
Isolation of a Novel Thermophilic Methanogen and the Evolutionary History of the Class Methanobacteria
 , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling, Enrichment, Isolation, and Pure Culturing
2.2. Headspace Gas Analysis
2.3. Microscopy Observation
2.4. ToF-SIMS Analysis
2.5. DNA Extraction, Sequencing, and Analysis
2.6. Phylogenetic Analyses
2.7. Comparative Genomic and Ancestral Analyses
3. Results and Discussion
3.1. Enrichment and Isolation of a Thermophilic Methanogen from a Hot Spring
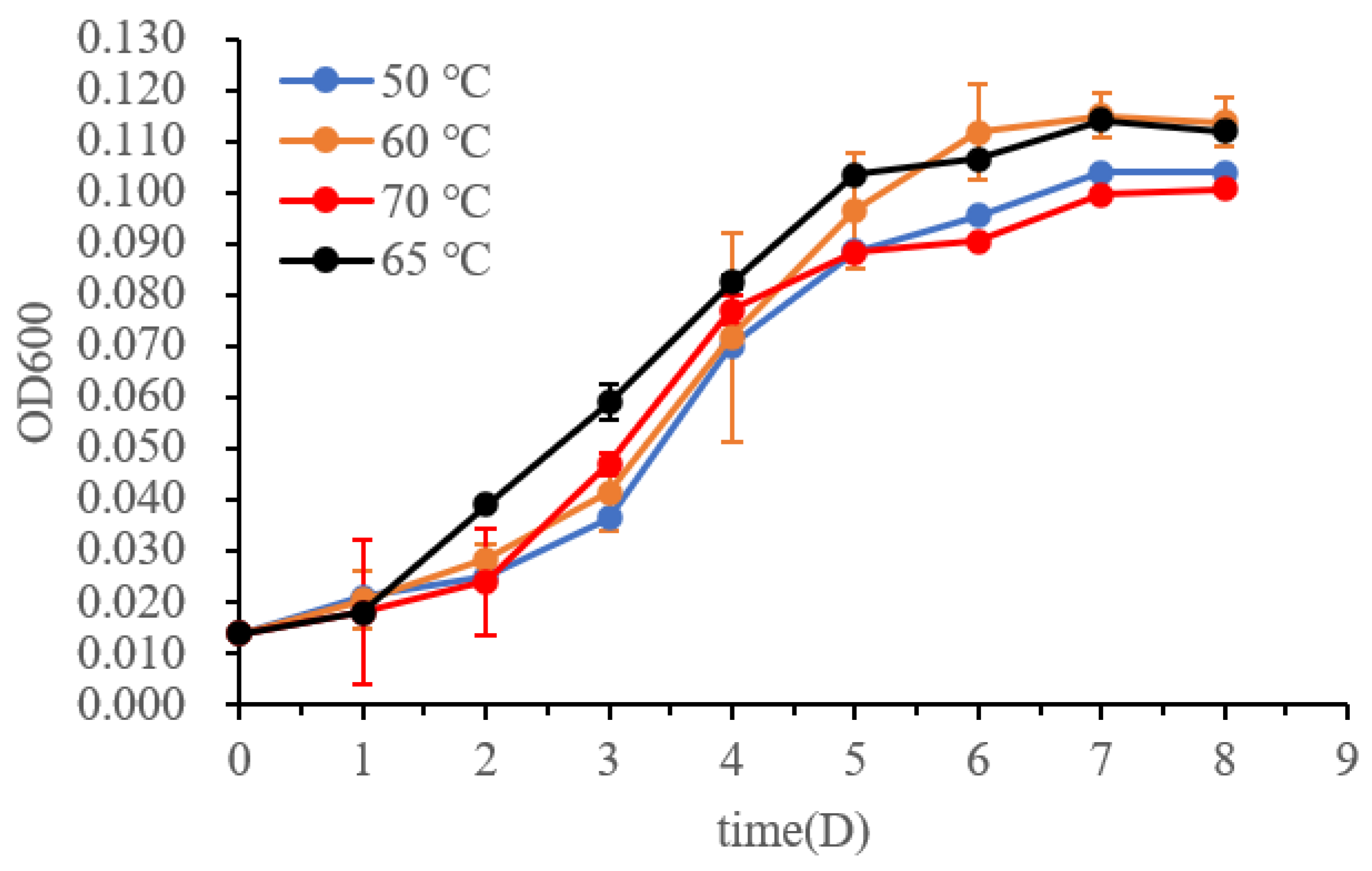
3.2. Morphology and Physiological Characteristics of Strain DL9LZB001
3.3. General Genome Characteristics of the Strain DL9LZB001
3.4. The Evolutionary History of the Class Methanobacteria
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ueno, Y.; Yamada, K.; Yoshida, N.; Maruyama, S.; Isozaki, Y. Evidence from fluid inclusions for microbial methanogenesis in the early Archaean era. Nature 2006, 440, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Battistuzzi, F.U.; Feijao, A.; Hedges, S.B. A genomic timescale of prokaryote evolution: Insights into the origin of methanogenesis, phototrophy, and the colonization of land. BMC Evol. Biol. 2004, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Borrel, G.; Adam, P.S.; Gribaldo, S. Methanogenesis and the Wood-Ljungdahl Pathway: An Ancient, Versatile, and Fragile Association. Genome Biol. Evol. 2016, 8, 1706–1711. [Google Scholar] [CrossRef]
- Liu, Y.; Whitman, W.B. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann. N. Y. Acad. Sci. 2008, 1125, 171–189. [Google Scholar] [CrossRef] [PubMed]
- Mayumi, D.; Mochimaru, H.; Tamaki, H.; Yamamoto, K.; Yoshioka, H.; Suzuki, Y.; Kamagata, Y.; Sakata, S. Methane production from coal by a single methanogen. Science 2016, 354, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Thauer, R.K.; Kaster, A.K.; Seedorf, H.; Buckel, W.; Hedderich, R. Methanogenic archaea: Ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 2008, 6, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Kiehl, J.T.; Trenberth, K.E. Earth’s annual global mean energy budget. Bull. Am. Meteorol. Soc. 1997, 78, 197–208. [Google Scholar] [CrossRef]
- Berghuis, B.A.; Yu, F.B.; Schulz, F.; Blainey, P.C.; Woyke, T.; Quake, S.R. Hydrogenotrophic methanogenesis in archaeal phylum Verstraetearchaeota reveals the shared ancestry of all methanogens. Proc. Natl. Acad. Sci. USA 2019, 116, 5037–5044. [Google Scholar] [CrossRef]
- Evans, P.N.; Parks, D.H.; Chadwick, G.L.; Robbins, S.J.; Orphan, V.J.; Golding, S.D.; Tyson, G.W. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science 2015, 350, 434–438. [Google Scholar] [CrossRef]
- Wang, Y.; Wegener, G.; Hou, J.; Wang, F.; Xiao, X. Expanding anaerobic alkane metabolism in the domain of Archaea. Nat. Microbiol. 2019, 4, 595–602. [Google Scholar] [CrossRef]
