scholar] [CrossRef]
- Cleveland, M.A.; Hickey, J.M. Practical implementation of cost-effective genomic selection in commercial pig breeding using imputation. J. Anim. Sci. 2013, 91, 3583–3592. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Zhang, C.; Ali, N.; Gandeka, M. Highdensity SNP map facilitates fine mapping of QTLs and candidate genes discovery for Aspergillus favus resistance in peanut (Arachis hypogaea). Theor. Appl. Genet. 2020, 133, 2239–2257. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Zhang, J.; Wang, C.; Qin, X.; Zhang, Y.; Liu, J.; Pan, Z. Effective Quality Breeding Directions—Comparison and Conservative Analysis of Hepatic Super-Enhancers between Chinese and Western Pig Breeds. Biology 2022, 11, 1631. https://doi.org/10.3390/biology11111631
Zhang Y, Zhang J, Wang C, Qin X, Zhang Y, Liu J, Pan Z. Effective Quality Breeding Directions—Comparison and Conservative Analysis of Hepatic Super-Enhancers between Chinese and Western Pig Breeds. Biology. 2022; 11(11):1631. https://doi.org/10.3390/biology11111631
Chicago/Turabian StyleZhang, Yi, Jinbi Zhang, Caixia Wang, Xinxin Qin, Yuge Zhang, Jingge Liu, and Zengxiang Pan. 2022. "Effective Quality Breeding Directions—Comparison and Conservative Analysis of Hepatic Super-Enhancers between Chinese and Western Pig Breeds" Biology 11, no. 11: 1631. https://doi.org/10.3390/biology11111631
APA StyleZhang, Y., Zhang, J., Wang, C., Qin, X., Zhang, Y., Liu, J., & Pan, Z. (2022). Effective Quality Breeding Directions—Comparison and Conservative Analysis of Hepatic Super-Enhancers between Chinese and Western Pig Breeds. Biology, 11(11), 1631. https://doi.org/10.3390/biology11111631