- Hua, Z.S.; Wang, Y.L.; Evans, P.N.; Qu, Y.N.; Goh, K.M.; Rao, Y.Z.; Qi, Y.L.; Li, Y.X.; Huang, M.J.; Jiao, J.Y.; et al. Insights into the ecological roles and evolution of methyl-coenzyme M reductase-containing hot spring Archaea. Nat. Commun. 2019, 10, 4574. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wegener, G.; Williams, T.A.; Xie, R.; Hou, J.; Wang, F.; Xiao, X. A methylotrophic origin of methanogenesis and early divergence of anaerobic multicarbon alkane metabolism. Sci. Adv. 2021, 7, eabj1453. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.A.; Jungbluth, S.P.; Leu, A.O.; Evans, P.N.; Woodcroft, B.J.; Chadwick, G.L.; Orphan, V.J.; Amend, J.P.; Rappé, M.S.; Tyson, G.W. Divergent methyl-coenzyme M reductase genes in a deep-subseafloor Archaeoglobi. BioRxiv 2018. [Google Scholar] [CrossRef] [PubMed]
- Adam, P.S.; Borrel, G.; Brochier-Armanet, C.; Gribaldo, S. The growing tree of Archaea: New perspectives on their diversity, evolution and ecology. ISME J. 2017, 11, 2407–2425. [Google Scholar] [CrossRef]
- Bapteste, E.; Brochier, C.; Boucher, Y. Higher-level classification of the Archaea: Evolution of methanogenesis and methanogens. Archaea 2005, 1, 353–363. [Google Scholar] [CrossRef]
- Stetter, K.O. Methanothermus. In Bergey’s Manual of Systematics of Archaea and Bacteria; Wiley: Hoboken, NJ, USA, 2015; pp. 1–4. [Google Scholar]
- Boone, D.R. Methanothermobacter. In Bergey’s Manual of Systematics of Archaea and Bacteria; Wiley: Hoboken, NJ, USA, 2015; pp. 1–8. [Google Scholar]
- Miller, T.L. Methanobrevibacter. In Bergey’s Manual of Systematics of Archaea and Bacteria; Wiley: Hoboken, NJ, USA, 2015; pp. 1–14. [Google Scholar]
- Boone, D.R. Methanobacterium. In Bergey’s Manual of Systematics of Archaea and Bacteria; Wiley: Hoboken, NJ, USA, 2015; pp. 1–8. [Google Scholar]
- Miller, T.L. Methanosphaera. In Bergey’s Manual of Systematics of Archaea and Bacteria; Wiley: Hoboken, NJ, USA, 2015; pp. 1–6. [Google Scholar]
- Validation of the publication of new names and new combinations previously effectively published outside the IJSB. List No. 63. Int. J. Syst. Bacteriol. 1997, 47, 1274. [CrossRef]
- Nakamura, K.; Takahashi, A.; Mori, C.; Tamaki, H.; Mochimaru, H.; Nakamura, K.; Takamizawa, K.; Kamagata, Y. Methanothermobacter tenebrarum sp. nov., a hydrogenotrophic, thermophilic methanogen isolated from gas-associated formation water of a natural gas field. Int. J. Syst. Evol. Microbiol. 2013, 63, 715–722. [Google Scholar] [CrossRef]
- Leadbetter, J.R.; Crosby, L.D.; Breznak, J.A. Methanobrevibacter filiformis sp. nov., A filamentous methanogen from termite hindguts. Arch. Microbiol. 1998, 169, 287–292. [Google Scholar] [CrossRef]
- Cadillo-Quiroz, H.; Brauer, S.L.; Goodson, N.; Yavitt, J.B.; Zinder, S.H. Methanobacterium paludis sp. nov. and a novel strain of Methanobacterium lacus isolated from northern peatlands. Int. J. Syst. Evol. Microbiol. 2014, 64, 1473–1480. [Google Scholar] [CrossRef]
- Krivushin, K.V.; Shcherbakova, V.A.; Petrovskaya, L.E.; Rivkina, E.M. Methanobacterium veterum sp. nov., from ancient Siberian permafrost. Int. J. Sys.t Evol. Microbiol. 2010, 60, 455–459. [Google Scholar] [CrossRef]
- Xue, Y.; Xu, Y.; Liu, Y.; Ma, Y.; Zhou, P. Thermoanaerobacter tengcongensis sp. nov., a novel anaerobic, saccharolytic, thermophilic bacterium isolated from a hot spring in Tengcong, China. Int. J. Syst. Evol. Microbiol. 2001, 51, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Plugge, C.M. Anoxic Media Design, Preparation, and Considerations. Methods Enzymol. 2005, 397, 3–16. [Google Scholar] [PubMed]
- Gordon, D.F.; Stutman, M.; Loesche, W.J. Improved isolation of anaerobic bacteria from the gingival crevice area of man. Appl. Microbiol. 1971, 21, 1046–1050. [Google Scholar] [CrossRef]
- Yang, S.; Li, X.; Xiao, X.; Zhuang, G.; Zhang, Y. Sphingomonas profundi sp. nov., isolated from deep-sea sediment of the Mariana Trench. Int. J. Syst. Evol. Microbiol. 2020, 70, 3809–3815. [Google Scholar] [CrossRef] [PubMed]
- Vetriani, C. Archaea, Origin of. In Encyclopedia of Biodiversity, 2nd ed.; Levin, S.A., Ed.; Academic Press: Waltham, MA, USA, 2001; pp. 218–226. [Google Scholar]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Gardner, P.P.; Daub, J.; Tate, J.G.; Nawrocki, E.P.; Kolbe, D.L.; Lindgreen, S.; Wilkinson, A.C.; Finn, R.D.; Griffiths-Jones, S.; Eddy, S.R.; et al. Rfam: Updates to the RNA families database. Nucleic Acids Res. 2009, 37, D136–D140. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Infernal 1.0: Inference of RNA alignments. Bioinformatics 2009, 25, 1335–1337. [Google Scholar] [CrossRef]
- Hsiao, W.; Wan, I.; Jones, S.J.; Brinkman, F.S. IslandPath: Aiding detection of genomic islands in prokaryotes. Bioinformatics 2003, 19, 418–420. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A new view of the tree of life. Nat. Microbiol. 2016, 1, 16048. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez, R.L.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. BioRxiv 2018. [Google Scholar] [CrossRef]
- Csuros, M. Count: Evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics 2010, 26, 1910–1912. [Google Scholar] [CrossRef] [PubMed]
- Wasserfallen, A.; Nolling, J.; Pfister, P.; Reeve, J.; Conway de Macario, E. Phylogenetic analysis of 18 thermophilic Methanobacterium isolates supports the proposals to create a new genus, Methanothermobacter gen. nov., and to reclassify several isolates in three species, Methanothermobacter thermautotrophicus comb. nov., Methanothermobacter wolfeii comb. nov., and Methanothermobacter marburgensis sp. nov. Int. J. Syst. Evol. Microbiol. 2000, 50, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Zeikus, J.G.; Wolfe, R.S. Methanobacterium thermoautotrophicus sp. n., an anaerobic, autotrophic, extreme thermophile. J. Bacteriol. 1972, 109, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Kotelnikova, S.V.; Obraztsova, A.Y.; Gongadze, G.M.; Laurinavichius, K.S. Methanobacterium thermoflexum sp. nov. and Methanobacterium defluvii sp. nov., Thermophilic Rod-Shaped Methanogens Isolated from Anaerobic Digestor Sludge. Syst. Appl. Microbiol. 1993, 16, 427–435. [Google Scholar] [CrossRef]
- Winter, J.; Lerp, C.; Zabel, H.P.; Wildenauer, F.X.; König, H.; Schindler, F. Methanobacterium wolfei, sp. nov., a New Tungsten-Requiring, Thermophilic, Autotrophic Methanogen. Syst. Appl. Microbiol. 1984, 5, 457–466. [Google Scholar] [CrossRef]
- Oren, A. The Family Methanotrichaceae. In The Prokaryotes; Springer: Berlin/Heidelberg, Germany, 2014; pp. 297–306. [Google Scholar]
- Oren, A. The Family Methanosarcinaceae. In The Prokaryotes; Springer: Berlin/Heidelberg, Germany, 2014; pp. 259–281. [Google Scholar]
- Lyu, Z.; Lu, Y. Metabolic shift at the class level sheds light on adaptation of methanogens to oxidative environments. ISME J. 2018, 12, 411–423. [Google Scholar] [CrossRef]
- Hoedt, E.C.; Parks, D.H.; Volmer, J.G.; Rosewarne, C.P.; Denman, S.E.; McSweeney, C.S.; Muir, J.G.; Gibson, P.R.; Cuiv, P.O.; Hugenholtz, P.; et al. Culture- and metagenomics-enabled analyses of the Methanosphaera genus reveals their monophyletic origin and differentiation according to genome size. ISME J. 2018, 12, 2942–2953. [Google Scholar] [CrossRef]
- Lulchev, P.; Klostermeier, D. Reverse gyrase--recent advances and current mechanistic understanding of positive DNA supercoiling. Nucleic Acids Res. 2014, 42, 8200–8213. [Google Scholar] [CrossRef]
- Kandler, O. Zellwandstrukturen bei Methan-Bakterien. Naturwissenschaften 1979, 66, 95–105. [Google Scholar] [CrossRef]
- Borrel, G.; McCann, A.; Deane, J.; Neto, M.C.; Lynch, D.B.; Brugere, J.F.; O’Toole, P.W. Genomics and metagenomics of trimethylamine-utilizing Archaea in the human gut microbiome. ISME J. 2017, 11, 2059–2074. [Google Scholar] [CrossRef]
- Sprenger, W.W.; Hackstein, J.H.; Keltjens, J.T. The competitive success of Methanomicrococcus blatticola, a dominant methylotrophic methanogen in the cockroach hindgut, is supported by high substrate affinities and favorable thermodynamics. FEMS Microbiol. Ecol. 2007, 60, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Mausz, M.A.; Chen, Y. Microbiology and Ecology of Methylated Amine Metabolism in Marine Ecosystems. Curr. Issues Mol. Biol. 2019, 33, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Ametaj, B.N.; Zebeli, Q.; Saleem, F.; Psychogios, N.; Lewis, M.J.; Dunn, S.M.; Xia, J.G.; Wishart, D.S. Metabolomics reveals unhealthy alterations in rumen metabolism with increased proportion of cereal grain in the diet of dairy cows. Metabolomics 2010, 6, 583–594. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | 1 * | 2 * | 3 * | 4 * | 5 * | 6 * | 7 * | 8 * |
|---|---|---|---|---|---|---|---|---|
| Origin | Gas field | Sewage sludge | Anaerobic sludge | Anaerobic sludge | Anaerobic sludge | Sewage sludge and river sediment | Sewage sludge digester | Hot spring upper water and sediment |
| Cell size (diameter × length, μm × μm) | 0.5 × 3.5–10.5 | 0.35–0.6 × 3–7 | 0.40 × 7–20 | 0.36 × 1.4–6.5 | 0.42 × 3–6 | 0.35–0.5 × 2.5 | 0.3–0.4 × 3–3.5 | 0.436 × 2.80–6.43 |
| Filaments | – | + | + | + | – | + | + | + |
| Gram stain results | + | + | + | – | + | + | + | + |
| Optimum temperature (°C) | 70 | 65–70 | 55 | 57 | 60–65 | 55–65 | 65 | 60–65 |
| pH range for growth | 5.8–8.7 | 6.0–8.8 | 7.5–8.5 | 7.0–8.5 | 6.0–7.5 | 6.0–8.2 | 5.0–8.0 | 6.0–8.0 |
| Max NaCl concentration for growth (w/v, %) | 2 | n. d. | 3 | 3 | 2 | 1 | 3.5 | 1 |
| Growth with formate | – | – | + | – | + | + | – | + |
| Autotrophic | – | + | + | + | + | + | + | + |
| Dependent growth factors: | ||||||||
| Acetate | – | – | – | n. d. | – | – | – | – |
| Yeast extract | + | – | – | n. d. | – | – | – | – |
| Coenzyme M | n. d. | – | + | + | – | n. d. | – | – |
| Peptone | + | – | – | n. d. | – | n. d. | – | – |
| General Features | M. tengchongensis DL9LZB001 |
|---|---|
| Size (bp) | 1,674,288 |
| GC content (%) | 48.39 |
| Protein coding genes | 1802 |
| Genomic islands | 6 |
| CRISPR 1 sequences | 4 |
| Genes assigned to COG 2 categories | 1487 |
| tRNA | 36 |
| 5S rRNA | 3 |
| 16S rRNA | 2 |
| 23S rRNA | 2 |
| sRNA | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, Z.; Ding, J.; Wang, H.; Wan, J.; Chen, Y.; Liang, L.; Yu, T.; Wang, Y.; Wang, F. Isolation of a Novel Thermophilic Methanogen and the Evolutionary History of the Class Methanobacteria. Biology 2022, 11, 1514. https://doi.org/10.3390/biology11101514
Lv Z, Ding J, Wang H, Wan J, Chen Y, Liang L, Yu T, Wang Y, Wang F. Isolation of a Novel Thermophilic Methanogen and the Evolutionary History of the Class Methanobacteria. Biology. 2022; 11(10):1514. https://doi.org/10.3390/biology11101514
Chicago/Turabian StyleLv, Zhenbo, Jiaxin Ding, Heng Wang, Jiaxin Wan, Yifan Chen, Lewen Liang, Tiantian Yu, Yinzhao Wang, and Fengping Wang. 2022. "Isolation of a Novel Thermophilic Methanogen and the Evolutionary History of the Class Methanobacteria" Biology 11, no. 10: 1514. https://doi.org/10.3390/biology11101514
APA StyleLv, Z., Ding, J., Wang, H., Wan, J., Chen, Y., Liang, L., Yu, T., Wang, Y., & Wang, F. (2022). Isolation of a Novel Thermophilic Methanogen and the Evolutionary History of the Class Methanobacteria. Biology, 11(10), 1514. https://doi.org/10.3390/biology11101514